

Abstract
The estrogen receptors, ERα and ERβ, are ligand-regulated transcription factors that control gene expression programs in target tissues. The molecular events underlying estrogen action involve minimally two steps, hormone binding to the ER ligand-binding domain followed by coactivator recruitment to the ER·ligand complex; this ligand·receptor·coactivator triple complex then alters gene expression. Conceptually, the potency of an estrogen in activating a cellular response should reflect the affinities that characterize both steps involved in the assembly of the active ligand·receptor·coactivator complex. Thus, to better understand the molecular basis of estrogen potency, we developed a completely in vitro system (using radiometric and time-resolved FRET assays) to quantify independently three parameters: (a) the affinity of ligand binding to ER, (b) the affinity of coactivator binding to the ER·ligand complex, and (c) the potency of ligand recruitment of coactivator. We used this system to characterize the binding and potency of 12 estrogens with both ERα and ERβ. Some ligands showed good correlations between ligand binding affinity, coactivator binding affinity, and coactivator recruitment potency with both ERs, whereas others showed correlations with only one ER subtype or displayed discordant coactivator recruitment potencies. When ligands with low receptor binding affinity but high coactivator recruitment potencies to ERβ were evaluated in cell-based assays, elevation of cellular coactivator levels significantly and selectively improved their potency. Collectively, our results indicate that some low affinity estrogens may elicit greater cellular responses in those target cells that express higher levels of specific coactivators capable of binding to their ER complexes with high affinity.
Keywords: Coactivator Transcription, Estrogen, Fluorescence Resonance Energy Transfer (FRET), Hormones, Receptor Structure-Function, Cellular Potency, Coactivator Binding Affinity, Estrogen Ligands, Estrogen Receptor Subtypes, Ligand Binding Affinity
Introduction
Estrogens of diverse structure are used for many clinical needs and various health benefits. Ethynylestradiol is used for fertility regulation (1, 2), and the drug Premarin (3, 4), which contains a mixture of 10 structurally different equine estrogens, is widely prescribed for menopausal hormone replacement therapy. Non-steroidal estrogens, such as diethylstilbestrol, are used to suppress androgen production in the treatment of prostate cancer (5, 6), and soy isoflavone extracts that contain the phytoestrogen genistein are consumed by older women as estrogen supplement for its possible beneficial effect in relieving some of the post-menopausal symptoms (7, 8). In addition, estrogens selective for one of the estrogen receptor (ER)2 subtypes, ERβ, are under active investigation for the management of breast, prostate, and colon cancers and specific cases of cardiovascular and central nervous system disorders (9, 10). Little is known, however, about what structural features of a particular estrogen and what molecular interactions it undergoes during its course of action contribute to its potency and to its selectivity of action through the two ER subtypes, ERα and ERβ.
ERα and ERβ are members of the nuclear receptor (NR) family of ligand-regulated transcription factors that mediate the biological effects of estrogens (11). Upon binding to an estrogen agonist, such as estradiol (E2), ER undergoes a conformational change that positions the C-terminal helix (H12) to complete formation of a hydrophobic groove that is a docking site for coactivator proteins, such as SRC3 (steroid receptor coactivator 3). By forming a ligand·receptor·coactivator complex, coactivator binding provides critical structural and functional links between ER and the transcriptional machinery that mediates the cellular (and ultimately the physiological) effects of estrogens (Fig. 1A) (12).
FIGURE 1.

Functionally relevant interactions of estrogens with the ERs and SRCs. A, interactions in a cellular context. B, in vitro systems for analysis of RLA, RCA, and RRP.
The biological potency of a hormonal agent, such as an estrogen, obviously depends on both extrinsic and intrinsic factors. Extrinsic factors, such as adsorption, distribution, metabolism, and elimination, are difficult to predict in advance of in vivo studies and continue to pose challenges in drug development (13, 14). On the other hand, intrinsic factors, such as ligand binding and coactivator recruitment are more closely associated with a cellular response. Conceptually, as schematized in Fig. 1A, the potency of an estrogen in activating a cellular response should reflect on the affinities that characterize both steps involved in the assembly of the active ligand·receptor·coactivator triple complex: the affinity with which the ligand binds to the ER (Step 1) and the affinity with which the coactivator binds to the ER·ligand complex (Step 2) rather than simply the ligand binding affinity (Step 1) (Fig. 1A). In general, however, potency has been correlated only with ligand binding affinity, although it is appreciated that ligands with similar binding affinity but different structures stabilize different conformations of ER. These differences, in principle, could affect the affinity with which coactivators bind to the ER·ligand complex and thereby affect ligand potency.
We have sought to better understand how these two intracellular steps contribute to ligand potency by replicating these interactions in a completely in vitro system, where they can be studied quantitatively (schematized in Fig. 1B). The ligand binding affinity to ER (Step 1) was quantified by competitive radiometric binding assays, as is commonly done. To quantify coactivator binding affinity for the ER·ligand complex (Step 2), we followed the titration of coactivator to the preformed ER·ligand complex using our recently developed time-resolved fluorescence resonance energy transfer (tr-FRET) assay (15). As a surrogate measure of ligand potency, we determined by tr-FRET the progress of coactivator recruitment to ER during a ligand titration, a process that combines both steps, ligand binding by ER and coactivator recruitment by the ER·ligand complex, mimicking in vitro the process that takes place in target cells (cf. Fig. 1A). Because these three interactions are determined relative to 17β-estradiol (E2), we have termed them, respectively, relative ligand binding affinity (RLA), relative coactivator binding affinity (RCA), and relative recruitment potency (RRP).
In undertaking this study, we selected 12 estrogens in four sets (Fig. 2), all of which are considered nominally to be estrogen agonists: Group 1, physiological estrogens (17β-estradiol (17β-E2), estrone (E1), and estriol (E3) as well as the nominally “inactive estrogen” 17α-estradiol (17α-E2) and the contraceptive drug 17α-ethynylestradiol (EE2); Group 2, non-steroidal estrogens (diethylstilbestrol (DES) and dimethylstilbestrol (DMS), meso-hexestrol (meso-Hex), and dl-hexestrol (dl-Hex); Group 3, equine estrogens (equilin (Equ) and equilenin (Eqn); and Group 4, a soy phytoestrogen (genistein (Gen)). We examined how each ligand interacted with ERα and ERβ and the manner in which these ER·ligand complexes recruited the coactivator SRC3.
FIGURE 2.

Structures of the four classes of estrogens used in this study.
We find that E2 and EE2 showed good correlations between RLA, RCA, and RRP with both ERα and ERβ, whereas other estrogens showed a correlation among these parameters with only one ER subtype. We also found ligands having significant discordance between their ligand binding affinities and their coactivator recruitment activities, in some cases due to only minor differences in stereochemistry and substitution. To compare how the coactivator recruitment pattern of a ligand, measured in vitro, relates to ligand potency in a cellular response, we measured the potencies of some candidate estrogens in cell-based reporter gene assays; we selected ligands that were avid in recruiting SRC3 through ERβ and measured them in the presence and absence of co-transfected SRC3. Although quantitatively different, the potency measurements in cells showed the same general trends as found in the in vitro assays, and the addition of SRC3 amplified the potency of these compounds through ERβ. Thus, our data provide evidence that the biological activity of different estrogens derives not only from their binding affinity for the ERs but also from the affinity that their ligand·receptor complexes have for particular coactivators. It further indicates that some weakly binding estrogens might have their potency amplified in those target cells that express a particular combination of ER subtypes and coactivators.
EXPERIMENTAL PROCEDURES
17β-E2, 17α-E2, E1, E3, DES, EE2, meso-Hex, Equ, Eqn, and Gen were purchased from Sigma. DMS and dl-Hex were synthesized in our laboratory (16). The thiol-reactive fluorophore, 5-iodoacetamido fluorescein, and terbium-labeled streptavidin were obtained from Molecular Probes/Invitrogen (Eugene, OR). Thiol-reactive biotin derivative (MAL-dPEG4-biotin) was from Quanta BioDesign (Powell, OH). [3H]17β-E2 (specific activity, 118 Ci/mmol (4366 GBq/mmol)) was purchased from PerkinElmer Life Sciences.
Protein Expression, Purification, and Labeling of ERα-417, ERβ-369, and SRC3
The bacterial expression plasmids encoding His6 fusion proteins of ER ligand binding domains (LBDs), ERα-417 (amino acids 304–554) and ERβ-369 (amino acids 256–505), each with a single reactive cysteine at Cys417 or Cys369, respectively, and the nuclear receptor interaction domain of human SRC3 encompassing three NR boxes (amino acids 627–829) have been described previously (15, 17). ER LBDs and the SRC3 fragment were labeled with MAL-dPEG4-biotin and 5-iodoacetamido fluorescein, respectively (15).
