

Abstract
Adenovirus disrupts endosomal membranes during cell entry. The membrane lytic capsid protein VI (pVI) facilitates entry by fragmenting membranes. Although an N-terminal amphipathic α-helix (VI-Φ) possesses similar membrane affinity as pVI, truncated protein lacking VI-Φ, (VIΔ54) still possesses moderate membrane affinity. We demonstrate that incorporation of nickel-NTA lipids in membranes enhances increases the membrane affinity and the membrane lytic activity of VIΔ54. We also demonstrate that 3 predicted pVI α-helices within residues 54–114 associate with membranes, sitting roughly parallel to the membrane surface. His-tagged VIΔ54 is capable of fragmenting membranes similar to pVI and the VI-Φ peptide. Interestingly, neither VI-Φ nor His-tagged VIΔ54 can induce tubule formation in giant lipid vesicles as observed for pVI. These data suggest cooperativity between the amphipathic α-helix and residues in VIΔ54 to induce positive membrane curvature and tubule formation. These results provide additional details regarding the mechanism of nonenveloped virus membrane penetration.
Keywords: nonenveloped, adenovirus, protein VI, capsid, cell membrane
INTRODUCTION
Recent advances in our understanding of the mechanisms of nonenveloped virus membrane penetration have involved the identification of viral and cellular factors which mediate this process (Danthi et al., 2008; Farr, Zhang, and Tattersall, 2005; Kim et al., 2010; Maier et al., 2010; Wiethoff et al., 2005; Zhang et al., 2009). How these factors mediate membrane penetration is not as clearly understood as enveloped virus membrane fusion mechanisms, however. While enveloped virus membrane fusion during cell entry shares analogous features with normal cellular membrane fusion events, the mechanisms of nonenveloped virus membrane rupture appear, so far, to be fairly unique in the context of cell biology and may therefore serve as potential targets for antiviral interventions. Additionally, a better understanding of the mechanisms of nonenveloped virus membrane penetration could shed considerable light on viral pathogenesis.
Adenovirus is an excellent model to study the process of nonenveloped virus membrane penetration as considerable structural, biochemical and cell biological information exists regarding the mechanisms Ad uses to enter cells. To enter cells, Ads first bind to a primary attachment receptor via high affinity interactions between the virus fiber protein and the cell surface receptor (Chroboczek, Ruigrok, and Cusack, 1995). For subgroup A, C, E and F, this receptor is the coxsackievirus and adenovirus receptor (Roelvink et al., 1998). For subgroup B and D viruses, this receptor has been found to be CD46 (Segerman et al., 2003; Sirena et al., 2004; Wu et al., 2004), sialic acid (Arnberg et al., 2000; Johansson et al., 2007) CD80/CD86 (Short et al., 2004; Short et al., 2006) or heparin sulfate proteoglycans (Tuve et al., 2008). After attachment, secondary engagement of αv integrins by the RGD motif of the viral penton base protein triggers clathrin mediated endocytosis of virions (Nemerow, 2000).
For subgroup C viruses, disassembly of the viral capsid is initiated very soon after attachment, with the fiber reportedly released from the virion at the cell surface (Greber et al., 1993; Nakano et al., 2000). Endocytosis results in further disassembly of the Ad capsid proteins as additional proteins are shed from the virus (Greber et al., 1993). Uncoating correlates with acidification of the endosomal compartment (Greber et al., 1993; Wohlfart, Svensson, and Everitt, 1985) and is enhanced by interactions between the penton base and αv integrins (Wickham et al., 1994; Wickham et al., 1993). Studies employing a temperature sensitive mutant, Ad2ts1, which possesses a hyperstable capsid when produced at the nonpermissive temperature due to lack of proteolytic capsid maturation, have demonstrated that, although this virus can attach and trigger endocytosis, it’s failure to uncoat in endosomes prevents endosomal escape (Greber et al., 1996; Hannan et al., 1983). Additionally, while entry of wt Ad2 virions leads to eventual degradation of the interior capsid protein VI (pVI), by 2 hrs post infection, the precursor form of pVI present in the ts1 virions is not degraded during cell entry presumably because it is not released from the virion (Greber et al., 1996; Greber et al., 1993).
Adenovirus uncoating within endosomes is a requirement for viral membrane penetration so that protein VI can be released from the interior of the capsid (Smith and Nemerow, 2008; Wiethoff et al., 2005). Protein VI, present in the Ad capsid at 342–369 copies (Lehmberg et al., 1999; van Oostrum and Burnett, 1985), is expressed as a 250 residue preprotein which undergoes maturation cleavage by the virally encoded 23K cysteine protease before and after residues 34 and 239 respectively (Sung et al., 1983). Release of protein VI from the capsid interior was previously shown to be a requirement for in vitro membrane lytic activity of the Ad virion (Wiethoff et al., 2005). Furthermore, Ad penetration of endosomal membranes was shown to be inhibited in the presence of anti-protein VI antibodies capable of preventing membrane lytic activity of protein VI (Maier et al., 2010).
The molecular mechanism through which pVI disrupts membranes has not been completely characterized, but appears to be highly dependent upon an N-terminal amphipathic α-helix (Maier et al., 2010; Wiethoff et al., 2005). Deletion of this α-helix greatly reduces protein VI membrane affinity and membrane lytic activity, although these properties are not completely abolished in the truncated form of the protein (Maier et al., 2010). Furthermore, this N-terminal amphipathic α-helix appears to have affinity for lipid membranes identical to that of the mature form of protein VI. Disruption of membranes by protein VI involves the induction of positive curvature stress in the lipid bilayer. This curvature stress is thought to be supported by the superficial, oblique orientation of the N-terminal amphipathic α-helix of protein VI in the lipid bilayer (Maier et al., 2010).
