

Abstract
IL-23 and Th17 cells play important roles in host defense against systemic infections with extracellular bacteria and fungi, although their roles in immunity against localized skin infections are less well defined. Here, the contributions of IL-23 and Th17 cytokines in host defense against cutaneous Candida albicans infection were evaluated. Mice deficient in IL-23 or IL-17A demonstrated delayed healing and decreased IL-17A production after skin infection with C. albicans compared with wild-type mice or mice deficient in IL-12 or IL-22. Histologic examination revealed epidermal hyperplasia overlying infected dermis four days postinoculation in wild-type mice. In IL-23–deficient mice, fungal burden was greater in skin, neither IL-17A nor IL-22 mRNAs were expressed postinfection, and these mice demonstrated only minimal epidermal hyperplasia. Exogenous recombinant IL-17A injected at the site of skin infection promoted more rapid healing of candidiasis in both wild-type mice and mice deficient in IL-23 and IL-12. Taken together, these results demonstrate that IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal host defense against cutaneous candidiasis. In addition, recombinant IL-17A may serve as a potential therapy to enhance healing in individuals with chronic cutaneous candidiasis.
Cutaneous infections with the ubiquitous yeast Candida albicans are common throughout the world (1). Although many infections occur in warm, moist areas of the body, such as in the inguinal and inframammary folds, in otherwise healthy individuals, cutaneous candidiasis is also an important marker of innate or acquired immune dysfunction. For example, individuals with diabetes mellitus, HIV disease, chronic systemic corticosteroid usage, or chemotherapy-induced neutropenia are predisposed to cutaneous candidiasis (2–5). This diverse range of clinical settings associated with susceptibility to candidiasis fits with what is currently known about immune responses directed against C. albicans, as host defense against this pathogen involves complex coordination of both innate and acquired immune responses. Pattern recognition of organisms via TLRs (e.g., TLR2 and TLR4) and C-type lectin receptors (e.g., dectin-1, dectin-2, and mannose receptor), Th1 and Th17 number and function, and neutrophil number and function have all be reported to be critical pieces in controlling infection by C. albicans (6–10).
Regarding T cell studies, Th1 cells and IFN-γ production are believed to be critical in host defense against C. albicans (11–15). IL-12 production, priming of Th1 responses, and IFN-γ production were critical for optimal control of C. albicans infection (14, 15). In IFN-γR–deficient mice, Th2 responses were enhanced, which led to increased susceptibility to infection (15). These studies suggest that Th1 responses promote resistance to C. albicans infection, whereas Th2 cytokines are associated with increased susceptibility to infection.
Th17 cells play a role in host defense against C. albicans as well (16, 17). Th17 cells produce cytokines, principally IL-17A and IL-22, which mediate protection from infection at mucocutaneous surfaces (18, 19). Specifically, IL-17A stimulates neutrophil accumulation and function, whereas IL-22 stimulates proliferation and antimicrobial peptide production by epithelial cells, key defense strategies designed to combat infection. Notably, individuals with Job's syndrome (also known as hyper-IgE syndrome) have been found to have mutations in STAT3 that lead to a deficiency in Th17 cells, rendering them susceptible to chronic mucocutaneous infections with C. albicans (20–22). Furthermore, humans with mutations in dectin-1 or a protein downstream of dectin-1 signaling, CARD9, have defective IL-23 production, subsequent decreases in IL-17A production by Th17 cells, and increased susceptibility to chronic mucocutaneous candidiasis (23, 24). Collectively, these data suggest that both IL-12/Th1 responses and IL-23/Th17 responses contribute to host defense against C. albicans. It is unclear, however, which response predominates in combating cutaneous candidiasis.
To define further the specific roles of IL-12 and IL-23 as well as cytokines produced by Th17 cells (e.g., IL-17A and IL-22) in host defense against C. albicans, a cutaneous infection model in mice was established. This model involved the inoculation of C. albicans into the dorsal back skin of various strains of mice that are genetically deficient in key components of the IL-12/Th1 and IL-23/Th17 inflammatory pathways, and the ensuing healing rates, fungal burden, leukocyte infiltration, epidermal hyperplasia, and cytokine production were compared.
Materials and Methods
Mice
All breeding and experiments were undertaken with review and approval from the Portland Veterans Affairs Medical Center Institutional Animal Care and Use Committee. C57BL/6 wild-type (WT) mice, IL-12p35−/− mice, and IL-12/23p40−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME). Through material transfer agreements, IL-17A−/− mice were obtained from Dr. Yoichiro Iwakura (Center for Experimental Medicine, Institute of Medical Science, University of Tokyo, Japan) (25), and IL-22−/− mice and IL-23p19−/− mice were obtained from Dr. Wenjun Ouyang and Dr. Frederic de Sauvage (Genentech, South San Francisco, CA) (26, 27). All knockout mice had been bred for many years on a C57BL/6 background.
C. albicans
C. albicans (strain SC5314) was obtained from Dr. Janet Staab (The Johns Hopkins University, Baltimore, MD) and maintained in 1%yeast extract, 2% peptone, 2% dextrose (YPD). To generate pseudohyphae, an isolate of strain SC5314 was grown in YPD at 30°C with shaking for 16 h. Cells were adjusted to 5 × 106 cells/ml in YPD with 10% FBS and incubated at 37°C for 2 h. Microscopic examination confirmed that >95% of the cells converted to pseudohyphae.
