

Abstract
The BK channel is a Ca2+- and voltage-gated potassium channel with many important physiological functions. To identify proteins important to its function in vivo, we screened for Caenorhabditis elegans mutants that suppressed a lethargic phenotype caused by expressing a gain-of-function (gf) isoform of the BK channel α-subunit SLO-1. BKIP-1 (for BK channel interacting protein), a small peptide with no significant homology to any previously characterized molecules, was thus identified. BKIP-1 and SLO-1 showed similar expression and subcellular localization patterns and appeared to interact physically through discrete domains. bkip-1 loss-of-function (lf) mutants phenocopied slo-1(lf) mutants in behavior and synaptic transmission and suppressed the lethargy, egg-laying defect, and deficient neurotransmitter release caused by SLO-1(gf). In heterologous expression systems, BKIP-1 decreased the activation rate and shifted the conductance–voltage relationship of SLO-1 in a Ca2+-dependent manner and increased SLO-1 surface expression. Thus, BKIP-1 is a novel auxiliary subunit critical to SLO-1 function in vivo.
Introduction
The large-conductance and Ca2+/voltage-gated potassium channel (the BK channel) is almost ubiquitously expressed and performs many important physiological functions. In the nervous system, a major function of the BK channel is to downregulate neurotransmitter release at the presynaptic site (Robitaille et al., 1993; Hu et al., 2001; Wang et al., 2001; Raffaelli et al., 2004; Liu et al., 2007).
The central components of a BK channel are four α-subunits (known as Slo1). In addition, BK channels may contain auxiliary subunits. Several proteins in mammals have been identified as auxiliary subunits of the BK channel, including four β-subunits (β1–β4) (Knaus et al., 1994; Wallner et al., 1999; Xia et al., 1999, 2000; Brenner et al., 2000; Uebele et al., 2000; Weiger et al., 2000) and a MinK-related peptide, MiRP3 (Levy et al., 2008). In Drosophila, Slob (Zhou et al., 1999) and dSLIP1 (Xia et al., 1998), which are unrelated to either the β-subunits or MinK-related peptides, also regulate Slo1. The first β-subunit (β1) was identified through copurification with radiolabeled charybdotoxin (Knaus et al., 1994), whereas the remaining BK channel auxiliary/regulatory proteins were identified though homology searches (β2–β4) (Wallner et al., 1999; Xia et al., 1999, 2000; Behrens et al., 2000; Brenner et al., 2000; Uebele et al., 2000), yeast two-hybrid assays using the Drosophila Slo1 C terminal as bait (Slob and dSLIP1) (Schopperle et al., 1998; Xia et al., 1998), and analyses of cellular expression patterns (MiRP3) (Levy et al., 2008). These auxiliary/regulatory proteins may modulate several properties of the channel in heterologous expression systems, including apparent Ca2+ dependence, gating kinetics, surface expression, membrane current density, and sensitivity to pharmacological blockers (McManus et al., 1995; Wallner et al., 1999; Xia et al., 1999, 2000; Brenner et al., 2000; Meera et al., 2000; Uebele et al., 2000; Toro et al., 2006; Kim et al., 2007; Zarei et al., 2007; Levy et al., 2008). Knock-outs of the β1-subunit, β4-subunit, and Slob have been shown to affect blood pressure (Brenner et al., 2000; Plüger et al., 2000), neuronal excitability (Brenner et al., 2005), and resistance to starvation (Shahidullah et al., 2009), respectively. However, no auxiliary subunit has been implicated in the BK channel function of regulating neurotransmitter release.
Given that the BK channel has multiple important functions in vivo, there are likely unidentified auxiliary/regulatory proteins tuning BK channel functional properties. To search for such proteins, we used a genetic approach to screen for suppressors of the SLO-1 gain-of-function (gf) mutants in Caenorhabditis elegans. SLO-1 shares a high level of sequence homology with mammalian Slo1 (Wei et al., 1996). Our analyses of isolated mutants led to the identification of bkip-1 (for BK channel interacting protein), which encodes a small peptide with several effects on SLO-1. Expression pattern and mutant phenotype analyses suggest that BKIP-1 is likely important to SLO-1 function in most tissues that express SLO-1. Thus, BKIP-1 appears to be a BK channel auxiliary subunit of physiological importance.
Materials and Methods
Growth and culture of worms.
C. elegans hermaphrodites were grown on agar plates with a layer of OP50 Escherichia coli at room temperature (21–22°C) or inside an environmental chamber (21°C). Adult hermaphrodites were used for all analyses.
Mutant screening.
The integrated transgenic strain expressing Pslo-1::SLO-1(E350Q) and Pmyo-2::YFP in the wild-type genetic background was used for the mutant screen. The Pmyo-2::YFP transgene was included to express yellow fluorescent protein (YFP) in the pharynx to serve as a genetic marker. Synchronized L4-stage slo-1(gf) worms were treated with the chemical mutagen ethyl methanesulfonate (50 mm) for 4 h at room temperature. The F2 progeny were screened for animals that moved better than the original slo-1(gf) animals. Isolated mutants were grouped through complementation tests.
