

Abstract
Proteomics is a highly informative approach to analyze cancer-associated transformation in tissues. The main challenge to use a tissue for proteomics studies is the small sample size and difficulties to extract and preserve proteins. The choice of a buffer compatible with proteomics applications is also a challenge. Here we describe a protocol optimized for the most efficient extraction of proteins from the human breast tissue in a buffer compatible with two-dimensional gel electrophoresis (2D-GE). This protocol is based on mechanically assisted disintegration of tissues directly in the 2D-GE buffer. Our method is simple, robust and easy to apply in clinical practice. We demonstrate high quality of separation of proteins prepared according to the reported here protocol.
Keywords: 2D-GE, breast tissue, sample preparation, SBO4 buffer
Introduction
Proteins are main components of the organisms. They control many different and important functions related to the cell proliferation, differentiation and death. Changes in the protein expression and structure affect protein’s functions.1 A number of reports have shown that even small changes in protein functions may lead to a disease development.2 The ultimate role of proteins in tumorigenesis has also been reported.3 Proteins are primary targets of anti-cancer drugs and the source of cancer markers.4
Tissue is a perfect source for monitoring changes and modifications of proteins that are related to carcinogenesis. However, there are pitfalls in using tissues for proteome analysis. For the first, the breast tissue is not a homogenous substance, but consists of a number of various cells and extracellular matrix, such as stromal and blood cells, adipocytes, collagen, etc. For the second, non-protein elements are present in tissues. In the case of the breast tissue, the high content of lipids may have a strong impact on protein extraction and separation.
Therefore, several issues must be solved for an efficient preparation of proteins from tissues: 1) maximal efficiency of extractions and 2) removal of non-protein components must be achieved, and 3) extracted proteins have to be under controlled conditions to ensure their suitability for a proteomics study in terms of concentration and stability upon storage. Table 1 presents these 3 challenges, and suggests how they may be solved.
Table 1.
Sample preparation from the tissue for 2D-GE experiments.

For proteomics applications, proteins have to be solubilized in buffers compatible with separation techniques. For two-dimensional electrophoresis, it has to be a low-conductivity buffer, e.g. urea containing buffer. For shotgun applications, proteins are digested directly by a protease, and therefore have to be solubilized in a digestion-compatible buffer, e.g. ammonium bicarbonate buffer. It is known that the best solubilization of proteins requires strong detergents, e.g. SDS, in a buffer containing salts, such as Tris-HCl buffers and NaCl. Such solubilization conditions are not compatible with a direct analysis of extracted proteins by proteomics techniques. Protein purification procedures often lead to loses of proteins. Especially significant losses can be upon protein precipitation followed by a solubilization from a pellet, or upon an extensive dialysis. Therefore, there is a need for a robust and easy protocol for extraction of proteins, which would be compatible with protein separation by 2D-GE. Here we report a protocol which is easy to use and allows protein solubilization directly in a buffer compatible with 2D-GE.
Materials and Methods
Studied samples
Cell line K562 (human myeloid leukemia) was obtained from ATCC (Manassas, USA). As a control tissue, we used a commercially available fresh frozen chicken liver. Breast tissue was collected at Broomfield Hospital (Chelmsford, UK), under Ethical Permit 04/Q0303/28, issued by the North and Mid Essex Local Research Ethics Committee (Harlow, UK). Clinical samples were collected immediately upon surgery and stored on wet ice before being dissected by a pathologist. Samples of breast epithelial tissue were immediately frozen in liquid nitrogen. K562 cells were used as a control to determine an efficiency of proteins extraction and a quality of proteins separation, as compared to the tissue. Frozen chicken liver tissue was used as a model to optimize the protocol for tissue extraction in terms of exploring an impact of interfering detergents, salts and any other chemicals in used buffers and solutions. As frozen chicken liver is available commercially, it allowed performing a significant part of trials without using the human breast tissue. The final optimization and validation of the protocol was performed with an aliquot of the human breast tissue.
Composition of buffers
1% Triton X-100 buffer contained 1% Triton X-100, 20 mM Tris-HCl pH. 8.0, 150 mM NaCl and Protease Inhibitor Cocktail “Complete” (Roche) as recommended by the supplier. RIPA buffer contained phosphate buffer saline (PBS), 0,1%, SDS, 1% NP-40, and 0,5% sodium deoxycholate. Urea buffer contained 8 M urea, 2% (w\v) CHAPS, 50 mM DTT, and 0.8% (v\v) ampholytes.
Protein concentration measurement
Bradford reagent (Bio-Rad), 2D-Quant Kit (Amersham/GE Healthcare) and RC\DC Kit (Bio-Rad) were used as recommended by the suppliers.