Clinical course of C. albicans-injected mice
C. albicans pseudohyphae (5 × 106 cells) were injected into the deep dermis and superficial fat of the shaved dorsal region of recipient mice. In some experiments, mice were injected with a mixture of C. albicans pseudohyphae and 1 mg recombinant murine IL-17A (R&D Systems, Minneapolis, MN). Mice were assessed for presence of nodule, ulceration, erythema, and crust three times weekly thereafter by an evaluator blinded to the genotype of the mice. Disease was scored positively if any one of the four clinical features was present. In three separate experiments, at least 12 mice for each genetic background were studied for the clinical course experiments. In the experiments using recombinant murine IL-17A, a total of seven mice in each treatment group were studied in three separate experiments.
Immunohistochemistry
A dose of 5 × 104 C. albicans pseudohyphae was injected to improve recovery of intact skin tissue from infected skin. Four days postinfection, 4-mm punch biopsies were performed in the center of the C. albicans nodules. Sections from formalin-fixed, paraffin-embedded biopsies were stained. Blocking solutions, primary Abs, and secondary Abs are shown in Supplemental Table 1. For immunohistochemistry, sections were deparaffinized and hydrated by washing sections in xylene followed by a graded alcohol series. The sections were incubated in 10 mM citric acid (pH 6) at 95°C for 30 min to unmask Ags, and endogenous peroxidase activity was quenched by treating sections with 3% hydrogen peroxide for 5 min at room temperature. Sections were blocked for 60 min at room temperature, followed by incubation with primary Abs overnight at 4°C. Samples were washed and incubated at room temperature for 60 min with secondary Abs, followed by a 30-min incubation with streptavidin-conjugated HRP (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA), developed using diaminobenzidine substrate kit for peroxidase (Vector Laboratories), and counterstained with hematoxylin (Vector Laboratories). To quantify epidermal hyperplasia, defined as area/length, randomly chosen fields for each section were measured using Image Pro software (version 4.5; Media Cybernetics, Bethesda, MD). To quantify Gr-1+ cells, an observer blinded to the genotype of the mice graded Gr-1–stained specimens from 0 (few) to 3 (many). At least three mice per group were stained, and photographs were taken from three representative ×40 magnification fields per section. Average numbers of positive cells per picture were counted for quantification.
Fungal burden assay
Mice were injected with 5 × 106 pseudohyphae as described above, and 4-mm punch biopsies were taken at the injection site 4 d postinfection. The biopsies were weighed and then gently homogenized using a Dounce homogenizer. Homogenates were diluted, and 100-μl aliquots were plated onto YPD growth medium to permit Candida colony growth. Plates were incubated at room temperature for 2 d, and the numbers of CFU per gram of tissue were calculated. Experiments were repeated at least three times.
RNA isolation and quantitative real-time PCR
Infected skin was collected, placed into TRIzol (Invitrogen, Carlsbad, CA), and homogenized with a mechanical rotor for 1 min. Total RNA was isolated and purified using PureLink RNA Mini Kit (Invitrogen) with the protocol for using TRIzol with the PureLink RNA Mini Kit as recommended by the manufacturer. cDNA was prepared from 1 μg total RNA by reverse transcription using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). Reactions were diluted three times with water for all quantitative real-time PCR (qPCR) experiments. qPCR was performed on the MyiQ system (Bio-Rad) using TaqMan Gene Expression Master Mix with the following TaqMan primers and fluorescent probes from Applied Biosystems: Gapdh (catalog no. Mm9999991-5_g1), Il23a (catalog no. Mm00518984_m1), Il12b (catalog no. Mm01288990_m1), Il12a (catalog no. Mm01218136_m1), Il17a (catalog no. Mm01268754_m1), Il22 (catalog no. Mm01226722_g1), Ifnγ (catalog no. Mm00801778_m1), Il17f (catalog no. Mm00521423_m1), Il21 (catalog no. Mm00517640_m1), and Tnfα (catalog no. Mm00443258_m1). Expression of each transcript was calculated relative to Gapdh expression.
Protein isolation and ELISA
Infected skin was collected and protein was extracted using the Qproteome Mammalian Protein kit (Qiagen, Germantown, MD). Briefly, skin was placed in lysis buffer containing benzonuclease and a protease inhibitor solution and homogenized for 1 min with a mechanical rotor. Homogenates were then centrifuged, and the clear supernatants were collected. Mouse IL-17A and IL-22 ELISA kits (R&D Systems) and mouse IL-21 ELISA kit (eBioscience, San Diego, CA) were used according to the manufacturer's instructions.
Statistical analyses
The chi-square goodness of fit test was used to evaluate normality for all parameters. The F test and Bartlett test were used to evaluate equality of variance between two or more groups, respectively. Student t test, Welch's t test, or Mann–Whitney U test was used for comparison between WT and knockout mice. For testing equality of population means among three or more groups, Kruskal–Wallis test and Scheffe's F test were used. Kaplan–Meier method and log rank test were used for the analysis of clinical course. The p values ≤ 0.05 were considered significant.
Results
IL-23–deficient mice have impaired clearance of C. albicans skin infection
After inoculation of C. albicans within skin, WT mice developed visible skin nodules that took a median of 13 d to heal (Fig. 1A). By contrast, IL-23–deficient mice (IL-23p19−/− mice) and mice deficient in both IL-12 and IL-23 (IL-12/23p40−/− mice) developed skin lesions that took much longer to heal with a median of >15 d (p < 0.025 versus WT mice for both knockout strains) (Fig. 1A). Furthermore, 20 d postinfection, all WT mice had resolved the infection versus only 50% of the IL-12/23p40−/− mice and 62% of the IL-23p19−/− mice. IL-12–deficient (IL-12p35−/−) mice healed Candida lesions in a time span similar to that of WT mice (p < 0.19; Fig. 1A). IL-12/23p40−/− mice also demonstrated higher fungal burdens in skin compared with that in WT mice (p < 0.001; Fig. 1B), which was consistent with the clinical course (Fig. 1A). The average CFU/gram of tissue obtained from IL-23p19−/− mice was higher than that of WT mice, although this was not statistically significant (p < 0.066; Fig. 1B).