We now demonstrate that, although the N-terminal amphipathic α-helix of protein VI is sufficient to lyse membranes, a truncated version of protein VI lacking this α-helix, VIΔ54, can efficiently lyse membranes when membrane binding is enhanced through interactions between an N-terminal 6xHis tag and liposomes containing lipids with a NTA-Ni2+ headgroup. We further demonstrate that 3 additional predicted α-helices within residues 54–114 in protein VI also interact with the lipid bilayer with superficial and oblique orientations. This orientation is similar to that observed for the N-terminal amphipathic α-helix. Additionally, the N-terminal amphipathic α-helix has a similar capacity to induce positive membrane curvature as previously reported for protein VI (Maier et al., 2010). In contrast, VIΔ54 appears to lyse membranes via a mechanism which is less influenced by the presence of lipids which alter membrane curvature than either pVI or the N-terminal amphipathic α-helix. Interestingly, while pVI induces membrane tubulation upon fragmenting giant lipid vesicles (GLVs), neither the N-terminal amphipathic α-helix, nor VIΔ54 are able to induce membrane tubulation despite efficient fragmentation of GLVs. Thus, these data suggest that although protein VI interactions with membranes is driven largely by an N-terminal amphipathic α-helix, additional protein VI membrane interactions occur which influence the topology of the resulting membrane fragments. This data provides new insight into the interactions of a nonenveloped virus membrane lytic protein with membranes. Future studies will be necessary to understand the contributions of the different domains of protein VI to viral escape from endosomes during cell entry.
RESULTS
VIΔ54 has increased membrane lytic activity with DOGS-NTA-Ni liposomes
Previously we have shown that an N-terminal amphipathic α-helix in pVI is largely responsible for membrane lytic activity, however removing this helix does not completely abrogate pVI membrane lytic activity. A construct that lacks this helix, VIΔ54, can still disrupt membranes but with much lower efficiency (Maier et al., 2010). This decreased membrane lytic activity could be either due to decreased membrane binding or an inability to induce the membrane curvature observed with full length pVI. To determine the contributions of the N-terminal amphipathic α-helix-mediated membrane binding to membrane lytic activity of protein VI we artificially enhanced the membrane affinity of VIΔ54 including an N-terminal 6xHis-tagged tag and using liposomes containing 5 mol% of the Nickel-chelating phospholipid, DOGS-NTA-Ni. The binding of His-tagged pVI constructs to liposomes was assessed as previously described using changes in intrinsic tryptophan fluorescence upon association to membranes (Maier et al., 2010). By assuming that the relative change in tryptophan fluorescence upon binding liposomes is directly related to the amount of pVI bound to membranes, binding isotherms were generated and the fractional saturation of binding sites was plotted versus increasing lipid concentrations. Compared to untagged VIΔ54, binding of 6xHis-tagged VIΔ54 to DOGS-NTA-Ni containing liposomes displayed an apparent affinity much closer to that observed for pVI (Fig 1A) Addition of a 6xHis tag to pVI did not significantly enhance membrane affinity. To determine if this increased membrane binding correlates with increased membrane lytic activity, VIΔ54 was added to sulforhodamine B (sulfoB) loaded DOGS-NTA-Ni liposomes and dye release was then measured. Addition of VIΔ54 to sulfoB-loaded DOGS-NTA-Ni liposomes resulted in increased membrane lytic activity compared to liposomes without the nickel-chelating lipid (Fig 1B). To control for the influence of the N-terminal 6xHis-tag on observed membrane lytic activity, similar titrations with purified 6xHis-tagged penton base were performed. Tagged penton base did not induce significant SulfoB release over the same concentration range, suggesting that the His-tag was not responsible for the enhanced membrane lytic activity of VIΔ54 on DOGS-NTA-Ni liposomes. These results suggest that while the N-terminal amphipathic α-helix greatly enhances pVI membrane affinity, the remainder of the protein can also contribute to efficient membrane lysis.
Figure 1. The membrane affinity and lytic activity of VI 54 are enhanced by DOGS-NTA-Ni phospholipids.

(A) Membrane binding of the different pVI constructs. Increasing lipid concentrations were added to pVI constructs and changes in tryptophan fluorescence were used to determine the fractional saturation (θ) of pVI binding capacity as described in the Material and Methods. B) Protein VI membrane lytic activity was determined by measuring the release of Sulfo B from liposomes after incubation with increasing protein concentrations. (B) 6xHis-tagged pVI incubated with PC:PS:DOGS-NTA-Ni (70:25:5 mol%) (■) or PC:PS(70:25mol% ) (□).6xHis-tagged VIΔ54 incubated with PC:PS:DOGS-NTA-Ni (70:25:5 mol%) (△) or PC:PS (75:25 mol%) (▲) 6xHis-tagged penton base incubated with PC:PS:DOGS-NTA-Ni (70:25:5 mol%) (●).
Mapping membrane interacting domains within pVI
An N-terminal 80 amino acid domain in pVI (residues 34–114) possesses 85% α-helical content as assessed by circular dichroism (Maier et al., 2010). This domain is predicted to contain 4 α-helices (Fig 2A), and we have previously shown that the first amphipathic α-helix has an oblique membrane orientation. Since residues outside this N-terminal α-helix contribute to pVI lytic activity, we looked at the potential of the additional predicted α-helices in this 80 residue domain to associate with membranes. Helical wheel diagrams suggest that each of these putative α-helices have considerable amphipathy (Fig 2C-E). Additionally, predictions of the free energy of interfacial membrane partitioning, ΔGif, suggest that each of these helices would spontaneously associate with membranes although the magnitude of ΔGif for these 3 additional α helices is considerably smaller than that for the N-terminal α-helix (Fig 2B-E). To determine whether these α-helices interact with membranes, single tryptophan (Trp) mutations were introduced individually at 1 or 2 sites for each helix by replacing L67, L71, V80, L84 or V91 in the plasmid encoding VI114Δ which had previously had all 3 native Trp residues replaced by phenylalanine (Maier et al., 2010). Introduction of each of these mutations had no effect on membrane lytic activity or secondary structure as assessed by circular dichroism spectroscopy (data not shown). The depth of Trp penetration into lipid bilayers was determined by measuring quenching of Trp fluorescence by lipids that have bromine atoms covalently attached to their acyl tails at known distances from the center of the bilayer, zcf (Markello et al., 1985). Bromine quenching of Trp fluorescence is distance dependent, and therefore can be used to determine how deep into the bilayer a tryptophan residue penetrates (Chattopadhyay and London, 1987; Ladokhin, 1997).