Radiometric Competitive Binding Assay to Determine RLA
The dissociation constants (Kd) of estradiol binding to the ER LBDs were measured by saturation binding with [3H]17β-E2 and Scatchard plot analyses, as described previously, and were 0.2 and 0.5 nm for ERα and ERβ LBD, respectively (18). The RLAs of the remaining estrogens were determined by a competitive binding assay using 0.5 nm ERα or ERβ in the presence of 2 nm [3H]17β-E2 and various concentrations of unlabeled 17β-E2 and other estrogens. The concentrations of unlabeled 17β-E2 and other compounds required to reduce the binding of [3H]17β-E2 by 50% (IC50) were obtained from the displacement curves. The RLA values of the various estrogenic ligands were determined using Equation 1.
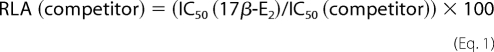 |
SRC3 Titration Assay to Determine RCA
Fluorescein-labeled SRC3 fragment (Fl-SRC3) was serially diluted at 3 times the required final concentration in buffer A (50 mm Tris (pH 7.9) containing 10% glycerol, 0.01% Nonidet P-40, 50 mm KCl, 2 mm β-mercaptoethanol, 2% dimethylformamide, and 0.3 mg/ml ovalbumin). 3× premixture of streptavidin-terbium (SA-Tb) and biotinylated ERα or ERβ LBD were prepared in buffer A. Ligand dilutions (3×) was made in buffer B (20 mm Tris (pH 7.9) and 100 mm NaCl containing 2% dimethylformamide). A 5-μl aliquot of SA-Tb-ERα or SA-Tb-ERβ mixture and a 5-μl aliquot of Fl-SRC3 were added first to the wells of a 96-well black microplate (Molecular Devices, Sunnyvale, CA), followed by the addition of 5-μl volumes of ligands. The final assay concentrations were 0.25 nm SA-Tb, 1 nm ERα LBD or 1 nm ERβ LBD, 25 μm ligands (except 1 μm for DES; see below), and the indicated concentrations of Fl-SRC3. Nonspecific binding was determined by parallel incubations that contained all of the components but without biotinylated ER LBD, which was used to correct for diffusion-enhanced FRET. The plates were mixed, protected from light, and incubated for 1 h at room temperature before being measured for tr-FRET. The FRET values obtained from the test samples (total FRET) were subtracted from the corresponding background diffusion-enhanced FRET (control), and the resulting specific FRET values were plotted against the log Fl-SRC3 concentrations. The concentration of SRC3 that gave 50% (EC50) of maximal binding in the presence of 17β-E2 and other ligands was obtained from the respective binding curves for both ERα and ERβ LBD. The RCA values of SRC3 for ERα or ERβ complexed with different ligands were determined using Equation 2.
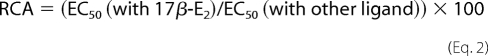 |
In addition, from the coactivator titration curves, the efficacy (i.e. the maximal FRET at saturating concentrations of SRC3) with which each ligand·ERα or ligand·ERβ complex recruited SRC3 relative to that of the 17β-E2·ER complex (relative recruitment efficacy (RRE)) was also determined by Equation 3.
 |
In preliminary SRC3 titration experiments, different ligand concentrations were tested with ERα to ensure that the resulting binding curves would show a clear saturation, such that measured EC50 values would be consistent between experiments. A concentration of 1 μm was sufficient for all ligands except E1, Equ, and Eqn, which required a concentration of 25 μm to give complete saturations. Because no significant differences were observed in the maximal binding (Bmax) and EC50 values of SRC3 binding when the titration assays were performed at 1 or 25 μm ligand concentrations, we used a ligand concentration of 25 μm for both the receptors in all but one experiment. The 25 μm concentration DES, however, displayed significant autofluorescence that interfered with FRET measurements. At 1 μm, the SRC3 binding curve to the ER·DES complexes gave complete saturation curves with minimal autofluorescence. Thus, in experiments with DES, the reference standard 17β-E2 was also used at 1 μm for both ERs (Fig. 4, compare E and F).
FIGURE 4.

Determination of affinity of SRC3 for ERα and ERβ LBDs complexed with various other ligands. The control-corrected SRC3 titration curves of specific tr-FRET values for ERα LBD (4; A) and ERβ LBD (4; B) in the presence of 25 μm of 17β-E2, Equ, Eqn, Gen, and 17α-E2; with 25 μm 17β-E2, DMS, meso-Hex, and dl-Hex for ERα (C) and ERβ (D); and with 1 μm 17β-E2 and DES for ERα (E) and ERβ (F). Data were analyzed as in Fig. 3. Assays were repeated three times with replicates, and the results from a representative experiment are shown. From the titration curves, the concentrations of SRC3 at 50% of maximal recruitment (EC50) in the presence of each of the ligands for both ERα and ERβ from the three different experiments were obtained and are listed as mean EC50 ± S.D. in Table 2. These values are also expressed relative to 17β-E2 as RCA values. The efficacies with which each ligand recruited SRC3 relative to 17β-E2 (RREs) to each ER are also listed in Table 2. Error bars, S.D.
Ligand Titration Assay to Determine RRP
A 3× concentration solution of the following reaction components was individually made: premixture of SA-Tb-ERα or SA-Tb-ERβ in buffer A, Fl-SRC3 in buffer A, and the indicated ligand dilutions in buffer B. A 5-μl aliquot of the SA-Tb-LBD premixture and a 5-μl aliquot of Fl-SRC3 were added first, followed by the addition of the indicated serially diluted ligands, and incubated for 1 h in the dark before measuring tr-FRET. Control wells had all of the components except biotinylated ER LBD. The final reaction concentrations were 0.25 nm SA-Tb, 1 nm ERα LBD or 1 nm ERβ LBD, 100 nm Fl-SRC3, and the indicated ligand concentrations.
The concentrations of 17β-E2 and other ligands required to give 50% (EC50) of SRC3 recruitment were obtained from each of the binding curves from both ERα and ERβ LBD, and the RRP values for other ligands were calculated using Equation 4.
tr-FRET Measurements
tr-FRET was measured on a Wallac Victor II plate reader (Molecular Devices, Sunnyvale, CA). SA-Tb, the donor, was excited at 340/80 nm. Emissions from the donor (D) and the acceptor fluorescein (A) were monitored at 495/20 and 520/25 nm, respectively, with a 100-μs delay. tr-FRET is expressed as A/D × 1000 (19).
ERE-Luciferase Reporter Gene Assay
U2OS cells were grown in MEM containing phenol red, 5% calf serum, and 100 μg/ml penicillin/streptomycin. Cells cultured at least 6 days in phenol red-free MEM supplemented with 5% charcoal-dextran stripped calf serum were seeded into 24-well plates (5 × 104 cells/well) and transfected with 0.5 μg of ERE-luciferase, 0.05 μg of full-length hERα or hERβ, and the internal control pCMV-β-gal (0.05 μg) in the presence and absence of 0.3 μg of pCMX-hSRC3 or pCMX empty vector (for experiments that did not require SRC3 expression) according to the published procedure (20). At 20 h post-transfection, cells were treated with increasing concentrations of E1, E3, Gen, or 17β-E2, and 24 h later, cells were harvested and assayed for luciferase and β-galactosidase activities.
Western Blot Analysis
Whole cell lysates (50 μg), prepared in radioimmune precipitation assay buffer from pCMX and pCMX-SRC3 transfected cells co-transfected with either ERα or ERβ, were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with antibodies to human SRC3 (sc-7216, Santa Cruz Biotechnology, Inc. (Santa Cruz, CA)) and β-actin (A5441, Sigma). Proteins were detected using the Pierce Fast Western blot Kit-West Femto (Thermo Scientific), according to the manufacturer's instructions.
RESULTS
Determination of RLA of ER Ligands
Competitive Radiometric Binding Assay
To probe ligand binding, Step 1 of the ER activation process (Fig. 1B), we measured the RLAs of the 12 estrogen agonists (Fig. 2) for both ER subtypes. The measured RLA values, listed in Table 1, show that meso-Hex, DES, and EE2 bind both ERα and ERβ with affinities higher than that of 17β-E2. DMS, on the other hand, bound preferentially with ERβ over ERα with respective RLAs of 237 ± 28 and 22.2 ± 4.3%. The RLAs of E1, E3, and Equ are much lower than that of 17β-E2 but are all comparable on both ERs. The ligands Eqn, Gen, and dl-Hex showed lower RLAs than 17β-E2 for both ERs, with some preferential binding to ERβ. The overall rank order of RLAs for the ERα LBD is meso-Hex > DES > EE2 > 17β-E2 > DMS > 17α-E2 > E3 > E1 = dl-Hex > Equ > Eqn > Gen, whereas the order for the ERβ LBD is meso-Hex > DES > DMS > EE2 > 17β-E2 > dl-Hex > E3 > Gen > 17α-E2 > E1 > Equ > Eqn.