FIGURE 1.

Optimal resolution of cutaneous C. albicans infection is dependent upon IL-23. A, Kaplan–Meier curves demonstrating the percentage of C. albicans-infected mice with disease, as defined by the presence of nodule, ulcer, erythema, or crust at the skin site of inoculation. This was assessed by an investigator (blinded to the genotype of the mice) three times weekly postinfection. In three separate experiments, at least 12 mice for each genetic background were evaluated. *p < 0.025 versus WT. B, Quantification of C. albicans fungal burden in skin 4 d after inoculation. Infected tissue was isolated, weighed, minced, and cultured on agar plates to determine CFU/gram skin tissue. **p < 0.025 versus WT.
Histologic examinations of WT mouse skin 4 d postinfection showed a large collection of fungal organisms deep within the dermis and superficial fat (Fig. 2A) surrounded by numerous Gr-1+ neutrophils (Fig. 2A). The number of CD3+ cells was decreased in IL-23p19−/− mice and IL-12p35−/− mice (p < 0.01 and p < 0.05 versus WT mice, respectively; Fig. 2A, 2B). There were less F4/80+ cells in IL-23p19−/− mice than in WT mice (p < 0.05; Fig. 2A, 2B). Fewer neutrophils infiltrated infection sites of IL-12/23p40−/− mice compared with that in WT mice (p < 0.05; Fig. 2A, 2B).
FIGURE 2.

Increased numbers of infiltrating cells in WT mouse skin compared with knockout mouse skin. A, H&E, CD3 (T cells), F4/80 (macrophages), and Gr-1 (neutrophils) staining of skin 4 d postinfection with C. albicans showing a large collection of fungal elements in the deep dermis and superficial fat. H&E and Gr-1 staining, original magnification ×10; scale bars, 0.2 mm. CD3 and F4/80 staining, original magnification ×40; scale bars, 0.025 mm. B, Quantification of CD3+ and F4/80+ cells and grading of Gr-1 staining in each of the four groups of mice shown in A. Data are expressed as mean ± SD. Bar graphs were analyzed with Kruskal–Wallis test and Scheffe's F test.*p < 0.05; **p < 0.01 versus WT.
C. albicans induces epidermal hyperplasia overlying infected dermis that is dependent on IL-23
Notably, histologic analyses of infected skin of WT mice also revealed prominent epidermal hyperplasia overlying the nidus of infection in the deep dermis compared with nonlesional skin of WT mice (mean ± SD of thickness = 47.7 ± 8.4 μm and 19.0 ± 3.0 μm, respectively, p < 0.001; Fig. 3A, 3B). In mice deficient in IL-23 (IL-23p19−/− mice) and mice deficient in both IL-12 and IL-23 (IL-12/23p40−/− mice), epidermal hyperplasia was much less prominent compared with WT epidermis (26.1 ± 4.5 μm and 23.1 ± 4.9 μm, respectively, p < 0.001 versus WT mice for both strains; Fig. 3A, 3B). In contrast, infected skin of IL-12–deficient mice (IL-12p35−/− mice) developed prominent epidermal hyperplasia that was similar to the epidermal hyperplasia observed in WT mice (Fig. 3A, 3B).
FIGURE 3.

C. albicans infection in skin induces epidermal hyperplasia that is dependent upon IL-23. A, H&E staining of mouse skin 4 d postinfection with C. albicans. Original magnification ×20; scale bars, 0.05 mm. B, Quantification of epidermal hyperplasia in each of the five groups of mice in A. Data are expressed as mean epidermal thickness (μm) ± SD. Data were analyzed using Kruskal–Wallis test and Scheffe's F test. *p < 0.001 versus WT.
C. albicans induces rapid upregulation of IL-12 and IL-23 expression within infected skin
To determine whether IL-12 and IL-23 expression was induced in skin after C. albicans infection, RNA was isolated from WT skin at various time points postinfection, and IL-23p19, IL-12p35, and IL-12/23p40 mRNAs were measured by qPCR. Prior to infection, mRNA levels for IL-23p19, IL-12p35, and IL-12/23p40 were absent or minimal (data not shown). In contrast, mRNA expression for all three subunits peaked at 24 h in WT mice after skin inoculation with C. albicans (data not shown), and thus all data presented are from this time point (Supplemental Fig. 1A). IL-23p19 mRNA levels were induced to high levels in WT mice, IL-12p35−/− mice, and IL-12/23p40−/− mice. IL-12p35 mRNA levels were induced to high levels in WT mice, IL-23p19−/− mice, and IL-12/23p40−/− mice. As an expected control, IL-23p19, IL-12p35, and IL-12/23p40 mRNA levels were absent in IL-23p19−/− mice, IL-23p35−/− mice, and IL-12/23p40−/− mice, respectively. Thus, the presence of IL-12 in IL-23p19−/− mice could not compensate for the lack of IL-23 in these mice, suggesting that IL-23 is a more critical determinant than IL-12 to the increased healing times observed in IL-23p19−/− mice. Confirmation of mRNA results was performed via protein analysis using immunohistochemistry. There were more IL-23p19+ cells in WT mice and IL-12p35−/− mice compared with that in IL-23p19−/− mice and IL-12/23p40−/− mice (Supplemental Fig. 1B, 1C). The majority of IL-23p19+ cells were also positive for F4/80, indicating that macrophages were the primary producers of IL-23 in this model (Supplemental Fig. 1D).