Two-dimensional gel electrophoresis
2D-GE was performed as described earlier.5 In brief, total amount of proteins loaded was from 50 μg to 100 μg per an IPGstrip. Proteins were solubilized in the rehydration buffer containing 8 M urea, 2% (w\v) CHAPS, 50 mM DTT, 0.8% (v\v) ampholytes (pH 3–10), and were loaded on the 18 cm NL-IPG strips pH 3–10, (GE Healthcare, Uppsala, Sweden). Strips were rehydrated in the urea-containing buffer for 12 hours. Isoelectrofocusing (IEF) was performed with stepwise increasing voltage as follows: 50 v for 10 min, 100 v for 30 min, 500 v for 1 h, 1000 v for 1 h and 5000 v for the time needed to reach 35,000 Vh. After isoelectrofocusing was completed, strips were equilibrated for 15 min as described earlier.6 12% PAGE was performed at 70 W/6 gels for 7 hours. Proteins were visualized by staining with 0.2% silver nitrate as described.7
Results and Discussion
Table 2 presents the optimized protocol for extraction of proteins from breast tissue in a solution compatible for 2D-GE. Briefly, the protocol consists of 3 following steps: 1) evaluation of a sample, 2) extraction of proteins, and 3) evaluation of a quantity of extracted proteins.
Table 2.
Detailed description of the protocol and procedure.
| Step by step protocol (overview) |
|
||
| Steps | Description | Reagents and tools | Troubleshoots |
| Evaluation of a sample | Clinical samples have to be evaluated by a pathologist. Quantity and quality of cellular, stromal and other histological features have to be evaluated. | Light microscope. | Selection of a non-appropriate sample will affect results of the experiment. |
| Extraction of proteins from the tissue | Tissue must be homogenized, proteins extracted and separated from the pellet. Sonication can be performed in steps, eg, 3 times of 10 min each. It is important that the sample is not heated upon extraction. Centrifugation can be repeated to improve separation from lipids and the pellet. | Urea buffer: 8 M urea, 2% (w\v) ChAPS, 50 mM DTT, 0.8% (v\v) ampholytes (pH range of ampholytes depends on used strips). Glass beads can be added in a proportion of 1/3 (beads/sample; v/v). Water-bath, sonicator, a centrifuge with cooling. | Particles, lipids and impurities in the sample lead to the distortions of protein separation during 2D-GE. During extraction procedure, it is important to control temperature. Especially during sonication and centrifugation. Urea may crystallize at low temperature (eg, +4 °C), while temperature higher than +20 °C may induce degradation processes in a sample. Shorter than 30 min centrifugation time may not be sufficient for efficient separation. |
| Evaluation of protein concentration | Optimal concentration of the protein required for achieving good quality of 2D gels. Usually, 80–100 μg of protein is enough to prepare one 2D maxi-gel (20 cm × 20 cm). | RC/DC kit or a similar kit. | Overload with proteins will make analysis of the separated proteins difficult due to very intense staining. If concentration is too low only few protein will be visualized in the 2D gels. |
The first step in this protocol is an evaluation of the sample by a pathologist. This is required for an estimation of the presence of the cellular and stromal components. Specifically, relative volume of malignant cells, tumor fibroblasts, adipocytes, cells of the immune system and vessels have to be evaluated. The sections have to be taken from the same sample that is prepared for 2D-GE. As a rule, the part of the malignant tumor cells has to be not less than 50%. It also has to be noted whether there are any areas of necrosis or macrophage infiltration, which should be excluded from proteomics analysis. The necrosis is often manifested as areas with destroyed tumor cells, fragments of cells and infiltration of macrophages. Macrophages can be identified by their specific staining pattern and morphology, eg, strong staining with Hematoxylin-Eosin and appearance as multiple dots in sections. Such evaluation has to be performed by a trained pathologist. The sections of the biopsies have to be stored for a validation study, when expression of proteins of interest identified by proteomics would be monitored by immunohistochemistry with specific antibodies in the sections of the same sample.
The first step in protein extraction from the tissue is selection of an optimal extraction buffer. We prepared experiments with buffers that contained different detergents and concentration (Fig. 1), because different detergents solubilize cells membranes with different efficiencies.8,9 Extraction conditions (time, treatments) were similar for all tested conditions. For composition of tested buffers, see the materials and methods section. The output of experiments was monitored by intensity of prepared and separated in 1D SDS-PAGE gels proteins, stained with coomassie blue R-250 (Sigma). To evaluate the maximal level of protein extraction, a similar sample was extracted using SDS. SDS provides maximal extraction, but is not compatible with 2D separation due to SDS interfering with isoelectrofocusing of proteins.10 We estimated the yield of proteins by comparing quantities of proteins extracted using our protocol and a direct boiling in 2.5% SDS-containing electrophoresis sample buffer, followed by 1D SDS-PAGE. An efficiency of protein extraction was evaluated as a quantity of proteins detected in 1D SDS-PAGE. We observed that the highest efficiency of protein recovery with 2D-GE-compatible solution was in the case of direct extraction with urea buffer (Fig. 1).