Figure 2. α-helices in the N terminal 80 residue domain of pVI are predicted to interact with the membrane.

A) predicted secondary structure of protein VI. Helical wheel diagrams of the 1st (B), 2nd (C), 3rd (D) and 4th (E) helix in the N terminal 80 residue domain of pVI are shown, with the hydrophilic residues in white and hydrophobic residues in black. The residues in each helix mutated to tryptophan have been boxed. The Free energy of partitioning into the lipid membrane interface, ΔGif, calculated using the Membrane Protein Explorer software (Snider et al., 2009) are shown for each helix with units of KJ/mol.
To measure the depth of Trp membrane penetration, the single Trp mutants were incubated with PS:PC:PC-Br liposomes (25:25:50 mol%) at a 1:100 ratio (protein:lipid), and Trp fluorescence was then measured. Tryptophan fluorescence of each of these mutants was quenched upon mixing with liposomes containing these brominated lipids suggesting that they interact the lipid bilayer. The distance from the center of the bilayer was calculated using both the parallax method and the distribution analysis (Table 1). Since the W67 and W71 residues in the 2nd helix, and W80 and W84 in the third helix would be 6 Å apart in an α-helix, yet they are positioned at depths which differ by only 1 Å in the membrane, it is likely that the 2nd and 3rd helices are positioned in an oblique orientation relative to the membrane surface and do not traverse the apolar region of the lipid bilayer similar to the orientation reported previously for the N-terminal amphipathic α-helix (Maier et al., 2010).
Table 1.
Depth of protein VI tryptophan residues penetration into the membrane
| Single Trp. Mutant | Zcf* (Å) |
|
|---|---|---|
| Parallax | Distribution Analysis | |
| W67 | 7.3 ± 0.5 | 8.1 ± 0.8 |
| W71 | 8.7 ± 0.3 | 8.7 ± 0.1 |
| W80 | 9.7 ± 0.1 | 9.1 ± 0.2 |
| W84 | 8.8 ± 0.3 | 8.1 ± 0.2 |
| W91 | 7.5 ± 0.0 | 8.4 ± 0.4 |
Zcf- the distance from the center of the bilayer
Induction of positive membrane curvature by the N-terminal amphipathic α-helix and VIΔ54
Protein VI disrupts membranes by inducing a positive curvature stress in membranes, however the domains responsible for this induction are not known. The shallow and oblique membrane orientation observed with the alpha helices within the N-terminal 80 residues is indicative of a protein that induces this type of curvature stress (Drin et al., 2007; Epand and Epand, 2000; Zimmerberg and Kozlov, 2006). However, the relative contributions of the N-terminal amphipathic α-helix and the helices between residues 54 and 114 to induction of positive membrane curvature are unknown. To determine this contribution, we examined the influence of lipids with a propensity to adopt positive (lysoPC) or negative (POPE) membrane curvature on the membrane lytic activity of the N-terminal amphipathic α-helix or VIΔ54. If the membrane lytic activity of either the N-terminal amphipathic α-helix or VIΔ54 involves the induction of positive membrane curvature, then we would expect the presence of lysoPC to enhance membrane lytic activity while POPE would inhibit membrane lytic activity. Similar to our previous observations with pVI, the N-terminal amphipathic α-helix membrane lytic activity is significantly enhanced in the presence of increasing amounts of lysoPC while this activity is reduced in the presence of POPE (Fig 3A). The membrane lytic activity of VIΔ54 is much less influenced by the inclusion of increasing amounts of lysoPC in liposomes, and only slightly influenced by the inclusion of POPE (Fig 3B). These data suggest that the N-terminal amphipathic α-helix contributes more significantly to protein VI induction of positive membrane curvature than the rest of the protein.
Figure 3. The amphipathic alpha helix peptide and VI 54 induce positive membrane curvature to lyse target membranes.

Increasing concentrations of VI 34–54 (A) or VIΔ54 (B) VI 34–54 (A) were incubated with POPC:POPS (75:25 mol%) (A) or POPC:POPS :DOGS-NTA-Ni (70:25:5 mol%) (B)(*) liposomes entrapping SulfoB and 26 in which some POPC was replaced with 5 (■) or 10 (□) mol% lysoPC or 5 (▲) or 25 (△) mol% of POPE .The %SulfoB released was determined as described in the Material and Methods
The N-terminal amphipathic α-helix and VIΔ54 cooperate to induce membrane tubule formation
Previously we observed that pVI membrane lytic activity involves the fragmentation of target membranes (Maier et al., 2010; Wiethoff et al., 2005). When added to fluorescently labeled giant lipid vesicles (GLV), this fragmentation also leads to the formation of tubular structures which likely possess significant membrane curvature stress. It was also shown that the ability of protein VI to induce tubule formation requires only the N-terminal 80 residues of pVI (Maier et al., 2010). To determine what domains in pVI are responsible for tubule formation we investigated the effects of N-terminal amphipathic α-helix and VIΔ54 on GLV morphology. The GLV membranes with or without 5 mol% DOGS-NTA-Ni were labeled with 5mol% fluoresceinylated lipid and observed by epifluorescence microscopy. Vesicles appear 5–50 μm in diameter and this morphology is unchanged upon addition of PBS (Fig 4A). As previously observed, the addition of full length pVI to GLVs at a protein:lipid molar ratio of 1:100, fragments 10 vesicles into smaller structures (Fig 4B) including tubules (arrows). However, although the N-terminal amphipathic α-helix is able to disrupt these GLVs, it fails to form the highly curved tubular structures observed with full length protein (Fig 4C). Incubating VIΔ54 with GLVs containing DOGS-NTA-Ni also results in membrane fragmentation without tubulation (Fig 4D). These data suggest that although the N-terminal amphipathic α-helix is sufficient to induce positive membrane curvature and lyse membranes, the highly curved membrane tubules observed upon pVI membrane lysis require additional elements within residues 54–114 of pVI to interact with membranes.