IL-17A and IL-22 expression in C. albicans-infected skin are dependent on IL-23
To investigate the downstream effects of activation of the IL-12/Th1 and IL-23/Th17 inflammatory pathways postinfection, mRNA expression levels of IL-17A, IL-17F, IL-22, IL-21, IFN-γ, and TNF-α were analyzed. In the absence of infection, mRNA amounts for all of six genes were absent or minimal in WT mice (data not shown). In contrast, mRNA levels of all gene products reached maximal levels by 48 h postinfection in WT mice (data not shown), and thus all data presented are from this time point (Fig. 4). IL-17A, IL-17F, and IL-22 mRNA were not induced in IL-23p19−/− mice or in IL-12/23p40−/− mice after C. albicans cutaneous infection (Fig. 4). IL-12p35−/− mice exhibited similar IL-17A and IL-22 mRNA expression as WT mice (Fig. 4). There were no differences in IL-21 mRNA expression among WT and knockout mice (Fig. 4). IFN-γ mRNA expression was markedly decreased in IL-12p35−/− mice and in IL-12/23p40−/− mice after C. albicans cutaneous infection (p < 0.001 and p < 0.01 versus WT mice, respectively; Fig. 4). Of note, IFN-γ mRNA was increased in IL-23–deficient mice compared with that in WT mice (p < 0.01; Fig. 4). In IL-12/23p40−/− mice, TNF-α mRNA expression was lower compared with that in WT mice (p < 0.05; Fig. 4). Taken together, these data indicated that IL-17A, IL-17F, and IL-22 mRNA production was completely dependent upon IL-23 and that IFN-γ mRNA production was largely dependent upon IL-12.
FIGURE 4.

Impaired production of IL-17A, IL-17F, and IL-22 in IL-23–deficient mice. IL-17A, IL-17F, IL-22, IL-21, IFN-γ, and TNF-α mRNA expression 48 h after C. albicans skin infection in WT and the various knockout mice as measured by qPCR. Each symbol indicates the amount of mRNA transcript as measured by arbitrary units (AU) of a single specimen, and horizontal bars denote the mean of scatterplots. Data were analyzed using Kruskal–Wallis test and Scheffe's F test. IL-17A: *p < 0.001 versus WT. IL-17F: *p < 0.001 versus WT. IL-22: *p < 0.001 versus WT. IFN-γ: *p < 0.01 for IL-23p19−/− and IL-12p35−/− versus WT; *p < 0.01 for IL-12/23p40−/− versus WT. TNF-α: *p < 0.01 versus WT.
Confirmation of mRNA results was performed via protein analyses using ELISA 48 h after C. albicans inoculation and immunohistochemistry 4 d after C. albicans inoculation. Both IL-17A and IL-22 protein levels as measured by ELISA were nearly absent in infected skin lesions of IL-23p19−/− and IL-12/23p40−/− mice (p < 0.025 versus WT mice for both cytokines and both strains of mice; Supplemental Fig. 2). Neither IL-17A+ nor IL-22+ cells were visualized in IL-23p19−/− mice and IL-12/23p40−/− mice (Fig. 5A, 5B), corroborating the mRNA and ELISA data. In contrast, both IL-17A+ and IL-22+ cells were readily observed in the dermis of infected WT and IL-12p35−/− mice (Fig. 5A, 5B). Of note, protein levels of IL-21, which is also expressed by Th17 cells, were equal among WT mice and all of the knockout mice, suggesting that IL-21 production was not dependent on either IL-23 or IL-12 (Supplemental Fig. 2). It should also be mentioned that IFN-γ protein levels as measured by ELISA among all of the samples were below the level of detection (data not shown). Next, we stained serial sections to detect double-positive cells. IL-17A was mainly produced by CD3+ cells; some Gr-1+ cells also produced IL-17A (Fig. 5C). IL-22 was primarily produced by CD3+ cells (Fig. 5C). Some IL-17A+ cells were also IL-22+ (Fig. 5C).
FIGURE 5.

Impaired protein production of IL-17A and IL-22 in IL-23–deficient mice. A, Anti–IL-17A Ab-stained or anti–IL-22 Ab-stained sections of skin 4 d postinfection with C. albicans. Original magnification ×40; scale bars, 0.025 mm. B, Quantification of IL-17A+ and IL-22+ cells in each of the four groups of mice shown in A. Bar graphs were analyzed using Kruskal–Wallis test and Scheffe's F test. IL-17A: *p < 0.05; **p < 0.01 versus WT. IL-22: *p * 0.05; **p < 0.001 versus WT. C, IL-17A, IL-22, and CD3, or IL-17A and Gr-1 staining of serial sections of WT mouse skin 4 d postinfection with C. albicans. Original magnification ×100; scale bars, 0.025 mm. Black arrows indicate cells positive for IL-17A, IL-22, and CD3 (top panels) and cells positive for IL-17A and Gr-1 (bottom panels). Green arrows indicate cells positive for IL-22 and CD3, but negative for IL-17A. Red arrows indicate cells positive for IL-17A and CD3, but negative for IL-22.
IL-17A–deficient mice but not IL-22–deficient mice have impaired clearance of C. albicans skin infection
Because IL-23–deficient mice had defective host defense against cutaneous C. albicans infection and had impaired production of IL-17A and IL-22, the specific roles of IL-17A versus IL-22 in combating cutaneous candidiasis were investigated further using IL-17A−/− and IL-22−/− mice. IL-17A−/− mice developed skin lesions that took longer to heal than those in WT mice with a median of >15 d (p < 0.025 versus WT mice; Fig. 6A). In contrast, IL-22−/− mice healed Candida lesions in a similar time span as that of WT mice (Fig. 6A). IL-17A−/− mice, but not IL-22−/− mice, demonstrated increased fungal burdens in skin compared with that in WT mice (p < 0.001; Fig. 6B), which is consistent with clinical course data. Four days postinfection, lesional mouse skin from IL-17A−/− and IL-22−/− exhibited fewer CD3+ cells than skin from WT mice (p < 0.01 and p < 0.05, respectively; Fig. 7A, 7B). However, both strains of knockout mice showed similar levels of Gr-1+ and F4/80+ cells compared with those in WT mice (Fig. 7A, 7B).