Analyses of locomotion speed and head-bending angle.
The mean locomotion speed was calculated from the distance traveled by individual worms over time based on images taken at 1 s intervals for 30 s (Chen et al., 2010). The head-bending angle was determined using an automated worm tracking and analysis system (Chen et al., 2010). Briefly, 13 marker points were placed at equal intervals along the spline of each worm, which was imaged at 15 frames/s, and the angle supplementary to the angle formed by the two straight lines connecting the marker points 1 and 2, and the marker points 2 and 3 was determined. Both the full-amplitude dorsal/ventral head-bending angle and the root mean square of head-bending angles were determined. While the former measures the full amplitude of the dorsal/ventral bending angle, the latter measures all bending activities, including small-amplitude oscillations.
Cloning of bkip-1.
bkip-1(zw2) was used for single nucleotide polymorphism-based genetic mapping (Davis et al., 2005). After mapping the mutation to a small interval, the candidate gene was identified by testing whether cosmids and PCR-amplified genomic DNA fragments covering this interval could reinstate the lethargic phenotype in bikp-1(lf);slo-1(gf). Full-length cDNA of the candidate gene was determined through 5′ and 3′ rapid amplification of cDNA ends (RACE). Molecular lesions of bkip-1 were identified by sequencing genomic DNA.
Analysis of expression pattern and subcellular localization.
The expression pattern of bkip-1 was determined by expressing green fluorescent protein (GFP) under the control of 3.9 kb bkip-1 promoter (Pbkip-1::GFP, wp794), whereas that of slo-1 was determined by expressing enhanced green fluorescent protein (EGFP) under the control of 5.2 kb slo-1 promoter (Pslo-1::EGFP, wp758). The plasmids were separately injected into the lin-15(n765) strain using a lin-15 rescue plasmid as a transformation marker. Cells expressing the fluorescent proteins were visualized and photographed with a Carl Zeiss Axiovert 200M fluorescence microscope (40× objective) with an apotome device (Carl Zeiss) for optical sectioning.
Subcellular localization of BKIP-1was determined by fusing GFP to its C terminus and expressing the fusion protein under the control of Pbkip-1 (Pbkip-1::BKIP-1::GFP, wp931). To determine whether SLO-1 protein expression or subcellular localization was altered in bkip-1 loss-of-function (lf) mutants, SLO-1 was labeled by inserting GFP into the linker region between two RCK domains (Jiang et al., 2002; Yusifov et al., 2008) and expressed under the control of Pslo-1 (Pslo-1::SLO-1::GFP, wp5). All constructs were injected independently into the lin-15(n765) strain together with a lin-15 rescue plasmid as a transformation marker. Strains expressing SLO-1::GFP fusions were integrated through γ-irradiation, backcrossed with wild-type animals three times, and crossed into bkip-1(lf). Epifluorescence of the fusion proteins in transgenic animals was visualized and photographed with a Nikon TE2000-U inverted microscope (40× or 60× objective) connected to the F-view II camera.
Bimolecular fluorescence complementation assay.
The DNA sequences encoding YFP N and C terminals (YFPa and YFPc) were amplified by PCR from pCE–BiFC–VN173 and pCE–BiFC–VC155 vectors (Hiatt et al., 2008), respectively, to make the following plasmids: Pslo-1::SLO-1::YFPa (wp840), Prab-3::BKIP-1::YFPc (wp813), Prab-3::BKIP-1▵1–13::YFPc (wp942), Prab-3::BKIP-1▵30–42::YFPc (wp943), Prab-3::BKIP-1▵51–60::YFPc (wp944), Pmyo-3::BKIP-1::YFPc (wp860), Pmyo-3::BKIP-1▵1–13::YFPc (wp945), Pmyo-3::BKIP-1▵30–42::YFPc (wp946), and Pmyo-3::BKIP-1▵51–60::YFPc (wp947). The plasmid encoding SLO-1::YFPa was first injected into lin-15(n765) to establish independent transgenic lines, with a rescuing lin-15 plasmid coinjected to serve as a transformation marker. A representative transgenic line thus obtained was then injected with a plasmid encoding one of the BKIP-1::YFPc fusions together with Pmyo-2::DsRED2 (wp568) or the dominant roller marker pRF4 as a transformation marker. Epifluorescence of transgenic worms was visualized and photographed as described above.
Coimmunoprecipitation.