Figure 4. Protein VI fragmentation of giant lipid vesicles.
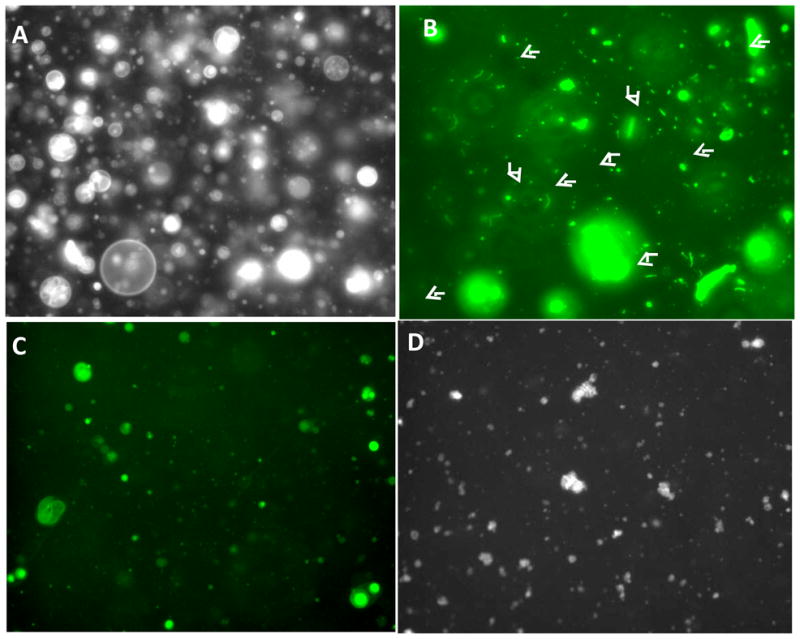
Fluorescein-DHPE labeled POPC:POPS (75:25 mol%) (A–C) or POPC:POPS:DOGS-NTA-Ni (70:25:5 mol% (D) giant lipid vesicles were incubated with PBS (A), pVI (B), VI 34–54 (C) or VIΔ54 (D) for 15 min. Tubule formation was visualized by epifluorescence microscopy. Arrows indicate tubular lipid structures formed in the presence of pVI
DISCUSSION
Currently, the molecular mechanisms of cell membrane disruption by capsid proteins of nonenveloped viruses are still poorly defined. While studies have demonstrated that reovirus μ1 protein (Ivanovic, et al. 2008) and picornavirus VP1/VP4 (Tosteson and Chow, 1997) form pores in membranes, much less is known regarding the mechanisms used by other nonenveloped viruses to penetrate cell membranes. To disrupt membranes during cell entry, release of protein VI from the interior of Ad capsids is required (Smith and Nemerow, 2008; Wiethoff et al., 2005). This protein fragments membranes by inducing positive membrane curvature (Maier et al., 2010). Fragmentation as a mode of Ad disruption of endosomal membranes is in agreement with the ability of Ad to facilitate the cytosolic translocation of co-endocytosed high molecular weight molecules such as 70 kDa dextrans (Prchla et al., 1995) antibody-toxin conjugates (FitzGerald et al., 1983) and even whole parvoviruses (Farr, Zhang, and Tattersall, 2005). Our current data further describe the contributions of an N-terminal amphipathic α-helix to pVI membrane fragmentation and identify additional domains within pVI which interact with membranes and likely contribute to membrane lysis.
Previously we have shown that pVI is responsible for Ad endosomal escape, and that an N-terminal amphipathic α-helix is the major determinant of membrane lytic activity (Maier et al., 2010). A key role for this N-terminal amphipathic α-helix in pVI membrane lytic activity was suggested to be the enhancement of pVI affinity for membranes since a peptide corresponding to VI34–53 had nearly identical membrane lytic activity and membrane affinity as pVI. Furthermore, a protein lacking residues 34–53, VIΔ54, possesses severely reduced membrane affinity and lytic activity. We further demonstrated that pVI and an 80 residue α-helical domain, VI114Δ fragment membranes by inducing positive membrane curvature (Maier et al., 2010). We now demonstate that, a peptide corresponding to the N-terminal amphipathic α-helix can fragment GLV membranes. Additionally, like pVI, the membrane lytic activity of the N-terminal amphipathic α-helix is enhanced or attenuated in the presence of lipids with a propensity to adopt positively or negatively curved membranes, respectively. Thus, many of the membrane lytic properties of protein VI appear to be contained within residues 34–53. However, the inability of the N-terminal amphipathic α-helix to induce membrane tubules upon fragmentation of GLVs led us to consider whether additional domains within protein VI could interact with membranes to facilitate this tubulation.
Our previous work found that the 80 residues of VI114Δ possess ~85% α-helical character (Maier et al., 2010). In addition to the N-terminal amphipathic α-helix, three additional α-helices are also predicted for this domain. These α-helices are also predicted to spontaneously associate with membrane interfaces as evidenced by the negative ΔGif values (Fig 2). These predictions were confirmed by our observation that these 3 helices associate with membranes, binding in shallow oblique orientations in the lipid bilayer. The magnitude of these ΔGif is considerably less than that for the N-terminal amphipathic α-helix which is in agreement with the severe reduction in membrane affinity observed for VIΔ54 compared to pVI. To examine the contributions of these additional α-helices to pVI membrane lytic activity, the affinity of VIΔ54 for membranes was artificially enhanced using Ni2+-NTA containing lipids in the target membrane and a 6xHis tag on the protein. The increased affinity of 6xHis-VIΔ54 for membranes correlated with an increase in membrane lytic activity which was more comparable to that observed for pVI. Although the affinity of the various forms of pVI for membranes correlates with membrane lytic activity this relationship appears to be non-linear since the 6xHis-VIΔ54 binding to Ni2+-NTA liposomes is less than pVI yet possesses similar membrane lytic activity.