FIGURE 6.

Optimal resolution of cutaneous C. albicans infection is dependent upon IL-17A but not IL-22. A, Kaplan–Meier curves demonstrating the percentage of C. albicans-infected mice with disease, as defined by the presence of nodule, ulcer, erythema, or crust at the skin site of inoculation. This was assessed by an investigator (blinded to the genotype of the mice) three times weekly postinfection. *p < 0.025 versus WT. B, Quantification of C. albicans fungal burden in skin 4 d after inoculation. Infected tissue was isolated, weighed, minced, and cultured on agar plates to determine CFU/gram skin tissue. *p < 0.001 versus WT.
FIGURE 7.

Higher numbers of CD3+ T cells in WT mouse skin compared with that in IL-17A−/− and IL-22−/− mice. A, H&E, CD3, F4/80, and Gr-1 staining of skin 4 d postinfection with C. albicans showing a large collection of fungal elements in the deep dermis and superficial fat. H&E and Gr-1 staining, original magnification ×10; scale bars, 0.2 mm. CD3 and F4/80 staining, original magnification ×40; scale bars, 0.025 mm. B, Quantification of CD3+ and F4/80+ cells and grading of Gr-1 staining in each of the four groups of mice shown in A. Data are expressed as mean ± SD. Bar graphs were analyzed with Kruskal–Wallis test and Scheffe's F test. *p < 0.025; **p, 0.001 versus WT.
C. albicans induces epidermal hyperplasia overlying infected dermis in mice deficient in either IL-17A or IL-22
Infected skin of IL-17A−/− and IL-22−/− mice showed prominent epidermal hyperplasia that was similar to WT mice (52.0 ± 10.9 μm and 43.8 ± 10.2 μm, respectively, versus 47.7 ± 8.4 μm in WT mice; Supplemental Fig. 3A, 3B). Thus, epidermal hyperplasia induced during cutaneous C. albicans infections was not dependent upon IL-17 or IL-22. Furthermore, these results suggest that the presence of prominent epidermal hyperplasia as seen in IL-17A−/− mice and IL-22−/− mice did not play a major role in contributing to the delayed Candida healing times as observed in IL-23p19−/− and IL-12/23p40−/− mice (Fig. 1A).
IL-22 is expressed in IL-17A−/− mice and IL-17A is expressed in IL-22−/− mice after cutaneous C. albicans infection
Induction of IL-23p19, IL-12/23p40, and IL-12p35 mRNA levels in response to cutaneous C. albicans infection was similar in WT, IL-17A−/−, and IL-22−/− mice (data not shown). IL-17A mRNA was induced similarly in IL-22−/− mice and WT mice (Fig. 8A). IL-22 mRNA expression, however, was significantly decreased in IL-17A−/− mice compared with that in WT mice (p < 0.001). As an expected control, IL-17A expression was absent in IL-17A−/− mice, and IL-22 expression was absent in IL-22−/− mice (p < 0.001). These data suggest that IL-17A contributes to regulation of IL-22 mRNA expression, but not vice versa. TNF-α mRNA expression was decreased in both IL-17A−/− mice and IL-22−/− mice (p < 0.01). In contrast with the differential mRNA induction of other cytokines, IL-17F, IFN-γ, and IL-21 mRNA expression was similar among IL-17A−/−, IL-22−/−, and WT mice (Fig. 8). To confirm the mRNA results, protein analyses using ELISA and immunohistochemistry were performed. The ELISA data demonstrated similar findings to the mRNA data where IL-17A protein levels were induced similarly in IL-22−/− mice and WT mice (Supplemental Fig. 4A). However, in contrast with the mRNA data, IL-22 protein levels were not significantly different between IL-17A−/− mice and WT mice. As expected, protein levels of IL-17A and IL-22 were at or below the level of detection in IL-17A−/− mice and IL-22−/− mice, respectively. IL-21 protein levels were similar in IL-17A−/−, IL-22−/−, and WT mice (Supplemental Fig. 4A). It should also be noted that IFN-γ protein levels as measured by ELISA among all of the samples were below the level of detection (data not shown). Finally, to confirm the mRNA and ELISA data, IL-22+ cells were readily observed in the dermis of infected IL-17A−/− mice, and IL-17A+ cells were readily observed in the dermis of infected IL-22−/− mice (Supplemental Fig. 4B, 4C).
FIGURE 8.

IL-22 expression is decreased in IL-17A−/− mice, whereas IL-17A expression is normal in IL-22−/− mice after cutaneous C. albicans infection. IL-17A, IL-17F, IL-22, IL-21, IFN-γ, and TNF-α mRNA expression at 48 h after C. albicans skin infection in WT and the various knockout mice as measured by qPCR. Each symbol indicates the amount of mRNA transcript (as measured in AU) of a single specimen, and horizontal bars denote the mean of scatterplots. Data were analyzed using Kruskal–Wallis test and Scheffe's F test. IL-17A and IL-22: *p < 0.001 versus WT. IL-17F and TNF-α: *p < 0.01 versus WT. IL-21 and IFN-γ: no differences versus WT.