TABLE 1.
RLAs of different estrogens to ERα and ERβ
The RLA of each ligand was calculated as the ratio of IC50 of 17β-E2 and IC50 of competitor, multiplied by 100. Each value is the mean ± S.D. of measurements obtained from three independent experiments. The ratios of RLA for ERβ/ERα are shown. A ratio greater than 1 indicates higher affinity for ERβ, and a ratio less than 1 indicates higher affinity for ERα. Numbers in brackets ([100] or [1]) are the base values on which the RLA values are calculated. A value of 100 represents a Kd = 0.2 nm for ERα and 0.5 nm for ERβ (18).
| Ligand name | RLA |
RLA β/α | |
|---|---|---|---|
| ERα | ERβ | ||
| 17β-E2 | [100] | [100] | [1.0] |
| E1 | 4.5 ± 0.7 | 4.0 ± 1.0 | 0.89 |
| E3 | 6.8 ± 0.7 | 11.6 ± 0.6 | 1.7 |
| EE2 | 194 ± 22 | 151 ± 27 | 0.78 |
| 17α-E2 | 13.7 ± 1.6 | 5.3 ± 1.8 | 0.39 |
| DES | 290 ± 56 | 278 ± 54 | 0.96 |
| DMS | 22.2 ± 4.3 | 237 ± 28 | 10.7 |
| Meso-Hex | 314 ± 44 | 697 ± 74 | 2.2 |
| dl-Hex | 4.4 ± 0.3 | 60.3 ± 5.3 | 13.7 |
| Equ | 2.3 ± 0.2 | 3.2 ± 0.3 | 1.4 |
| Eqn | 0.35 ± 0.04 | 2.0 ± 0.3 | 5.7 |
| Gen | 0.06 ± 0.01 | 7.4 ± 0.5 | 123 |
tr-FRET Assay
We recently reported the development of a sensitive tr-FRET based assay for evaluating, in a rapid and quantitative manner, the interaction between SRC3 and the LBDs of ERα and ERβ (15). Briefly, this assay is constituted of the human ERα LBD (amino acids 304–554) mutated to include a single reactive cysteine (at position 417) that was indirectly labeled with SA-Tb (fluorescent donor) via biotinylation and the nuclear receptor interaction domain (NRID) fragment of SRC3 labeled directly with fluorescein (fluorescent acceptor). Human ERβ LBD (amino acids 256–505) containing only one reactive cysteine at position 369, generated as described previously (17), was similarly labeled with SA-Tb. Excitation of Tb at 340 nm results in emission at 495 nm. If the fluorescein-labeled SRC3 and the SA-Tb-labeled ER are close to one another, as would be the case after recruitment by agonist-bound ER, the energy from the excited state of the Tb complex is transferred to fluorescein, which emits at 520 nm (15, 19). By monitoring the degree of FRET as the ratio of acceptor emission intensity (A, at 520 nm) to donor emission intensity (D, at 495 nm), expressed as A/D × 1000, we could measure quantitatively the ligand-dependent binding of SRC3 to the LBD of ERα or ERβ. A notable aspect of our coactivator assays is that we have used a ∼200-amino acid segment of SRC3 encompassing all three of the LXXLL motifs that constitute the NRID of the coactivator rather than smaller peptides containing only one LXXLL motif, as have typically been used in coactivator peptide recruitment assays (21–23).
Determination of RCA for Various ER·Ligand Complexes; tr-FRET SRC3 Titration Assay
We measured the affinity with which the SRC3 NRID fragment bound to conformations of ERα and ERβ stabilized by the binding of each of the 12 estrogens at full saturation (Fig. 1B, Step 2). For this, we performed a coactivator titration assay in which increasing concentrations of fluorescein-labeled SRC3 (fluorescence acceptor) were added to a fixed concentration of ERα or ERβ LBD (both at 1 nm), saturated with each of these agonist ligands (25 μm for all except DES, which was used at a concentrations of 1 μm; see “Experimental Procedures”). The ligand-specific interaction of SRC3 with each of the ER·estrogen complexes was measured by quantifying the increase in the tr-FRET signal as a function of increasing SRC3 concentration.
The results with the first set of ligands, 17β-E2, E1, E3, and EE2, show that there was a concentration-dependent and ligand-specific increase in the tr-FRET signal, reflecting the binding of SRC3 to the various ERα·ligand (Fig. 3A) and ERβ·ligand (Fig. 3B) complexes. To correct for background or diffusion-enhanced FRET, a fluorescence control, observed in the absence of biotinylated ER LBDs (controls in Fig. 3, A and B), was subtracted from the total FRET values; the resulting specific FRET values are shown in Fig. 3, C and D (and in all subsequent figures).
FIGURE 3.

Determination of affinity of SRC3 for ERα and ERβ LBDs complexed with 17β-E2, E1, E3, EE2, and TOT (coactivator titration assay to determine RCA). The tr-FRET assays involving terbium-labeled ER LBDs (fluorescence donor) and fluorescein-labeled NRID fragment of SRC3 (fluorescence acceptor) were performed in the presence of saturating concentration of various ligands, as detailed under “Experimental Procedures.” Total tr-FRET values obtained with increasing concentrations of fluorescein-labeled SRC3 with 1 nm Tb-labeled LBD of ERα (A) or ERβ (B) in the presence of 25 μm 17β-E2, E1, E3, EE2, or TOT or in their absence (Apo) and in the absence of ER LBDs (control) were plotted against the log SRC3 concentrations. Data were analyzed by nonlinear regression with an equation for the sigmoidal dose response (variable slope) in GraphPad Prism 4. The control values (representing the diffusion-enhanced FRET) in A and B were subtracted from the total FRET values, and the resulting specific tr-FRET values were analyzed as in A and B and are shown in C and D. Each assay in these sets was performed in replicates, and the results from a representative experiment are shown. The concentrations of SRC3 at 50% of maximal binding (EC50) to both ERs were determined (GraphPad Prism analyses) from the binding curves (specific FRET versus log SRC3) from the three independent experiments and are listed as mean EC50 ± S.D. in Table 2. These values are also expressed relative to 17β-E2 as RCA values. The maximal FRET units (efficacy) measured with SRC3 at saturation with ERα or ERβ bound to different ligands are expressed relative to 17β-E2·ERα or 17β-E2·ERβ complex (RREs), which were set to 100% (Table 2). Error bars, S.D.
We observed complete saturation at the highest SRC3 concentrations with both ER subtypes for all of the estrogens tested. The concentrations of SRC3 NRID at half-maximal binding (EC50), a measure of the apparent affinity of SRC3 for ERs in the presence of 17β-E2, were found to be 0.88 ± 0.05 and 0.76 ± 0.04 nm for ERα and ERβ LBDs, respectively. Each of these values was arbitrarily set to 100%, and the RCAs for both ERα and ERβ in the presence of other ligands were determined from their respective EC50 values and are shown in Table 2. These binding affinities of SRC3 to both ERα and ERβ were each monitored in three sets of experiments and were highly reproducible.
TABLE 2.