Although the pVI membrane interacting region between residues 54–114 appears to contribute to pVI membrane disruption, how this region contributes is still unclear. Unlike pVI and a peptide corresponding to the N-terminal amphipathic α-helix, the membrane lytic activity of 6xHis-tagged VIΔ54 does not appear to be as strongly influenced by the inclusion of lipids which alter the propensity for positive or negative membrane curvature. Thus, it is possible that VIΔ54 is able to more strongly induce positive membrane curvature in the absence of lysolipids. We observed that neither the N-terminal amphipathic α-helix, nor 6xHis-tagged VIΔ54 can induce membrane tubulation independently. Our previous results indicate that VI114Δ, containing the 4 α-helices, is capable of inducing membrane tubulation. Together, these data suggest that although both the N-terminal amphiphathic α-helix and VIΔ54 alone can induce positive membrane curvature, an intact VI114Δ membrane-interacting domain is necessary to form membrane tubules. One possible explanation could be that an intact domain from residues 34–114 can stabilize the highly curved structures induced upon pVI membrane binding, either through cooperative influences on membrane curvature or through protein-protein interactions which would not occur in either the N-terminal peptide or VIΔ54 alone. Further studies are required to gain a better understanding of this phenomenon.
Cooperativity between an N-terminal amphipathic α-helix which induces positive membrane curvature and additional helical domains which stabilize these curved membranes is not unprecedented. A similar cooperativity is observed with the COPII proteins Sar1p, and scaffolding proteins Sec23/24p and Sec13/31p (Lee et al., 2005). During COPII vesicle formation an N-terminal amphipahtic α-helix in Sar1p induces positive membrane curvature, which is then recognized and stabilized by the additional coat proteins Sec23/24p and Sec13/31p. The same is true for other proteins involved in membrane fission events such as those involving the N-BAR domain containing proteins, endophilin and amphiphysin (Low et al., 2008; Masuda et al., 2006). Topologically, membrane fission through induction of positive membrane curvature is very similar to the membrane fragmentation performed by Ad pVI. This similarity appears to be only conceptual, as pVI does not share any obvious sequence similarity with these proteins.
Although the importance of membrane tubulation during Ad cell entry has yet to be defined, overexpression of the mRFP-pVI1–239 in mammalian cells was recently shown to result in the protein associating with dynamic tubular membrane structures (Wodrich et al., 2010). Furthermore, studies with proteins which deform membranes have shown that the formation of tubular structures in vitro can be used as a marker for a protein’s ability to mediate membrane fission in vivo (Lee et al., 2005). Therefore the cooperativity observed between the N-terminal amphipathic α-helix and the 3 additional helices to form tubular structures in vitro might be important for pVI to fragment the endosomal membrane during cell entry. Further experiments will be required to define a role for these pVI domains in endosomal escape of adenovirus during cell entry.
In summary, we have demonstrated that N-terminal amphipathic α-helix of pVI can fragment membranes via induction of positive membrane curvature in a manner similar to pVI. We have also identified additional residues in pVI which interact with membranes in an analogous fashion as the N-terminal amphipathic α-helix. Interactions of these additional regions of pVI with membranes appear to be important for the full phenotypic properties of pVI membrane fragmentation and tubulation in vitro. Further studies which address the mechanisms of protein VI will therefore have to consider the contributions of the entire N-terminal 80 residues of pVI to Ad endosomal escape.
MATERIAL AND METHODS
1-Palmitoyl,2-oleoylphosphatidylcholine (POPC), 1-palmitoyl,2-oleoylphosphatidylserine (POPS), 1-palmitoyl,2-oleoylphosphatidylethanolamine (POPE), 1-palmitoyl-2-stearoyl(6′,7′-dibromo)-sn-glycero-3-phosphocholine 1-palmitoyl-2-stearoyl(9′,10′-dibromo)-sn-glycero-3-phosphocholine, 1-palmitoyl-2-stearoyl(11′,12′-dibromo)-sn-glycero-3-phosphocholine and 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) (DOGS- NTA-Ni) were purchased from Avanti Polar Lipids. α-Lysophosphatidylcholine (lysoPC) were from Sigma, and N-fluoresceinyl-1, 2-sn-dihexadecylphosphatidylethanolamine (FITC-DHPE) and sulforhodamine B from Invitrogen. All other reagents were from FisherBiotech.
Generating pVI single tryptophan mutants
To generate pVI containing single tryptophan residues, mutations were introduced in pET15bVI-N, a construct encoding residues 34–114. This region of pVI has 3 native tryptophans. To obtain single tryptophan mutans in the 2nd, 3rd and 4th predicted helices, the 3 native tryptophans were first mutated to phenylalanine and various hydrophobic residues in these helices were then mutated to tryptophan using the QuickChange II site-directed mutagenesis kit (Strategene, La Jolla, CA). The no tryptophan construct was generated using the following primers. Altered nucleotides are indicated in bold.
VIW59F 5′ GGCAGCAAGGCCTTTAACAGCAGCACAGG 3′,
VIW41F 5′ GCTGGGGCTCGCTGTTTAGCGGCATTAAAAATTTCG 3′,
VIW37F 5′ ACAAGGCCTTCAGCTTTGGCTCGCTGTGGAGC 3′
The single tryptophan mutants were generated using the following primers:
L67W 5’ ACAGGCCAGATGTGGAGGGATAAGTTGAAAG 3’
V80W 5’ AAAATTTCCAACAAAAGTGGGTAGATGGCCTG 3’
L84W 5’ GGTGGTAGATGGCTGGGCCTCTGGCATTAGC 3’
V91W 5’ TCTGGCATTAGCGGGTGGGTGGACTGGCCAAC 3’
The mutations were confirmed by sequencing, and the plasmids were used to overexpress proteins in E. coli.