Recombinant murine IL-17A delivered at the site of skin infection promotes more rapid healing of candidiasis in IL-12/23p40−/− mice
To determine whether exogenous IL-17A could promote healing in mice deficient in both IL-12 and IL-23, we injected a mixture of Candida pseudohyphae and recombinant murine IL-17A into mouse skin on day 0. In a series of three independent experiments performed with a total of seven mice in each treatment group, 14% of WT mice were healed 12 d postinfection, whereas no IL-12/23p40−/− mice were healed at this time point (Fig. 9A). In contrast, 43% of WT mice infected in the presence of exogenous IL-17A were healed and 29% of IL-12/23p40−/− mice infected in the presence of exogenous IL-17A were healed on day 12 (Fig. 9A). Similar trends were observed 19 d postinfection (Fig. 9B). WT mice infected in the presence of exogenous IL-17A healed more quickly (86%) compared with WT mice infected without exogenous IL-17 (57%), and IL-12/23p40−/− mice infected in the presence of exogenous IL-17A healed more quickly (57%) compared with IL-12/23p40−/− mice infected without exogenous IL-17 (29%) (Fig. 9B).
FIGURE 9.
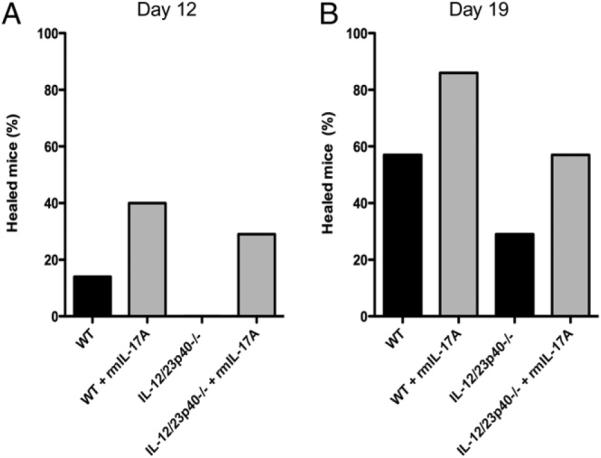
Administration of exogenous recombinant IL-17A at the onset of infection promotes more rapid healing of cutaneous candidiasis in both WT and IL-12/23p40−/− mice. A, The percentage of mice healed 12 d postinfection. B, The percentage of mice healed 19 d postinfection. Data shown were generated after completing three independent experiments using a total of seven mice in each group.
Discussion
The observations reported here demonstrate that host defense against a cutaneous C. albicans challenge is dependent on IL-23 and IL-17A but not on IL-12 and IL-22. Importantly, all of the mice that demonstrated delayed healing of cutaneous candidiasis, that is IL-23p19−/−, IL-12/23p40−/−, and IL-17A−/− mice (Figs. 1A, 5A), shared a deficiency of IL-17A (Figs. 4, 7) and increased fungal burdens in skin (Fig. 1B, 1C and Fig. 5B, 5C). In contrast, mice deficient in IL-12p35, which had levels of IL-17A that were similar to WT mice, had no impairment in healing time. Furthermore, of the Th17 cytokines, IL-17A was shown to be a much more critical determinant for host defense than IL-22, as IL-22–deficient mice did not exhibit delayed healing or increased fungal burdens (Fig. 5). Also of note, IL-22−/− mice expressed relatively normal levels of IL-17A, which likely accounted for their ability to clear C. albicans from skin effectively (Fig. 7). Importantly, administration of exogenous recombinant murine IL-17A directly at the sites of skin infection promoted more rapid healing of candidiasis in both WT and IL-12/23p40−/− mice (Fig. 9). Taken together, these results demonstrate the importance of IL-17A, whose production was dependent upon IL-23, but not IL-12 or IL-22, as a key mediator for an effective immune response against C. albicans within skin.
These findings have direct implications for human disease because they help provide an explanation for the increased susceptibility to C. albicans in patients who suffer from Job's syndrome, chronic mucocutaneous candidiasis, and HIV/AIDS (2, 23, 24, 28). It has recently been described that defective immune responses in Job's syndrome result from a defect in the ability to make Th17 cells due to mutations in STAT3, a transcription factor essential for Th17 cell differentiation (20, 21, 29). In addition, individuals with chronic mucocutaneous candidiasis have T cells that produce low amounts of Th17 cytokines (18, 23, 24). Patients with HIV/AIDS have also been demonstrated to have defects in Th17 responses (30). Thus, these current findings demonstrating that IL-17A is essential for host defense against cutaneous C. albicans infections provide a mechanism by which Th17 dysregulation leads to increased susceptibility to C. albicans infections among individuals with these disparate diseases.
We showed here that both CD3+ T cells and Gr-1+ neutrophils produced IL-17A (Fig. 5), but we cannot rule out the possibility that NK cells and γδ T cells also produced this cytokine. A recent study has demonstrated that IL-17A, which mediated neutrophil recruitment against cutaneous Staphylococcus aureus infection in mice, is produced by resident γδ T cells located in mouse epidermis (31). Thus, IL-17A–producing γδ T cells may also play a role in cutaneous C. albicans infection. Regardless of the source, IL-17A signaling through the IL-17RA receptor activates both the NF-κB and MAPK intracellular pathways within a variety of cell types (32). IL-17A regulates the production of G-CSF, which in turn promotes the proliferation of promyelocytes and maturation of neutrophils. This homeostatic mechanism plays an important role in normal physiology and in host defense against bacterial infections (33). The results shown here do not support an additional major role for IL-17F (Fig. 8), which is closely related to IL-17A and is also produced by Th17 cells (34). A previous report demonstrated that IL-17F levels were elevated in IL-17A−/− mice (35). Thus, IL-17F homodimers, which have similar affinity for the IL-17R, do not appear to compensate for the loss of IL-17A and associated impaired healing of cutaneous candidiasis.