Figure 1.
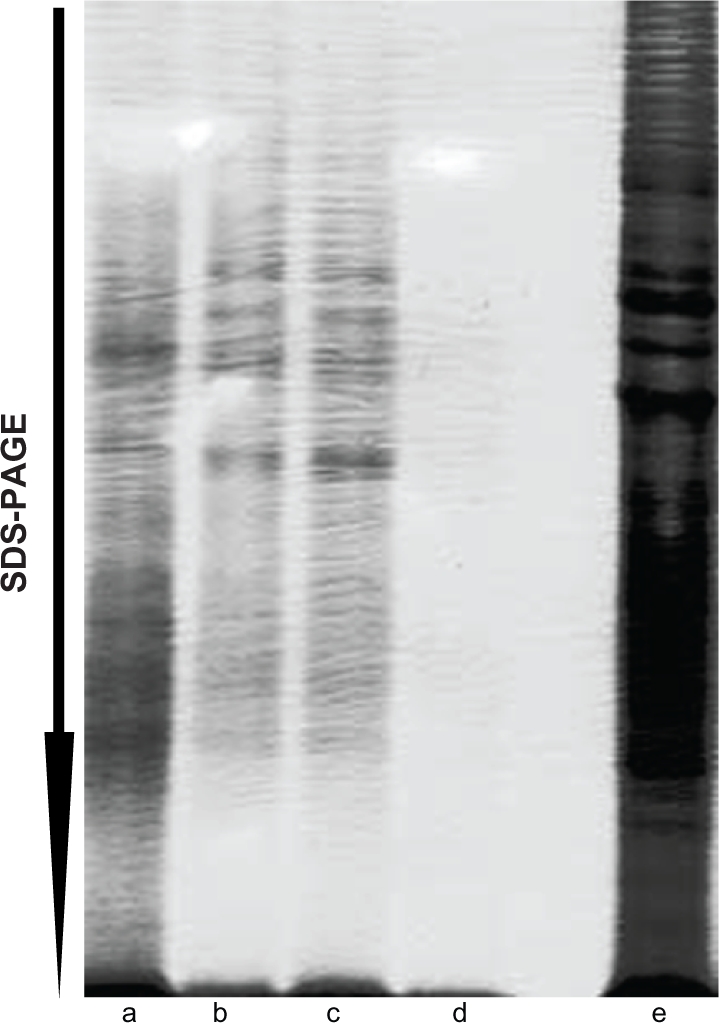
Optimization of protein solubilization and recovery in urea-containing solution. 1D SDS-PAGE of proteins prepared by direct extraction (a, e), or by extraction, precipitation and re-solubilization (b–d). Proteins were prepared by a) direct extraction in the urea-containing solution. For other extraction/precipitation/re-solubilization, proteins were b) extracted in 1% Triton X100-containing buffer, c) in RIPA buffer, d) in 1% SDS, followed by precipitation with 20% acetic acid and 40% methanol, and solubilization in the urea-containing solution. e) As a control of maximal extraction, proteins were extracted directly with 1% SDS. Separated proteins were stained with Coomassie Briliant Blue R-250.
We performed tests to find optimal conditions for extraction, separation and clarification of the proteins extracted from the tissue (Fig. 2). We optimized further condition of the extraction using a tissue sample (frozen chicken liver). This tissue was available commercially, and allowed extensive optimization experiments. The best results were achieved by combining disruption of the tissue with glass beads, sonication on ice and prolonged time of the centrifugation (Table 2). The use of glass beads significantly improved disintegration of tissue, as compared to vortexing or extraction by end-to-end mixing (data not shown). Sonication up to 30 min was found to be important for an efficient separation of the lipid fraction that was observed as a layer on the top of the protein extract. Shorter sonication times were less efficient (data not shown). Prolonged centrifugation for 30 min, as compared to often used 10–15 min, was found to be more efficient for separation of lipids and formation of a pellet (data not shown).
Figure 2.

Mechanical disintegration enhanced extraction of proteins. Extraction of proteins from fresh frozen chicken liver was monitored after extraction of proteins after sonication only (a–c), after treatment with glass beads only (d–f; vortex only) and after sonication with glass beads (g–i). Lanes a, d and g show proteins extracted directly with SDS. Lanes b, e and h show separated proteins extracted with the urea-containing buffer, and lanes c, f and g show proteins extracted with SDS from the pellet after urea-buffer extraction.