Purification of recombinant proteins
Recombinant proteins were expressed in BL21(DE3) cells. Cultures inoculated with overnight culture were grown at 37 °C, until they r eached an optical density at 600 nm of 1.0. The NaCl concentration was then increased by adding an additional 0.9 g NaCl/L, and protein expression was induced by adding 1 mM IPTG (isopropyl-α-d-thiogalactopyranoside) for 1 h. Cells were pelleted, resuspended in cell lysis buffer (1% Triton X-100, 25 mM phosphate, 150 mM NaCl pH 7.5, 0.5 mg/ml lysozyme, 0.1 mg/ml DNAse and 1 mM PMSF (phenylmethylsulfonyl fluoride)), and soluble protein was isolated by centrifugation at 13,000 × g for 15 min at 4 °C. Recombinant proteins were purified with Talon cobalt resin using the manufacturer's protocol (BD Biosciences). Proteins were extensively dialyzed into 25 mM Phosphate, 150 mM NaCl, and 10% (v/v) glycerol pH 7.5 before flash freezing in liquid nitrogen. Aliquots were stored at − 80 °C until use.
Determining pVI in vitro membrane lytic activity
Liposomes containing POPC:POPS (75:25 mol%), or PC:PS:DOGS-NTA-Ni (70:25:5 mol%) and entrapped 100 mM sulforhodamine B (SulfoB) (Molecular Probes) were generated as previously described (Maier et al., 2010; Wiethoff et al., 2005). Liposomes containing entrapped SulfoB were separated from free dye using a Sephadex G-75 column, preequilibrated with 25 mM HEPES, 150 mM NaCl buffer pH 7.5 (HBS). The liposome concentration was determined using a phosphate assay as previously described (Fiske and Subbarrow, 1925).
Membrane lytic activity of recombinant pVI was determined by measuring SulfoB fluorescence dequenching upon release from liposomes. The liposomes were diluted in HBS to a final concentration of 10 μM. Different concentrations of pVI were then added to the liposomes and incubated for 20 min at 37 °C. Fluorescence intensity was measured using the Cary Eclipse fluorescence spectrophotometer (Varian) with the excitation wavelength of 575 nm and emission wavelength of 590 nm. One hundred percent dye release was determined by adding Triton X-100 to the liposomes at a final concentration of 0.5% (w/v). The percentage of SulfoB released was calculated using the formula % SulfoB released = 100 × [Fmeas − F0)/(Ftx100 − F0)], where Fmeas is the maximum fluorescence intensity measured, F0 is fluorescence intensity in absence of protein, and Ftx100 is the fluorescence intensity in the presence of 0.5% Triton X-100. Analysis of pVI binding to membranes
Binding to liposomes was assessed by monitoring changes in pVI intrinsic tryptophan fluorescence upon titration with increasing amounts of liposomes (POPC:POPS 75:25 mol%). This approach is routinely used for monitoring interactions between proteins and ligands, membranes or other proteins and relies on the assumption that the fractional spectral change in tryptophan fluorescence correlates directly with the amount of protein bound to its substrate (Eftink, 1997). The fluorescence emission spectra from 300–480 nm of pVI in HBS and 37 °C was obtained by selective excitation of tryptophan at 295 nm. The protein is diluted in HBS such that trace amounts of glycerol remaining from protein storage buffer (<0.5%) do not influence pVI membrane binding. Increasing amounts of liposomes were added to pVI with mixing for 3 min and additional spectra were obtained. Spectra of buffer or an equivalent amount of liposomes alone were subtracted from the spectra of each protein/lipid mixture to obtain corrected spectra. The spectral center of mass, Iλ, for the emission spectra were determined using the Carey Eclipse software. Assuming that this spectral change in tryptophan fluorescence correlates with the amount of protein bound, the fractional saturation of binding sites, θ, was calculated using the following equation: , where Iλ (obs) is the spectral center of mass for each protein/lipid ratio and Iλ (0) and Iλ (max) is the spectral center of mass for protein alone and the protein in the presence of saturating amounts of liposomes, respectively. Plotting θ versus protein/lipid molar ratios yielded the resulting binding isotherms.
Analysis of pVI membrane penetration using Giant Lipid Vesicles (GLV)
GLV were generated as described previously (Akashi et al., 1996), by mixing POPC, POPS and FITC-DHPE (70:25:5 mol ratio) or POPC, POPS, DOGS-NTA-Ni, FITC-DHPE (65:25:5:5 mol ratio) in chloroform. A thin lipid film was then generated on a glass tube by evaporating the chloroform with a stream of nitrogen gas. Residual chloroform was removed by placing the tube under vacuum for 6 h. The lipid film was then prehydrated with a stream of water saturated nitrogen gas for 25 min, followed by rehydration in 6 ml of HBS containing 0.1 M sucrose. The tube was then sealed with parafilm and incubated overnight at 37 °C. GLVs wer e harvested as a flocculate near the top of the solution the next day and quantified by phosphate assay as described above. Typical preparations of GLVs are polydisperse with vesicle diameters ranging from 5 to 50 μm. To visualize pVI membrane lytic activity, recombinant pVI was incubated with GLV at a 1:100 (lipid:protein) ratio in HBS with 0.1 M glucose, on a glass slide. After 15 min the samples were analyzed by using an epifluorescence microscope.