Results shown here using a cutaneous C. albicans infection model are supported by other mouse studies of C. albicans infection in other tissues. IL-17AR–deficient mice had reduced survival rates after systemic Candida challenge (36). In that study, IL-17AR–deficient mice had increased C. albicans burden in the kidneys and impaired neutrophil influx, which resulted in reduced survival compared with WT mice. In addition, another study using a mouse model of oral candidiasis demonstrated that mice deficient in Th17 cells developed more severe disease than Th1 cell-deficient mice (17). Similar to the current study, this study also found that IL-17A was a more critical determinant for host defense against oral C. albicans infection than IL-22, as IL-22–deficient mice were only mildly susceptible to oral disease (17).
It should be mentioned that when epidermal hyperplasia was measured above the C. albicans dermal abscess, IL-23–deficient mice had significantly decreased epidermal thickening compared with that in WT mice or IL-12–deficient mice. These findings indicate that epidermal hyperplasia was mediated by IL-23 and not by IL-12. We identified F4/80+ macrophages as a source of IL-23 (Supplemental Fig. 1); we cannot rule out production by cutaneous dendritic cells. We also observed that C. albicans infection readily induced epidermal hyperplasia in IL-22−/− mice (Fig. 6), suggesting that epidermal hyperplasia observed in response to C. albicans cutaneous infection is not entirely dependent upon IL-22. This is a somewhat surprising finding given that injections of recombinant IL-23 into murine skin induced epidermal hyperplasia in WT mice but failed to do so in mice deficient in IL-22 (26). Thus, IL-23–mediated epidermal hyperplasia induced by a cutaneous pathogen is likely more complex than epidermal hyperplasia induced by recombinant IL-23, involving cytokines other than IL-22. A recent study revealed that injection of IL-21 into mouse skin could also induce epidermal thickening (37). Therefore, IL-21 production was studied here, but no significant differences in IL-21 production were detected among WT and various knockout mice after C. albicans infection, suggesting that IL-21 was not the key mediator for epidermal hyperplasia in this model (Figs. 4B, 7B). Other studies have demonstrated that IL-19, IL-20, and IL-24 can induce epidermal hyperplasia directly (38). IL-23 injection into mouse skin induced IL-19 and IL-24 but not IL-20 mRNA (39). Thus, it could be that multiple inflammatory cytokines downstream of IL-23, such as IL-19 and IL-24, contribute to epidermal hyperplasia induced by cutaneous C. albicans infection.
From a clinical point of view, the findings shown here suggest that mechanisms that enhance IL-23 and/or IL-17A responses may help prevent or treat C. albicans cutaneous infections in humans. This may be particularly important in the development of future immunomodulatory therapy and vaccine development against C. albicans, especially for those patients with altered Th17 responses that suffer from chronic candidiasis. Conversely, therapeutic blockade of IL-23/IL-17A–mediated immune responses may result in increased skin infections with C. albicans. For example, ustekinumab is an mAb directed against IL-12/23p40 and has recently been approved for use in individuals with moderate-to-severe psoriasis (40, 41). Similar drugs that target IL-23 alone or IL-17A are also in clinical development for psoriasis, rheumatoid arthritis, and other T cell-mediated inflammatory diseases. Our results suggest that patients treated with these drugs should be closely monitored for cutaneous infections with C. albicans and perhaps other pathogens that are controlled by IL-23/IL-17A–mediated immune responses. They also suggest that individuals with chronic candidiasis may benefit from exogenous recombinant IL-17A delivered directly into sites of skin infection.
Supplementary Material
Acknowledgments
This work was supported in part by a Veterans Affairs Merit Award (to A.B.), National Institutes of Health Grants 1 R21 AR054495-01A1 (to A.B.) and 1 R01 AI078910 (to L.S.M.), and the Naito Foundation (to S.K.).
Abbreviations used in this paper
- AU
arbitrary units
- qPCR
quantitative PCR
- WT
wild type
- YPD
1% yeast extract, 2% peptone, 2% dextrose
Footnotes
The online version of this article contains supplemental material.
Disclosures The authors have no financial conflicts of interest.
References
- 1.Holland SM, Vinh DC. Yeast infections—human genetics on the rise. N. Engl. J. Med. 2009;361:1798–1801. doi: 10.1056/NEJMe0907186. [DOI] [PubMed] [Google Scholar]
- 2.Ramos-E-Silva M, Lima CM, Schechtman RC, Trope BM, Carneiro S. Superficial mycoses in immunodepressed patients (AIDS) Clin. Dermatol. 2010;28:217–225. doi: 10.1016/j.clindermatol.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Marr KA. Fungal infections in oncology patients: update on epidemiology, prevention, and treatment. Curr. Opin. Oncol. 2010;22:138–142. doi: 10.1097/CCO.0b013e328335a755. [DOI] [PubMed] [Google Scholar]
- 4.Zilberberg MD, Shorr AF. Fungal infections in the ICU. Infect. Dis. Clin. North Am. 2009;23:625–642. doi: 10.1016/j.idc.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Soysa NS, Samaranayake LP, Ellepola AN. Diabetes mellitus as a contributory factor in oral candidosis. Diabet. Med. 2006;23:455–459. doi: 10.1111/j.1464-5491.2005.01701.x. [DOI] [PubMed] [Google Scholar]
- 6.Netea MG, Van Der Graaf CA, Vonk AG, Verschueren I, Van Der Meer JW, Kullberg BJ. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J. Infect. Dis. 2002;185:1483–1489. doi: 10.1086/340511. [DOI] [PubMed] [Google Scholar]
- 7.van de Veerdonk FL, Marijnissen RJ, Kullberg BJ, Koenen HJ, Cheng SC, Joosten I, van den Berg WB, Williams DL, van der Meer JW, Joosten LA, Netea MG. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe. 2009;5:329–340. doi: 10.1016/j.chom.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 8.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, Reis e Sousa C. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 9.Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, Verbeek JS, Ruland J, Tybulewicz V, Brown GD, et al. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J. Exp. Med. 2009;206:2037–2051. doi: 10.1084/jem.20082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richardson M, Rautemaa R. How the host fights against Candida infections. Front. Biosci. 2009;14:4363–4375. doi: 10.2741/3533. [DOI] [PubMed] [Google Scholar]