RCA and RRE of ERα and ERβ bound to various estrogenic ligands
The EC50 values were obtained from the coactivator titration curves of SRC3 (NRID fragment) recruitment by the LBDs of ERα and ERβ in the presence of 17β-E2 and other estrogens (Figs. 3 and 4). The RREs were determined as the ratio of maximal FRET of SRC3 recruitment with other ligand-ER complexes divided by maximal FRET with 17β-E2·ER complex multiplied by 100. The RCAs with different ligands were calculated as the ratio of EC50 of SRC3 recruitment by 17β-E2 divided by the EC50 in the presence of other ligands, multiplied by 100. The EC50 and RCA and RRE values are the mean ± S.D. of three independent experiments performed in replicates for each assay point. The β/α ratio indicates the preferential binding affinity of SRC3 for ERβ over ERα. Numbers in brackets ([100] or [1]) are the base values on which the RCA and RRE values are calculated. The actual EC50 values are given to the left.
| Ligand name | ERα |
ERβ |
RCA β/α | ||||
|---|---|---|---|---|---|---|---|
| EC50 | RCA | RRE | EC50 | RCA | RRE | ||
| nm | nm | ||||||
| 17β-E2 | 0.88 ± 0.05 | [100] | [100] | 0.76 ± 0.04 | [100] | [100] | [1.0] |
| E1 | 29.6 ± 1.5 | 3.00 ± 0.15 | 78.0 ± 2.8 | 2.7 ± 0.08 | 28.1 ± 1.8 | 83 ± 2.5 | 9.4 |
| E3 | 0.92 ± 0.07 | 95.7 ± 7.4 | 102 ± 3.4 | 0.51 ± 0.01 | 149 ± 6.7 | 103 ± 2.0 | 1.6 |
| EE2 | 0.87 ± 0.03 | 101 ± 8.2 | 100 ± 3.8 | 0.98 ± 0.06 | 77.6 ± 5.8 | 96 ± 1.6 | 0.77 |
| 17α-E2 | 1.70 ± 0.04 | 51.8 ± 4.6 | 98 ± 4.1 | 0.38 ± 0.05 | 200 ± 9.2 | 98 ± 2.5 | 9.2 |
| DES | 0.71 ± 0.03 | 104.2 ± 6.8 | 99 ± 1.7 | 0.97 ± 0.05 | 85.6 ± 6.8 | 99 ± 1.9 | 0.82 |
| DMS | 3.3 ± 0.23 | 26.7 ± 2.0 | 98 ± 5.7 | 0.79 ± 0.03 | 96.2 ± 4.9 | 117 ± 5.4 | 3.6 |
| meso-Hex | 0.90 ± 0.04 | 97.8 ± 6.8 | 93 ± 3.8 | 0.93 ± 0.05 | 81.7 ± 6.9 | 88 ± 5.5 | 0.83 |
| dl-Hex | 2.1 ± 0.1 | 41.9 ± 4.2 | 99 ± 3.8 | 0.84 ± 0.02 | 90.0 ± 8.8 | 102 ± 4.1 | 2.1 |
| Equ | 27.3 ± 1.0 | 3.2 ± 0.3 | 77 ± 3.2 | 1.6 ± 0.03 | 47.5 ± 4.2 | 105 ± 3.8 | 14.8 |
| Eqn | 62.8 ± 1.6 | 1.4 ± 0.1 | 72 ± 3.0 | 2.4 ± 0.18 | 31.7 ± 1.6 | 108 ± 4.8 | 22.6 |
| Gen | 9.5 ± 0.3 | 9.2 ± 0.1 | 89 ± 4.0 | 2.1 ± 0.18 | 36.2 ± 3.2 | 104 ± 4.0 | 4.0 |
The results show that RCAs of SRC3 for ERα and ERβ bound to E3 or EE2 are comparable with those of the ERα and ERβ complexes with 17β-E2, indicating that SRC3 has similar affinities for ERs bound to these ligands. The lower RCAs seen with E1-bound ERα and ERβ indicate that the SRC3 binds with lower affinity to E1-bound receptor complexes, probably a consequence of the differing hydrogen bonding capacity of a 17-keto- versus 17β-hydroxysteroid (see “Discussion”).
The base-line binding in Fig. 3C shows that ERα LBD has no affinity for SRC3 either in the unliganded state (Apo) or when bound by TOT. By contrast, the ERβ LBD displayed low but significant affinity for SRC3 in the ligand-free state (Fig. 3D), indicating that unliganded ERβ but not ERα exists in a partially active conformation. Such ligand-independent association between apo-ERβ and coactivators, which could be blocked by TOT (Fig. 3D), has been observed by others (21) and is probably a molecular manifestation of the greater basal activity of ERβ that is sometimes observed (24).
From Fig. 3, C and D, it is also evident that the maximal FRET values observed when E3- and EE2-bound ERs recruited SRC3 (i.e. the efficacy) to both ERs were nearly identical to those achieved with the 17β-E2 complexes, with RRE values of 102 ± 3.4 and 100 ± 3.8 for ERα and 103 ± 2.0 and 96 ± 1.6% for ERβ, respectively. The E1·ER complexes, however, gave maximum FRET values for SRC3 recruitment that were significantly and reproducibly less, with RRE values of 78 ± 2.8 and 83 ± 2.5% that of 17β-E2 with ERα and ERβ LBDs, respectively (Table 2). Because it seems unlikely that at saturation SRC3 would be recruited to fewer sites with E1·ERs than with the other ER·ligand complexes, the lower FRET value at saturation with E1·ER is most likely a manifestation of a difference in geometry or positioning with which the SRC3 is being bound to this complex. In a sense, it might be considered to represent a difference in “efficacy” with which this ligand is able to recruit SRC3.
Fig. 4, A and B, shows the SRC3 binding curves to ERα and ERβ LBDs complexed with Equ, Eqn, Gen, or 17α-E2. Although SRC3 bound to the ERβ complexes of Equ, Eqn, and Gen with 2–3-fold lower affinity than to the 17β-E2·ERβ complex, the RCAs for ERβ in the presence of these three ligands were significantly higher than those for ERα (RCAs of 1.5–9.0% for ERα versus 31–200% for ERβ). There is, in fact, no obvious relationship between the ERα or ERβ RCA and RLA values for these three compounds. With ERβ, the maximum FRET signals of SRC3 with these three ER ligand complexes were comparable with those observed with the ERβ·17β-E2 complexes, indicating that the ERβ·SRC3 complexes with these ligands are similar in conformation to that formed with 17β-E2. With ERα, however, Equ, Eqn, and Gen displayed RREs of about 77, 72, and 89% that of 17β-E2, respectively, indicative of differences in complex geometry that reflect their differing RCA values but not their RLA values. The estradiol epimers, 17β-E2 and 17α-E2, make an interesting pair in terms of coactivator binding. SRC3 binds with higher affinity to the ERα complex with the 17β-E2 but to the ERβ complex with 17α-E2. On the other hand, because they give comparable maximum FRET signals, both estradiol epimers appear to form similar ligand·receptor·coactivator complexes (Table 2).
The SRC3 binding curves to ERα and ERβ bound to DMS, meso-Hex, and dl-Hex (Fig. 4, C and D) show that, with the exception of meso-Hex on ERβ, all have comparable RRE values, indicating that they form similar complexes on both ER subtypes (meso-Hex on ERβ showed RRE of 88 ± 5.5% compared with 17β-E2). The RCAs for SRC3 binding to the ERβ complexes of these three compounds are comparable with that of the 17β-E2 complex, and the RCAs for ERα cover only a 4-fold range (Table 2). With DES, the SRC3 binding curves to both ERα and ERβ (Fig. 4, E and F) are very similar to those with 17β-E2, both in terms of RRE values and RCAs (Table 2). The rank order of RCA of SRC3 NRID for ERα in the presence of different estrogens is DES = EE2 = 17β-E2 = meso-Hex > E3 > 17α-E2 > dl-Hex > DMS > Gen > Equ > E1 > Eqn, and the order for ERβ is 17α-E2 > E3 > 17β-E2 > DMS > dl-Hex > DES > meso-Hex > EE2 > Equ > Gen > Eqn > E1.
Determination of RRP for Ligand Recruitment of SRC3 through ERα and ERβ; tr-FRET Ligand Titration Assay
As an in vitro measure of estrogen “potency,” we used an SRC3 recruitment assay with ligand titration. This experimental protocol is the same as that used in the CARLA or coactivator recruitment ligand assays originally described by Wahli and co-workers (25). The convenience of monitoring SRC3 recruitment by tr-FRET rather than by classical pull-down assays, however, makes our version of this assay sufficiently convenient that quantitative measurements can be obtained.
For this assay, a 100 nm concentration of fluorescein-labeled SRC3 was selected from the previous experiments because it provided a good compromise between a minimum nonspecific FRET and nearly maximal specific FRET with both ERα or ERβ (1 nm). Each assay set was performed with 17β-E2 as a reference standard, and the diffusion-enhanced FRET-corrected binding curves for these ligand titrations were determined. All of the compounds induced concentration-dependent, ligand-specific, and saturable SRC3 recruitment.