To confirm that the presented protocol is suitable for human clinical samples, we used breast tumors and histologically normal adjacent tissues. Proteins from tumors samples were separated in 10% acryl-amide gels prepared according to the described above protocol, and stained with silver nitrate to detect separated proteins. The 2D gel images showed no distortions in separation of proteins (distribution of high or low molecular mass proteins, or preferences in pI of proteins) (Fig. 3). Repeats of sample preparation confirmed reproducibility and good quality of protein extraction and separation in 2D-GE. We estimated that the yield of proteins was in the range of 80% to 100%. It is also important to mention that the extracted proteins were mostly soluble proteins not associated with cellular structures, eg, cytoskeleton (pellet). Thus, validation experiments with human breast tumors and histologically normal tissue confirmed efficiency of the described here protocol for protein extraction.
Figure 3.

No disturbance of protein separation in 2D gel electrophoresis of clinical samples prepared according to the described protocol. Various quantities of proteins prepared from human breast tumors separated by 2D-GE. Note lack of distortions in protein migration. Protein quantities were 100 μg (a), 85 μg (b) and 70 μg (c). Gradient of pH and migration of molecular mass markers are shown. Gels were stained with silver.
Conclusion
Here we report a protocol suitable for an efficient extraction of proteins from breast tissue. The high extraction and preservation of proteins was achieved by using glass beads for disruption and extraction in the urea-containing buffer. Sonication and prolonged time of centrifugation allowed removing contaminants and small particles that can distort protein separation. This is especially valid for removal of lipids. The proposed protocol may also be used for other tissues with minimal optimization.
Acknowledgments
We are grateful to Dr. Elena Ossipova and Dr. Kie Kasuga for comments and discussion. We are grateful to the patients that consented to donate their tissues, the surgeons (Professor Paul Sauven and Dr. Simon Smith) and the pathologist (Dr. Khalid-Al Janabi) for their expert help. This work was supported by grants from the Swedish Cancer Research Foundation, the Swedish Research Council, RTN EpiPlast Carcinoma, the Swedish Institute, KI/KBC to S.S, and by the Helen Rollason Charity support.
Footnotes
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
References
- 1.Bartsch R, Ziebermayr R, Zielinski C, et al. Triple-negative breast cancer. Wien Med Wochenschr. 2010;160(7–8):174–81. doi: 10.1007/s10354-010-0773-6. [DOI] [PubMed] [Google Scholar]
- 2.Wu C, Zhang Y. Active DNA demethylation: many roads lead to Rome. v Nat Rev Mol Cell Biol. 2010;11(9):607–20. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen G, Ravid K. Polyploidy: mechanisms and cancer promotion in hematopoietic and other cells. Adv Exp Med Biol. 2010;676:105–22. doi: 10.1007/978-1-4419-6199-0_7. [DOI] [PubMed] [Google Scholar]
- 4.Steinman S, Wang J, Bourne P, Yang Q, Tang P. Expression of cytokeratin markers, ER-existing invasive ductal carcinoma (IDC) of the breast. Ann Clin Lab Sci. 2007;37:127–34. [PubMed] [Google Scholar]
- 5.Zakharchenko O, Greenwood C, Lewandowska A, Hellman U, Alldridge L, Souchelnytskyi S. Meta-data analysis as a strategy to evaluate individual and common features of proteome changes in breast cancer. Cancer Genomics and Proteomics. 2010. in press. [PubMed]
- 6.Jia M, Souchelnytskyi N, Hellman U, et al. Proteome profiling of immortalization-to-senescence transition of human breast epithelial cells identified MAP2K3 as a senescence promoting protein which is down regulated in human breast cancer. Proteomics Clin Appl. 2010;4 doi: 10.1002/prca.201000006. in press. [DOI] [PubMed] [Google Scholar]
- 7.Shevchenko A, Wilm M, Vorm O, et al. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 8.Mills EN, Freedman RB. Two-dimensional electrophoresis of membrane proteins. Factors affecting resolution of rat-liver microsomal proteins. Biochim Biophys Acta. 1983 Oct 12;734(2):160–7. doi: 10.1016/0005-2736(83)90114-1. [DOI] [PubMed] [Google Scholar]
- 9.Schuck S, Honsho M, Ekroos K, Shevchenko A, Simons K. Resistance of cell membranes to different detergents. Proc Natl Acad Sci U S A. 2003;100(10):5795–800. doi: 10.1073/pnas.0631579100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perdew GH, Schaup HW, Selivonchick DP. The use of a zwitterionic detergent in two-dimensional gel electrophoresis of trout liver microsomes. Anal Biochem. 1983;135(2):453–5. doi: 10.1016/0003-2697(83)90711-x. [DOI] [PubMed] [Google Scholar]