- 11.Romani L, Mocci S, Bietta C, Lanfaloni L, Puccetti P, Bistoni F. Th1 and Th2 cytokine secretion patterns in murine candidiasis: association of Th1 responses with acquired resistance. Infect. Immun. 1991;59:4647–4654. doi: 10.1128/iai.59.12.4647-4654.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fidel PL, Jr., Lynch ME, Sobel JD. Candida-specific Th1-type responsiveness in mice with experimental vaginal candidiasis. Infect. Immun. 1993;61:4202–4207. doi: 10.1128/iai.61.10.4202-4207.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gozalbo D, Gil ML. IFN-gamma in Candida albicans infections. Front. Biosci. 2009;14:1970–1978. doi: 10.2741/3356. [DOI] [PubMed] [Google Scholar]
- 14.d'Ostiani CF, Del Sero G, Bacci A, Montagnoli C, Spreca A, Mencacci A, Ricciardi-Castagnoli P, Romani L. Dendritic cells discriminate between yeasts and hyphae of the fungus Candida albicans. Implications for initiation of T helper cell immunity in vitro and in vivo. J. Exp. Med. 2000;191:1661–1674. doi: 10.1084/jem.191.10.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cenci E, Mencacci A, Del Sero G, d'Ostiani CF, Mosci P, Bacci A, Montagnoli C, Kopf M, Romani L. IFN-gamma is required for IL-12 responsiveness in mice with Candida albicans infection. J. Immunol. 1998;161:3543–3550. [PubMed] [Google Scholar]
- 16.Matsuzaki G, Umemura M. Interleukin-17 as an effector molecule of innate and acquired immunity against infections. Microbiol. Immunol. 2007;51:1139–1147. doi: 10.1111/j.1348-0421.2007.tb04008.x. [DOI] [PubMed] [Google Scholar]
- 17.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eyerich K, Foerster S, Rombold S, Seidl HP, Behrendt H, Hofmann H, Ring J, Traidl-Hoffmann C. Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. J. Invest. Dermatol. 2008;128:2640–2645. doi: 10.1038/jid.2008.139. [DOI] [PubMed] [Google Scholar]
- 19.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N. Engl. J. Med. 2009;361:888–898. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 20.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008;205:1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minegishi Y. Hyper-IgE syndrome. Curr. Opin. Immunol. 2009;21:487–492. doi: 10.1016/j.coi.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 23.Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, Venselaar H, Elbers CC, Johnson MD, Cambi A, Huysamen C, et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 2009;361:1760–1767. doi: 10.1056/NEJMoa0901053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glocker EO, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, Tahami F, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 26.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 27.Ghilardi N, Kljavin N, Chen Q, Lucas S, Gurney AL, De Sauvage FJ. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J. Immunol. 2004;172:2827–2833. doi: 10.4049/jimmunol.172.5.2827. [DOI] [PubMed] [Google Scholar]
- 28.Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O'Connell AC, Puck JM. Hyper-IgE syndrome with recurrent infections—an autosomal dominant multisystem disorder. N. Engl. J. Med. 1999;340:692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 29.Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T (H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J. Allergy Clin. Immunol. 2008;122:181–187. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, Scheinberg P, Price DA, Hage CA, Kholi LM, et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood. 2008;112:2826–2835. doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest. 2010;120:1762–1773. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J. Biol. Chem. 1998;273:27467–27473. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 33.Ley K, Smith E, Stark MA. IL-17A-producing neutrophil-regulatory Tn lymphocytes. Immunol. Res. 2006;34:229–242. doi: 10.1385/IR:34:3:229. [DOI] [PubMed] [Google Scholar]
- 34.Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M, Goldman SJ, Dunussi-Joannopoulos K, Williams CM, Wright JF, Fouser LA. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J. Immunol. 2007;179:7791–7799. doi: 10.4049/jimmunol.179.11.7791. [DOI] [PubMed] [Google Scholar]
- 35.von Vietinghoff S, Ley K. IL-17A controls IL-17F production and maintains blood neutrophil counts in mice. J. Immunol. 2009;183:865–873. doi: 10.4049/jimmunol.0804080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 2004;190:624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 37.Caruso R, Botti E, Sarra M, Esposito M, Stolfi C, Diluvio L, Giustizieri ML, Pacciani V, Mazzotta A, Campione E, et al. Involvement of interleukin-21 in the epidermal hyperplasia of psoriasis. Nat. Med. 2009;15:1013–1015. doi: 10.1038/nm.1995. [DOI] [PubMed] [Google Scholar]
- 38.Sa SM, Valdez PA, Wu J, Jung K, Zhong F, Hall L, Kasman I, Winer J, Modrusan Z, Danilenko DM, Ouyang W. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J. Immunol. 2007;178:2229–2240. doi: 10.4049/jimmunol.178.4.2229. [DOI] [PubMed] [Google Scholar]
- 39.Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, Lucian L, Geissler R, Brodie S, Kimball AB, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J. Exp. Med. 2006;203:2577–2587. doi: 10.1084/jem.20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, Li S, Dooley LT, Gordon KB, PHOENIX 1 study investigators Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1) Lancet. 2008;371:1665–1674. doi: 10.1016/S0140-6736(08)60725-4. [DOI] [PubMed] [Google Scholar]
- 41.Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, Guzzo C, Hsu MC, Wang Y, Li S, et al. PHOENIX 2 study investigators Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2) Lancet. 2008;371:1675–1684. doi: 10.1016/S0140-6736(08)60726-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.