Although, in this assay, the maximum FRET value for most ligands was equivalent to that for 17β-E2, ligands having the lower RCA values, such as E1, Equ, Eqn, and Gen, gave maximum FRET values on the A/D × 1000 scale that were somewhat lower (∼70–90%) than for 17β-E2, particularly on ERα (data not shown). Unlike in the coactivator titration assay, in which the affinity and “efficacy” of SRC3 binding to the different ER·ligand complexes were measured and where a maximum FRET value less than that for 17β-E2 was interpreted as indicating a geometric difference in the conformation of the ER-coactivator complex, in this ligand titration assay, submaximal saturating FRET values indicate simply that 100 nm SRC3 is insufficient to bind fully to the ligand·ER complex, most notably for those complexes that have lower RCA values. We have noted this difference in our prior work, in which we studied ligand titrations with thyroid hormone receptor·retinoid X receptor heterodimers (19). Therefore, rather than using higher concentrations of SRC3 in assays with the low RCA compounds, we have simply normalized the FRET signal to span from 0 to 100%; these are shown in Figs. 5, A–F. The concentration of 17β-E2 that promoted 50% of SRC3 recruitment (EC50) was set 100% for both ERs (4.3 ± 0.3 nm for ERα and 8.0 ± 0.5 nm for ERβ), and the EC50 and RRP of each of the ligands are listed in Table 3.
FIGURE 5.

Determination of ligand potency for recruitment of SRC3 (ligand titration assay to determine RRP). In these assays, the recruitment of fluorescein-labeled SRC3 (100 nm) to the terbium-labeled LBDs of ERα or ERβ (1 nm) was evaluated as a function of increasing concentrations of different estrogenic compounds. The binding curves of control diffusion-enhanced FRET-corrected specific FRET values were normalized (with maximal tr-FRET values from 17β-E2 and different ligands set equal to 100%) and plotted against the log ligand concentrations, and the data were analyzed by nonlinear regression with an equation for the sigmoidal dose response (variable slope) in GraphPad Prism 4. Each experiment was repeated three times with replicates, and the results of a representative experiment are shown. The concentrations of each ligand that promoted 50% of maximal SRC3 recruitment (EC50) were obtained from the binding curves of three experiments and are expressed as mean EC50 ± S.D. in Table 3, and these values are also expressed relative to 17β-E2 as RRP values. Binding curves with various ligands are as follows: in the presence of 17β-E2, EE2, DMS, DES, and meso-Hex with ERα (A) and ERβ (B); in the presence of 17β-E2, E1, E3, Equ, Eqn, and Gen with ERα (C) and ERβ (D); and in the presence of 17β-E2, 17α-E2, and dl-Hex with ERα (E) and ERβ (F). Error bars, S.D.
TABLE 3.
RRP of various estrogens for SRC3 recruitment to ERα and ERβ
From the ligand titration curves (Fig. 5), the concentrations of 17β-E2 and other ligands to promote SRC3 recruitment by 50% were determined. The RRPs of various estrogenic ligands were calculated as the ratio of EC50 of 17β-E2 divided by EC50 of other ligands, multiplied by 100. Each experiment was repeated three times with replicates, and the indicated EC50 and RRP values represent the mean ± S.D. of six measurements. The Hill coefficients were determined by curve fitting using nonlinear regression with an equation for the sigmoidal dose response (variable slope) in GraphPad Prism 4. The β/α ratio indicates preferential ligand recruitment potency for ERβ over ERα. Numbers in brackets ([100] or [1]) are the base values on which the RRP values are calculated. The actual EC50 values are given to the left.
| Ligand name | ERα |
ERβ |
RRP β/α | ||||
|---|---|---|---|---|---|---|---|
| EC50 | RRP | Hill coefficient | EC50 | RRP | Hill coefficient | ||
| nm | nm | ||||||
| 17β-E2 | 4.3 ± 0.3 | [100] | 1.98 ± 0.03 | 8.0 ± 0.5 | [100] | 1.90 ± 0.02 | [1.0] |
| E1 | 74.7 ± 3.7 | 5.8 ± 0.5 | 1.0 ± 0.02 | 36.5 ± 4.0 | 21.9 ± 2.1 | 1.40 ± 0.01 | 3.8 |
| E3 | 13.8 ± 0.8 | 31.2 ± 0.8 | 1.3 ± 0.02 | 5.8 ± 0.3 | 137.9 ± 8.3 | 1.70 ± 0.03 | 4.4 |
| EE2 | 4.3 ± 0.4 | 100 ± 8.0 | 1.98 ± 0.02 | 9.5 ± 0.4 | 84.2 ± 6.2 | 1.80 ± 0.01 | 8.7 |
| 17α-E2 | 21.3 ± 1.0 | 20.2 ± 1.5 | 1.1 ± 0.01 | 27.6 ± 1.5 | 29.0 ± 1.1 | 1.20 ± 0.01 | 1.4 |
| DES | 30.7 ± 1.1 | 14.0 ± 0.7 | 2.0 ± 0.02 | 138 ± 12 | 5.8 ± 0.4 | 1.90 ± 0.02 | 0.41 |
| DMS | 10.4 ± 1.0 | 41.3 ± 1.1 | 1.6 ± 0.01 | 9.0 ± 0.7 | 88.9 ± 5.1 | 2.0 ± 0.02 | 2.2 |
| Meso-Hex | 13.2 ± 0.9 | 32.6 ± 1.6 | 2.0 ± 0.02 | 109.6 ± 8.0 | 7.3 ± 0.3 | 1.90 ± 0.01 | 0.22 |
| dl-Hex | 103.5 ± 6.1 | 4.20 ± 0.31 | 0.80 ± 0.01 | 101.5 ± 8.6 | 7.9 ± 0.6 | 1.80 ± 0.01 | 1.9 |
| Equ | 82 ± 6.2 | 5.2 ± 0.3 | 0.90 ± 0.01 | 17.7 ± 0.8 | 45.2 ± 3.1 | 1.10 ± 0.01 | 8.7 |
| Eqn | 398 ± 19 | 1.1 ± 0.1 | 1.10 ± 0.01 | 31.8 ± 1.7 | 25.2 ± 1.5 | 1.20 ± 0.01 | 22.9 |
| Gen | 1201 ± 62 | 0.36 ± 0.02 | 1.2 ± 0.02 | 10.1 ± 0.8 | 79.2 ± 4.6 | 1.50 ± 0.02 | 220 |
The measured RRPs of the first set of ligands, EE2, DES, DMS, and meso-Hex (Fig. 5, A and B), indicate that the RRP of EE2 is equivalent to that of 17β-E2 in recruiting SRC3 to both ERs, whereas DES and meso-Hex, on the other hand, display weaker RRPs (5–30%). DMS, on the other hand, was found to be more potent on ERβ than on ERα, with RRPs of 90 and 42%, respectively. Notably, ligand ranking by RRP is quite different from ranking by RLA because DES and meso-Hex are the highest affinity ligands (RLAs; Table 1).
Shown in Fig. 5, C and D, are the coactivator recruitment curves with ERα and ERβ for titrations with E1, E3, Equ, Eqn, and Gen. All ligands in this set are more potent on ERβ than ERα, with respective ERβ versus ERα RRPs of 22 versus 6%, 138 versus 31%, 45 versus 5.0%, 25 versus 1%, and 79 versus 0.36%, which generally reflects the higher binding affinity of these ligands for ERβ (Table 1); the 220-fold ERβ potency preference of Gen is particularly striking. Of note, however, E3 shows a 1.4-fold greater potency in recruiting SRC3 to ERβ than does 17β-E2, despite having a RLA of only 11.6%, most likely reflecting its very high RCA value for ERβ (RCA = 149). The titration curves in Fig. 5, E and F, show that the SRC3 recruitment potencies of 17α-E2 and dl-Hex are lower with both ERα and ERβ, with RRP values of 20 versus 4% and 29 versus 8.0%, respectively. The rank order of ligand potency for ERα based on SRC3 recruitment is 17β-E2 = EE2 > DMS = meso-Hex > E3 > 17α-E2 > DES > E1 > Equ > dl-Hex > Eqn > Gen, and with ERβ, the order is E3 > 17β-E2 > DMS > EE2 > Gen > Equ > 17α-E2 > Eqn > E1 > dl-Hex > meso-Hex > DES (Table 3).
In the ligand titration assays for measurement of RRP values, we noted that the binding curves were steeper than was typical for simple, non-cooperative binding interactions, and, in fact, we determined the EC50 values by fitting the curves with variable Hill coefficients, which are listed in Table 3. It is notable that with both ERα and ERβ, Hill coefficients varied from 0.8 to 2.0, with many values being above 1.5 (see “Discussion”).