Determining tryptophan depth of membrane penetration
Quenching of tryptophan fluorescence by brominated phospholipids was used to determine the depth of tryptophan penetration into the lipid bilayer(Chattopadhyay and London, 1987; Ladokhin, 1997). Liposomes containing 25 mol% POPS, 25 mol% POPC and 50 mol% brominated phosphatidylcholine (Br2-PC) were made as described above. Recombinant pVI single tryptophan mutants were incubated for 10 min at 37 °C with brominated liposomes at a 1:100 (protein:lipid) ratio in HBS pH 7.5. The intensity of tryptophan fluorescence was measured at 325 nm upon excitation at 295 nm. The differences in quenching tryptophan fluorescence by the (6,7)-, (9,10)-, (11,12)-Br2-PC was used to calculate the location of the residue in the bilayer using two methods: the parallax method (Chattopadhyay and London, 1987) and distribution analysis (Ladokhin, 1997). In the parallax method, the depth of the tryptophan residue was calculated using the formula:
where Zcf represents the distance of the fluorophore from the center of the bilayer, Lcl is the distance of the shallow quencher from the center of the bilayer, Lcl is the distance between the shallow and deep quencher, F1 is the fluorescence intensity in the presence of the shallow quencher, F2 is the fluorescence intensity in the presence of the deep quencher, and C is the concentration of quencher in molecules/Å2. In the distribution analysis the depth of tryptophan residue was calculated by fitting the data to the equation:
where F0 represents the fluorescence intensity in the absence of the brominated phospholipids, Fh is the intensity measured as a function of the distance from the center of the lipid bilayer to the quencher h, c(h) is the concentration of the different quenchers, S is the area under the curve (measurement of quenching efficiency), σ is the dispersion (a measure of the distribution of the depth in the bilayer), hm is the most probable position of the fluorophore in the membrane, and h is the average bromine distances from the center of the bilayer, based on X-ray diffraction and taken to be 10.8, 8.3, and 6.3 Å for (6,7)-, (9,10)- and (11,12)-Br2-PC respectively. When equal concentrations of the Br-lipids are used, the c(h) value is unity.
Acknowledgments
The authors would like to acknowledge financial support from the NIH to C.M.W (AI082430, and a subcontract from HL054352 (awarded to Glen R. Nemerow)), and O.M. (AI007508).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Akashi K, Miyata H, Itoh H, Kinosita K., Jr Preparation of giant liposomes in physiological conditions and their characterization under an optical microscope. Biophys J. 1996;71(6):3242–50. doi: 10.1016/S0006-3495(96)79517-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnberg N, Edlund K, Kidd AH, Wadell G. Adenovirus type 37 uses sialic acid as a cellular receptor. J Virol. 2000;74(1):42–8. [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay A, London E. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry. 1987;26(1):39–45. doi: 10.1021/bi00375a006. [DOI] [PubMed] [Google Scholar]
- Chroboczek J, Ruigrok RW, Cusack S. Adenovirus fiber. Curr Top Microbiol Immunol. 1995;199 ( Pt 1):163–200. doi: 10.1007/978-3-642-79496-4_10. [DOI] [PubMed] [Google Scholar]
- Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol. 2008;82(1):161–72. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drin G, Casella JF, Gautier R, Boehmer T, Schwartz TU, Antonny B. A general amphipathic alpha-helical motif for sensing membrane curvature. Nat Struct Mol Biol. 2007;14(2):138–46. doi: 10.1038/nsmb1194. [DOI] [PubMed] [Google Scholar]
- Eftink MR. Fluorescence methods for studying equilibrium macromolecule-ligand interactions. Methods Enzymol. 1997;278:221–57. doi: 10.1016/s0076-6879(97)78013-3. [DOI] [PubMed] [Google Scholar]
- Epand RM, Epand RF. Modulation of membrane curvature by peptides. Biopolymers. 2000;55(5):358–63. doi: 10.1002/1097-0282(2000)55:5<358::AID-BIP1009>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Farr GA, Zhang LG, Tattersall P. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc Natl Acad Sci U S A. 2005;102(47):17148–53. doi: 10.1073/pnas.0508477102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiske CH, Subbarrow Y. The colorimetric determination of phosphorous. J Biol Chem. 1925;66:374–389. [Google Scholar]
- FitzGerald DJ, Padmanabhan R, Pastan I, Willingham MC. Adenovirus-induced release of epidermal growth factor and pseudomonas toxin into the cytosol of KB cells during receptor-mediated endocytosis. Cell. 1983;32(2):607–17. doi: 10.1016/0092-8674(83)90480-4. [DOI] [PubMed] [Google Scholar]
- Greber UF, Webster P, Weber J, Helenius A. The role of the adenovirus protease on virus entry into cells. Embo J. 1996;15(8):1766–77. [PMC free article] [PubMed] [Google Scholar]
- Greber UF, Willetts M, Webster P, Helenius A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 1993;75(3):477–86. doi: 10.1016/0092-8674(93)90382-z. [DOI] [PubMed] [Google Scholar]
- Hannan C, Raptis LH, Dery CV, Weber J. Biological and structural studies with an adenovirus type 2 temperature-sensitive mutant defective for uncoating. Intervirology. 1983;19(4):213–23. doi: 10.1159/000149363. [DOI] [PubMed] [Google Scholar]
- Johansson SM, Nilsson EC, Elofsson M, Ahlskog N, Kihlberg J, Arnberg N. Multivalent sialic acid conjugates inhibit adenovirus type 37 from binding to and infecting human corneal epithelial cells. Antiviral Res. 2007;73(2):92–100. doi: 10.1016/j.antiviral.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Kim IS, Trask SD, Babyonyshev M, Dormitzer PR, Harrison SC. Effect of mutations in VP5 hydrophobic loops on rotavirus cell entry. J Virol. 2010;84 (12):6200–7. doi: 10.1128/JVI.02461-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin AS. Distribution analysis of depth-dependent fluorescence quenching in membranes: a practical guide. Methods Enzymol. 1997;278:462–73. doi: 10.1016/s0076-6879(97)78024-8. [DOI] [PubMed] [Google Scholar]
- Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122(4):605–17. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Lehmberg E, Traina JA, Chakel JA, Chang RJ, Parkman M, McCaman MT, Murakami PK, Lahidji V, Nelson JW, Hancock WS, Nestaas E, Pungor E., Jr Reversed-phase high-performance liquid chromatographic assay for the adenovirus type 5 proteome. J Chromatogr B Biomed Sci Appl. 1999;732(2):411–23. doi: 10.1016/s0378-4347(99)00316-3. [DOI] [PubMed] [Google Scholar]
- Low C, Weininger U, Lee H, Schweimer K, Neundorf I, Beck-Sickinger AG, Pastor RW, Balbach J. Structure and dynamics of helix-0 of the N-BAR domain in lipid micelles and bilayers. Biophys J. 2008;95(9):4315–23. doi: 10.1529/biophysj.108.134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier O, Galan DL, Wodrich H, Wiethoff CM. An N-terminal domain of adenovirus protein VI fragments membranes by inducing positive membrane curvature. Virology. 2010;402(1):11–9. doi: 10.1016/j.virol.2010.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markello T, Zlotnick A, Everett J, Tennyson J, Holloway PW. Determination of the topography of cytochrome b5 in lipid vesicles by fluorescence quenching. Biochemistry. 1985;24(12):2895–901. doi: 10.1021/bi00333a012. [DOI] [PubMed] [Google Scholar]
- Masuda M, Takeda S, Sone M, Ohki T, Mori H, Kamioka Y, Mochizuki N. Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. EMBO J. 2006;25(12):2889–97. doi: 10.1038/sj.emboj.7601176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano MY, Boucke K, Suomalainen M, Stidwill RP, Greber UF. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J Virol. 2000;74(15):7085–95. doi: 10.1128/jvi.74.15.7085-7095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemerow GR. Cell receptors involved in adenovirus entry. Virology. 2000;274(1):1–4. doi: 10.1006/viro.2000.0468. [DOI] [PubMed] [Google Scholar]
- Prchla E, Plank C, Wagner E, Blaas D, Fuchs R. Virus-mediated release of endosomal content in vitro: different behavior of adenovirus and rhinovirus serotype 2. J Cell Biol. 1995;131(1):111–23. doi: 10.1083/jcb.131.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelvink PW, Lizonova A, Lee JG, Li Y, Bergelson JM, Finberg RW, Brough DE, Kovesdi I, Wickham TJ. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J Virol. 1998;72(10):7909–15. doi: 10.1128/jvi.72.10.7909-7915.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerman A, Atkinson JP, Marttila M, Dennerquist V, Wadell G, Arnberg N. Adenovirus type 11 uses CD46 as a cellular receptor. J Virol. 2003;77(17):9183–91. doi: 10.1128/JVI.77.17.9183-9191.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short JJ, Pereboev AV, Kawakami Y, Vasu C, Holterman MJ, Curiel DT. Adenovirus serotype 3 utilizes CD80 (B7.1) and CD86 (B7.2) as cellular attachment receptors. Virology. 2004;322(2):349–59. doi: 10.1016/j.virol.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Short JJ, Vasu C, Holterman MJ, Curiel DT, Pereboev A. Members of adenovirus species B utilize CD80 and CD86 as cellular attachment receptors. Virus Res. 2006;122(1–2):144–53. doi: 10.1016/j.virusres.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirena D, Lilienfeld B, Eisenhut M, Kalin S, Boucke K, Beerli RR, Vogt L, Ruedl C, Bachmann MF, Greber UF, Hemmi S. The human membrane cofactor CD46 is a receptor for species B adenovirus serotype 3. J Virol. 2004;78(9):4454–62. doi: 10.1128/JVI.78.9.4454-4462.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JG, Nemerow GR. Mechanism of adenovirus neutralization by Human alpha-defensins. Cell Host Microbe. 2008;3(1):11–9. doi: 10.1016/j.chom.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Snider C, Jayasinghe S, Hristova K, White SH. MPEx: a tool for exploring membrane proteins. Protein Sci. 2009;18(12):2624–8. doi: 10.1002/pro.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung MT, Cao TM, Lischwe MA, Coleman RT. Molecular processing of adenovirus proteins. J Biol Chem. 1983;258(13):8266–72. [PubMed] [Google Scholar]
- Tuve S, Wang H, Jacobs JD, Yumul RC, Smith DF, Lieber A. Role of cellular heparan sulfate proteoglycans in infection of human adenovirus serotype 3 and 35. PLoS Pathog. 2008;4(10):e1000189. doi: 10.1371/journal.ppat.1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oostrum J, Burnett RM. Molecular composition of the adenovirus type 2 virion. J Virol. 1985;56(2):439–48. doi: 10.1128/jvi.56.2.439-448.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham TJ, Filardo EJ, Cheresh DA, Nemerow GR. Integrin alpha v beta 5 selectively promotes adenovirus mediated cell membrane permeabilization. J Cell Biol. 1994;127(1):257–64. doi: 10.1083/jcb.127.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 1993;73(2):309–19. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J Virol. 2005;79 (4):1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodrich H, Henaff D, Jammart B, Segura-Morales C, Seelmeir S, Coux O, Ruzsics Z, Wiethoff CM, Kremer EJ. A Capsid-Encoded PPxY-motif Facilitates Adenovirus Entry. PLoS Pathog. 2010;6(3):e1000808. doi: 10.1371/journal.ppat.1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfart CE, Svensson UK, Everitt E. Interaction between HeLa cells and adenovirus type 2 virions neutralized by different antisera. J Virol. 1985;56(3):896–903. doi: 10.1128/jvi.56.3.896-903.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E, Trauger SA, Pache L, Mullen TM, von Seggern DJ, Siuzdak G, Nemerow GR. Membrane cofactor protein is a receptor for adenoviruses associated with epidemic keratoconjunctivitis. J Virol. 2004;78(8):3897–905. doi: 10.1128/JVI.78.8.3897-3905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Agosto MA, Ivanovic T, King DS, Nibert ML, Harrison SC. Requirements for the formation of membrane pores by the reovirus myristoylated micro1N peptide. J Virol. 2009;83(14):7004–14. doi: 10.1128/JVI.00377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerberg J, Kozlov MM. How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol. 2006;7(1):9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]