Effect of Co-expression of SRC3 on Ligand Potency and Efficacy, ERα and ERβ Transient Transfection Assays, and Determination of Relative Cellular Potency (RCP)
To examine how the estrogen potencies measured from the in vitro tr-FRET SRC3 recruitment assay compare with the potencies with which estrogens elicit a cellular response, we evaluated the transactivation activities of some candidate ligands in reporter gene assays performed in the presence and absence of co-transfected SRC3. For this, we selected E1, E3, and Gen (along with 17β-E2 as the reference control) because the three ligands have relatively low RLAs for both ERs but have moderate to high SRC3 recruitment activities, particularly with ERβ, both in terms of RCA and RRP values. U2OS cells were used for this purpose because of their low endogenous SRC3 levels (26); also, when these ER-negative cells are transiently transfected with an ERE-driven reporter and ERα and ERβ expression plasmids, they show low basal reporter gene activity but respond well to 17β-E2. After transfection with 0.05 μg of hERα or hERβ expression plasmid, the U2OS cells had comparable levels of ERα or ERβ proteins, about 1500 receptors/cell, as determined by ligand equilibrium binding assays (data not shown). Transfection with 0.3 μg of pCMV-hSRC3 resulted in a 5–6-fold increase in SRC3 levels in these cells (Fig. 6I).
FIGURE 6.

Transient transfection assay. A–H, dose response to 17β-E2, E1, E3, and Gen of ERE-luciferase activity in U2OS cells transiently transfected with expression plasmids for ERα or ERβ with (solid rectangle) and without (solid circle) co-transfected SRC3 was determined as described under “Experimental Procedures.” The luciferase activity obtained at the maximal dose of 17β-E2 with ERα or ERβ in the presence of co-transfected SRC3 was set at 100%. Data were analyzed by nonlinear regression with an equation for the sigmoidal dose response (variable slope) in GraphPad Prism 4. Each data point represents the mean ± S.D. (error bars) of three experiments performed in replicates. The table below each panel shows the concentrations of the ligand (nm) at 50% of maximal response (EC50) in mediating ERE-luciferase either in the presence or absence of co-transfected SRC3 plasmid. The EC50 response of ERα or ERβ to 17β-E2 in the presence of SRC3 was set equal to 100%, and the RCPs for each ligand calculated from their respective EC50 values are shown. The RLA, RCA, and RRP values measured for each of the estrogens tested (obtained from Tables 1–3) are shown in each panel. I, Western blot analysis. Whole cell lysates (50 μg) prepared from SRC3-untransfected (lane 1), SRC3-transfected plus ERα-cotransfected (lane 2), or ERβ-cotransfected (lane 3) U2OS cells were run in a SDS-PAGE and transferred to nitrocellulose membrane. Membrane was probed with antibodies against SRC3 and β-actin as described under “Experimental Procedures.”
In all cases, the addition of SRC3 increased the maximum reporter gene response about 2-fold, and the response to different ligands was expressed as a percentage of the activity observed at the maximum 17β-E2 concentration with ERα or ERβ in the presence of added SRC3 (Fig. 6). The concentrations of 17β-E2 required to give 50% of maximal activation of ERα- or ERβ-dependent luciferase activity (i.e. the EC50) in the presence of SRC3 was set equal to 100%, and the RCP values of the other ligands in the presence and absence of SRC3 were calculated from the corresponding EC50 values; these are shown as percentage values in the tables below each panel of Fig. 6. Overall, the results indicate that the enhanced RRPs of E1, E3, and Gen for ERβ, measured from the in vitro coactivator recruitment assays (RRPs of 22, 138, and 79%, respectively), are reflected in RCP measured from the reporter gene assays when SRC3 levels are elevated but only to some degree (RCPs of 10, 26, and 4.8%, respectively; Fig. 6, D, F, and H), possibly due to the involvement of other extrinsic factors influencing the cell-based response (see “Discussion”). Nevertheless, there are significant trends in the cell-based potency measurements (RCP values) that do reflect the differing RRP values of these compounds.
We found that for these ligands, SRC3 co-transfection caused a greater improvement in the RCPs with ERβ than with ERα, consistent with the higher RCA of SRC3 for the ERβ complexes with these ligands. Thus, the addition of SRC3 levels increased the RCP of E1, E3, and Gen through ERβ by 2.6-, 3.3-, and 4.0-fold, respectively, whereas when these ligands acted through ERα, the addition of SRC3 caused either no increase or only a 1.5- and 1.3-fold increase in RCP, respectively. As a consequence, the potency selectively of these three ligands for ERβ over ERα increased significantly when SRC3 levels were increased, improving from 1.6-, 1.3-, and 15-fold for E1, E3 and Gen, respectively, in the absence of additional SRC3, to 4.9-, 2.9-, and 48-fold with additional SRC3. Taken together, the results indicate that the parameters of RCA and RRP measured from in vitro coactivator recruitment assays show better correlation to potency measurements from cell-based assays when the cellular levels of SRC3 are higher.
DISCUSSION
Different estrogen preparations, both natural and synthetic, are currently used as pharmaceuticals, but despite these widespread uses, there are no comprehensive studies that have examined how the structure and receptor binding affinity of an estrogen affect the binding affinity of coactivators to the ligand complex with the two ER subtypes and how these two distinct binding affinities contribute to the transactivation properties of ERs and ultimately to the potency of various estrogens acting through ERα and ERβ. For this reason, we developed an in vitro system through which we can quantify the parameters of ligand binding affinity for ERα and ERβ (RLA values) and coactivator binding affinity to complexes of these ERs (RCA values), and we studied these parameters with 12 representative estrogen agonists from four classes. We then examined how these parameters might be related to ligand potency, measured both in vitro (RRP values) and in cells (RCA values). Key to this study was the use of an optimized tr-FRET assay involving engineered ER LBDs mutated to contain a single reactive cysteine for fluorophore labeling and the use of the complete NRID fragment of SRC3 containing three LXXLL motifs (15). This approach provided a reliable method to quantify receptor-specific ER-coactivator interaction in a highly sensitive and reproducible manner. Unlike previous studies of ER-coactivator interactions, which used a synthetic peptide containing a single LXXLL motif as the coactivator component (21–23), our use of a complete NRID fragment of SRC3 in the present study resulted in binding curves displaying clear saturation for all of the estrogens and revealed considerable variations in affinities. The ERα and ERβ LBDs and SRC3 NRID peptide fragments used in the present study have been extensively characterized in a number of our previous studies that demonstrated that these fragments displayed functional characteristics very similar to full-length ERs in terms of ligand binding and ligand-specific conformational and receptor dimerization changes as well as agonist/antagonist-regulated coactivator recruitment profiles (15, 17, 18, 27–29). The subnanomolar sensitivity with which our tr-FRET assay measures SRC3-ER interaction, together with nearly stoichiometric binding of SRC3 to ER at equilibrium with an EC50 of 0.88 nm for ERα and 0.76 nm for ERβ in the coactivator titration assay, supports the fact that the ER LBDs and SRC3 NRID fragments that we have used retain most if not all of the receptor-coactivator interaction properties of full-length ER and SRC3 proteins.
Relationships Between RLA, RCA, RRP, and RCA Values with Individual Ligands and with the Two ER Subtypes
We looked very hard for correlations among the RLA, RCA, and RRP values from the in vitro assays and RCP from the cellular assay, but there were no statistically robust global relationships among these three parameters, either pairwise or in combination. Nevertheless, we found for some individual ligands good correlations among all three parameters, whereas with others there were correlations between RLA and RCA but not with RRP. There were also examples of ligands showing unusual relationships, with some ligands having low RLAs being able to recruit SRC3 efficiently; this gives them RCA and RRP values higher than expected based on ligand binding affinity alone. Importantly, the two ER subtypes differ substantially in these relationships.
Among the high affinity ligands like 17β-E2, only EE2 on ERα and EE2 and DMS on ERβ showed good correlations between RLA, RCA, and RRP, with all three parameters being in the higher range. Although some of these ligands bound to both ERs with significantly higher RLA values than did 17β-E2 (DMS on ERβ and EE2, meso-Hex, and DES on both ERs), their ligand-induced RCA and RRP values were not higher than those of 17β-E2. The ligands meso-Hex and DES bound to both ERs with very high affinity (RLA of 300–700%), and the resulting ligand·ER complexes had high RCA values (80–100%), indicating that SRC3 bound to their complexes with high affinity; curiously, however, their measured RRPs were significantly lower (33 and 14% for ERα and 6 and 7% for ERβ, respectively).
Among ligands with low affinity for ERα, DMS, E1, Equ, Eqn, and Gen showed a good correlation between RLA, RCA, and RRP, all showing relatively low values for all three indices. On the other hand, although the ligands in this group had relatively low RLA values, their RCA and RRP values with ERβ were higher than their RLAs (Tables 1–3). With ERα, these ligands did not show such relationships. The epimeric estrogen, 17α-E2, had RLA values of 14 and 5% for ERα and ERβ, but the RCAs and RRPs were 52 and 200% and 20 and 29%, respectively. Most unusual was E3, which has modest affinity for ERβ (RLA 12%), but the E3 complex with ERβ has very high affinity for SRC3 (RCA 149%), and E3 is potent in recruitment assays (RRP 138%); nothing is as striking for E3 with ERα. Previously, we have shown that the binding of coactivator to the ERα·ligand and thyroid hormone receptor·thyroid hormone complex stabilizes the ligand-receptor interaction by decreasing the dissociation rate of ligand from the receptor (19, 30). Thus, it is possible that in the context of the ligand·ER·SRC3 ternary complex, SRC3 preferentially strengthens the binding of ligands like E3 to ERβ, which further contributes to increased SRC3-ER interaction. Previous studies reporting the RLA values of 17α-E2, E3, DES, meso-Hex, and GEN for binding to full-length human ERα and rat ERβ expressed in insect cells (31, 32) agree well with our measured RLA values for these estrogens; however, somewhat higher RBAs for 17α-E2, E1, Equ, and Eqn for binding to both human full-length ERs made in insect cells were reported in another study (33).
Collectively, these results suggest that the relationships between RLA, RCA, the efficacy with which each ligand·receptor complex recruited coactivator (RRE), and ligand recruitment of coactivator (RRP) are both ligand- and receptor subtype-specific, with some estrogens showing good correlations, whereas others with either high or low RLAs demonstrate discordant coactivator recruitment activities with both ERs (see “Estrogen Receptor Structural Correlations” for further discussion). Such differences also indicate that both ERs undergo distinct conformational changes upon binding to different estrogens but also that the conformational changes that each ER undergoes upon binding to a given estrogen could be different. Indeed, it has recently been shown that the regional dynamics of ERα and ERβ LBDs complexed with various ER modulators, including 17β-E2, DES, and GEN, are quite distinct between the two ER subtypes, as probed by hydrogen-deuterium exchange mass spectrometry (34, 35).
Building a Mechanistic Basis for Understanding the Potency and ER Subtype Selectivity of Different Estrogens in a Cellular Context
We carried out the transient transfection experiments with the candidate ligands, E1, E3, and Gen, to examine how the in vitro ligand potencies measured from the tr-FRET assays would relate to the potencies with which the estrogens mediate a cellular response. These ligands represent a class of estrogens with relatively low RLA values but with greater RCA and RRP values in the in vitro SRC3 binding and recruitment assays, particularly with ERβ. Because RCA and RRP values are the function of SRC3 binding to ligand-bound ER, we expected that the cellular potencies of these ligands acting through ERβ would be enhanced if the intracellular levels of SRC3 were elevated, and indeed they were; the increase was most apparent with ERβ, but the magnitude of the increase was relatively modest.
The leveling effect that we found on the potency enhancements of these compounds with SRC3 through ERβ in the reporter gene assays is not entirely surprising because, compared with the in vitro assays that use purified components, the cellular context is more complex, and it involves potentially other coactivators and multifactorial, dynamic components required to mediate gene expression as well as potential differences in cell uptake and possible metabolic transformations of the ligands. Nevertheless, we found some significant correlations between the in vitro RRP and the cell-based RCP values for the three ligands we selected (E1, E3, and Gen) that became apparent with ERβ when cellular levels of SRC3 were increased by co-transfection. This ERβ potency amplification also resulted in a marked increase in their ERβ potency selectivity. Our findings are consistent with previous, more limited reporter gene assays in HeLa cells stably expressing ERα or ERβ in the absence of any added coactivators, which demonstrated the ERβ-selective activities for E3 and E1 (36). Our in vitro tr-FRET and reporter gene assays, however, provide further details about the molecular mechanism by which the ERβ preferences of E3 and E1 are manifested through the preferential recruitment of SRC3 to these ligand-bound ERβ complexes.
Estrogen Receptor Structural Correlations with Affinities and Cooperativity
The crystal structures of ER LBD complexes with ligands and SRC coactivators provide a basis to understand certain aspects of our findings: the limited correlation between RLA and RCA and cooperativity in coactivator recruitment assays.
Because ER crystal structures show that the ligand (in a pocket fully within the LBD) and the coactivator (in a surface groove of the LBD) are bound at structurally distinct sites, it is not surprising that the ligand structural features that affect RLA can be discordant with the ER-LBD conformational features that affect RCA. The two ligands that have the most discordant RLA and RCA values are E3 (7 and 96 for ERα and 12 and 150 for ERβ) and 17α-E2 (14 and 52 for ERα and 5 and 200 for ERβ). These two ligands differ from 17β-E2 only in the nature of the hydroxyl groups in the d-ring, which are proximal to helix-11 in the LBD. Although these ligands are not directly in contact with either the bound coactivator or with helix-12 (37), whose position enables or disables SRC binding, it is clear from other ER structural studies that ligand contact with helix-11 can affect the positioning or dynamics of helix-12 and thereby affect coactivator binding (31, 37, 38). Thus, it appears that the extra 16α-hydroxyl group in E3 and the epimeric 17-hydroxy of 17α-E2 enable these ligands to interact with helix-11 in a manner that engenders an LBD conformation that is particularly favorable for binding SRC despite their modest RLA values. In this regard, it is of note that estriol appears to have some preferential binding to ERβ (31), and an E3 stereoisomer, 16β,17β-epiestriol, is found to be more active through ERβ than ERα in cell-based reporter gene assays (24). Thus, despite its relatively low RLA, E3 may be capable of eliciting strong biological effects in target cells expressing high amounts of ERβ and SRC3. On the other hand, it is less likely that the 17α-hydroxy group of the E2 epimer (17α-E2) can form a good hydrogen bond with His524/His476 in ERα/ERβ, based on a crystal structure of a related steroid (39).
There are also structural reasons to expect that ligand binding to the ER might show positive cooperativity. The ER LBDs form dimers, and through our earlier FRET studies on dimer thermodynamic and kinetic stability, we have shown that ligand binding strengthens the dimer interface (18). As is well known from oxygen binding to hemoglobin as well as many other examples, communication across interfaces in protein-protein multimers is the key molecular event underlying cooperative behavior in ligand binding. In our measurement of RRP through ligand titration experiments, cooperativity could also be engendered because the assay is actually measuring the recruitment of a fragment of SRC3, the NRID, that encompasses the three possible LXXLL motifs for interaction with the ER dimer, an interaction that has, in many cases, a Kd value of less than 1 nm. Thus, the multimeric SRC3·ER dimer interaction could enhance cooperativity. In addition, we have found that coactivator binding provides additional stabilization of ER conformations (29) and greatly slows the rate of ligand dissociation (30). Nevertheless, it is of note that the binding of the selective estrogen receptor modulator (SERM) hydroxytamoxifen to ERα shows some positive cooperativity (40), although this complex is unable to recruit SRC coactivators.
CONCLUSION
In this study, we have probed the molecular basis for the potency of various estrogens. Our studies were enabled by a convenient and robust in vitro tr-FRET assay system that we developed, through which we have carefully quantified the parameters of ligand binding affinity for ERα and ERβ and SRC3 coactivator binding affinity to the complexes that these ERs formed with 12 representative estrogen agonists from four classes and examined how these might be related to ligand potency in SRC3 recruitment. We then related some of these results to potencies measured through ERα and ERβ in a cell-based assay, where the levels of SRC3 were also elevated.
We found that the relationship between ligand binding affinity and coactivator binding to ER·ligand complex is both ligand- and ER subtype-specific, and we found that the cellular potencies of estrogens correlated better with a combination of RCA and RRP than RLA. Thus, we demonstrate the importance of evaluating estrogen ligand potencies in the context of coactivators, and we also provide evidence that some weakly binding estrogens might be able to elicit considerable biological activity in target cells that express the proper combination of ER subtype and coactivator levels. In this regard, it is particularly notable that elevated levels of SRC3 can cause a preferential increase in the potency of certain ligands through ERβ.
This work was supported, in whole or in part, by National Institutes of Health Grants R37 DK015556, P01 AG024387, and P50 AT006268 (to J. A. K.). This work was also supported by National Research Service Awards F30 ES016484-01 and T32 GM070421 (to J. R. G.).
- ER
- estrogen receptor
- ERE
- estrogen response element
- NR
- nuclear receptor
- tr-FRET
- time-resolved FRET
- RLA
- relative ligand binding affinity
- RCA
- relative coactivator binding affinity
- RRP
- relative recruitment potency
- E2
- estradiol or 17β-estradiol
- 17α-E2 and 17β-E2
- 17α- and 17β-estradiol, respectively
- E1
- estrone
- E3
- estriol
- EE2
- 17α-ethynylestradiol
- DES
- diethylstilbestrol
- DMS
- dimethylstilbestrol
- Equ
- equilin
- Eqn
- equilenin
- meso-Hex
- meso-hexestrol
- dl-Hex
- dl-hexestrol
- Gen
- genistein
- LBD
- ligand binding domain
- Fl-SRC3
- fluorescein-labeled SRC3 fragment
- SA-Tb
- streptavidin-terbium
- RRE
- relative recruitment efficacy
- NRID
- nuclear receptor interaction domain
- RCP
- relative cellular potency
- SRC
- steroid receptor coactivator.
REFERENCES
- 1. Check J. H. (2006) Clin. Exp. Obstet. Gynecol. 33, 71–77 [PubMed] [Google Scholar]
- 2. Check M. L., Check J. H., Kaplan H. (2004) Clin. Exp. Obstet. Gynecol. 31, 299–301 [PubMed] [Google Scholar]
- 3. Berrodin T. J., Chang K. C., Komm B. S., Freedman L. P., Nagpal S. (2009) Mol. Endocrinol. 23, 74–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walf A. A., Frye C. A. (2008) Neuroreport 19, 789–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brawer M. K. (2006) Rev. Urol. 8, Suppl. 2, S35–S47 [PMC free article] [PubMed] [Google Scholar]
- 6. Scherr D. S., Pitts W. R., Jr. (2003) J. Urol. 170, 1703–1708 [DOI] [PubMed] [Google Scholar]
- 7. Marini H., Minutoli L., Polito F., Bitto A., Altavilla D., Atteritano M., Gaudio A., Mazzaferro S., Frisina A., Frisina N., Lubrano C., Bonaiuto M., D'Anna R., Cannata M. L., Corrado F., Adamo E. B., Wilson S., Squadrito F. (2007) Ann. Intern. Med. 146, 839–847 [DOI] [PubMed] [Google Scholar]
- 8. Williamson-Hughes P. S., Flickinger B. D., Messina M. J., Empie M. W. (2006) Menopause 13, 831–839 [DOI] [PubMed] [Google Scholar]
- 9. Minutolo F., Macchia M., Katzenellenbogen B. S., Katzenellenbogen J. A. (2009) Med. Res. Rev. 1002/med.20186 [DOI] [PubMed] [Google Scholar]
- 10. Zhao C., Dahlman-Wright K., Gustafsson J. A. (2008) Nucl. Recept. Signal. 6, e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heldring N., Pike A., Andersson S., Matthews J., Cheng G., Hartman J., Tujague M., Ström A., Treuter E., Warner M., Gustafsson J. A. (2007) Physiol. Rev. 87, 905–931 [DOI] [PubMed] [Google Scholar]
- 12. Smith C. L., O'Malley B. W. (2004) Endocr. Rev. 25, 45–71 [DOI] [PubMed] [Google Scholar]
- 13. Grow D. R. (2002) Obstet. Gynecol. Clin. North Am. 29, 425–436 [DOI] [PubMed] [Google Scholar]
- 14. Ruggiero R. J., Likis F. E. (2002) J. Midwifery Womens Health 47, 130–138 [DOI] [PubMed] [Google Scholar]
- 15. Gunther J. R., Du Y., Rhoden E., Lewis I., Revennaugh B., Moore T. W., Kim S. H., Dingledine R., Fu H., Katzenellenbogen J. A. (2009) J. Biomol. Screen. 14, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kilbourn M. R., Arduengo A. J., Park J. T., Katzenellenbogen J. A. (1981) Mol. Pharmacol. 19, 388–398 [PubMed] [Google Scholar]
- 17. Kim S. H., Tamrazi A., Carlson K. E., Daniels J. R., Lee I. Y., Katzenellenbogen J. A. (2004) J. Am. Chem. Soc. 126, 4754–4755 [DOI] [PubMed] [Google Scholar]
- 18. Tamrazi A., Carlson K. E., Daniels J. R., Hurth K. M., Katzenellenbogen J. A. (2002) Mol. Endocrinol. 16, 2706–2719 [DOI] [PubMed] [Google Scholar]
- 19. Jeyakumar M., Webb P., Baxter J. D., Scanlan T. S., Katzenellenbogen J. A. (2008) Biochemistry 47, 7465–7476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou H. B., Carlson K. E., Stossi F., Katzenellenbogen B. S., Katzenellenbogen J. A. (2009) Bioorg. Med. Chem. Lett. 19, 108–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bramlett K. S., Wu Y., Burris T. P. (2001) Mol. Endocrinol. 15, 909–922 [DOI] [PubMed] [Google Scholar]
- 22. Iannone M. A., Simmons C. A., Kadwell S. H., Svoboda D. L., Vanderwall D. E., Deng S. J., Consler T. G., Shearin J., Gray J. G., Pearce K. H. (2004) Mol. Endocrinol. 18, 1064–1081 [DOI] [PubMed] [Google Scholar]
- 23. Liu J., Knappenberger K. S., Kack H., Andersson G., Nilsson E., Dartsch C., Scott C. W. (2003) Mol. Endocrinol. 17, 346–355 [DOI] [PubMed] [Google Scholar]
- 24. Barkhem T., Carlsson B., Nilsson Y., Enmark E., Gustafsson J., Nilsson S. (1998) Mol. Pharmacol. 54, 105–112 [DOI] [PubMed] [Google Scholar]
- 25. Lascombe I., Beffa D., Rüegg U., Tarradellas J., Wahli W. (2000) Environ. Health Perspect. 108, 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Monroe D. G., Johnsen S. A., Subramaniam M., Getz B. J., Khosla S., Riggs B. L., Spelsberg T. C. (2003) J. Endocrinol. 176, 349–357 [DOI] [PubMed] [Google Scholar]
- 27. Kim S. H., Tamrazi A., Carlson K. E., Katzenellenbogen J. A. (2005) Mol. Cell Proteomics 4, 267–277 [DOI] [PubMed] [Google Scholar]
- 28. Tamrazi A., Carlson K. E., Katzenellenbogen J. A. (2003) Mol. Endocrinol. 17, 2593–2602 [DOI] [PubMed] [Google Scholar]
- 29. Tamrazi A., Carlson K. E., Rodriguez A. L., Katzenellenbogen J. A. (2005) Mol. Endocrinol. 19, 1516–1528 [DOI] [PubMed] [Google Scholar]
- 30. Gee A. C., Carlson K. E., Martini P. G., Katzenellenbogen B. S., Katzenellenbogen J. A. (1999) Mol. Endocrinol. 13, 1912–1923 [DOI] [PubMed] [Google Scholar]
- 31. Kuiper G. G., Carlsson B., Grandien K., Enmark E., Häggblad J., Nilsson S., Gustafsson J. A. (1997) Endocrinology 138, 863–870 [DOI] [PubMed] [Google Scholar]
- 32. Kuiper G. G., Lemmen J. G., Carlsson B., Corton J. C., Safe S. H., van der Saag P. T., van der Burg B., Gustafsson J. A. (1998) Endocrinology 139, 4252–4263 [DOI] [PubMed] [Google Scholar]
- 33. Bhavnani B. R., Tam S. P., Lu X. (2008) Endocrinology 149, 4857–4870 [DOI] [PubMed] [Google Scholar]
- 34. Dai S. Y., Burris T. P., Dodge J. A., Montrose-Rafizadeh C., Wang Y., Pascal B. D., Chalmers M. J., Griffin P. R. (2009) Biochemistry 48, 9668–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dai S. Y., Chalmers M. J., Bruning J., Bramlett K. S., Osborne H. E., Montrose-Rafizadeh C., Barr R. J., Wang Y., Wang M., Burris T. P., Dodge J. A., Griffin P. R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 7171–7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Escande A., Pillon A., Servant N., Cravedi J. P., Larrea F., Muhn P., Nicolas J. C., Cavaillès V., Balaguer P. (2006) Biochem. Pharmacol. 71, 1459–1469 [DOI] [PubMed] [Google Scholar]
- 37. Shiau A. K., Barstad D., Loria P. M., Cheng L., Kushner P. J., Agard D. A., Greene G. L. (1998) Cell 95, 927–937 [DOI] [PubMed] [Google Scholar]
- 38. Nettles K. W., Sun J., Radek J. T., Sheng S., Rodriguez A. L., Katzenellenbogen J. A., Katzenellenbogen B. S., Greene G. L. (2004) Mol. Cell 13, 317–327 [DOI] [PubMed] [Google Scholar]
- 39. Hsieh R. W., Rajan S. S., Sharma S. K., Greene G. L. (2008) Steroids 73, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sasson S., Notides A. C. (1988) Mol. Endocrinol. 2, 307–312 [DOI] [PubMed] [Google Scholar]