Hemagglutinin (HA)-tagged BKIP-1 and Myc-tagged SLO-1 were cloned into the pIRES2–mCherry and pIRES2–EGFP vectors (Clontech), respectively, to generate the following plasmids: BKIP-1::HA–IRES2–mCherry (wp856), BKIP-1▵1–13::HA–IRES2–mCherry(wp939), BKIP-1▵30–42::HA–IRES2–mCherry (wp940), BKIP-1▵51–60::HA–IRES2–mCherry (wp941), SLO-1::Myc–IRES2–EGFP(wp857), SLO-1▵371–1140::Myc–IRES2–EGFP (wp932), and SLO-1▵1–370::Myc–IRES2–EGFP(wp933). HEK293 cells were cultured in DMEM with 10% FBS and transiently transfected with Lipofectamine 2000 (Invitrogen). Cells were harvested 48 h after transfection and lysed in 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 150 mm NaCl, 1 mm CaCl2, and 62.5 mm Tris, pH 6.8, plus protease inhibitor (Roche). The supernatants of cell lysates were incubated with Myc antibody (Santa Cruz Biotechnology) for 3 h at 4°C and immunoprecipitated with protein A/G PLUS agarose (Santa Cruz Biotechnology) for 2 h at 4°C. Immune complexes were separated on 8–16% SDS-PAGE gels and probed with HA antibody (NeoMarkers).
Surface biotinylation.
Biotinylation assays were performed using the Cell Surface Protein Isolation kit (Pierce). Surface proteins were biotinylated 48 h after the transfection, precipitated with neutrAvidin–agarose beads, and eluted with SDS sample buffer (1% SDS, 50 mm DTT, 10% glycerol, and 62.5 mm Tris, pH 6.8). Total lysate or biotinylated proteins were separated by 4–12% SDS-PAGE, and the blots were detected as described above.
Recording of postsynaptic currents.
Evoked postsynaptic currents (ePSCs) were recorded from the C. elegans neuromuscular junction (NMJ) as described previously (Liu et al., 2005, 2007). The recording pipette solution contained the following (in mm): 120 KCl, 20 KOH, 5 Tris, 0.25 CaCl2, 4 MgCl2, 36 sucrose, 5 EGTA, and 4 Na2ATP, pH 7.2. Two external solutions with different [Ca2+] (5 mm and 500 μm) were used. The external solution with the higher [Ca2+] contained the following (in mm): 140 NaCl, 5 KCl, 5 CaCl2, 5 MgCl2, 11 dextrose, and 5 HEPES, pH 7.2. This solution was modified by reducing CaCl2 to 500 μm and increasing NaCl to 145 mm to make the external solution with the lower [Ca2+].
Xenopus oocyte expression.
Capped cRNAs were synthesized using the mMessage mMachine kit (Ambion). Approximately 50 nl of cRNA of either slo-1 alone (0.5 ng/nl) or slo-1 (0.5 ng/nl) plus bkip-1 (0.5 ng/nl) was injected into each oocyte using a Drummond Nanoject II injector (Drummond Scientific). Inside-out patches were obtained from the oocyte 2–3 d after cRNA injection. Macroscopic currents induced by voltage steps (−80 to +160 mV in 20 mV increments, 50, 100, or 300 ms duration) were amplified with a Multiclamp 700B amplifier (Molecular Devices) and acquired with the Clampex software (version 10.2; Molecular Devices). Data were sampled at 10 kHz after filtering at 2 kHz.
Composition of the pipette solution was 140 mm K+ gluconate, 1 mm Mg2+ gluconate, and 5 mm HEPES, pH 7.2. Three solutions, pH 7.2, that differed in free [Ca2+] were used to perfuse the cytoplasmic side of the patches. All cytoplasmic solutions contained 140 mm K+ gluconate and 10 mm HEPES. Besides, other components were added to adjust free [Ca2+] (1 mm Ca2+ gluconate for 1 mm [Ca2+], 0.1 mm Ca2+ gluconate for 100 μm [Ca2+], and 3.39 mm Ca2+ gluconate plus 5 mm HEDTA for 10 μm free [Ca2+]). Free [Ca2+] was calculated using online software (http://maxchelator.stanford.edu).
Data analysis.
Amplitudes of ePSCs were measured with the Clampfit software (Molecular Devices). Mean amplitude of the two largest ePSCs from each experiment was used for statistical analysis. Peak macroscopic currents from isolated oocyte patches were determined and used to plot the conductance–voltage (G–V) relationship. Graphing and statistical analyses were performed with Origin (version 8.0; OriginLab). Either unpaired t test or ANOVA (followed by Bonferroni's post hoc tests) was used for statistical comparisons. p < 0.05 is considered statistically significant. All values are expressed as mean ± SE. n is the number of patches or muscle cells analyzed.
Results
Mutation in bkip-1 suppressed the lethargic phenotype caused by SLO-1(gf)
To perform a forward genetic screen for molecules related to SLO-1 function, we created a SLO-1(gf) isoform by mutating glutamate 350 to glutamine (E350Q). E350 is the same residue that was changed to lysine in a previously reported slo-1(gf) mutant (Davies et al., 2003) and the equivalent of mouse Slo1 E321, which contributes to one of the two negative rings at the entrance to the intracellular vestibule of the BK channel (Brelidze et al., 2003). When tested in Xenopus oocytes, SLO-1(E350Q) shifted the half-maximal voltage for channel activation (V50) toward more hyperpolarized membrane potentials by ∼40 mV (Fig. 1A). Expression of SLO-1(E350Q) in worms under the control of slo-1 promoter (Pslo-1) caused a distorted locomotion waveform and greatly reduced locomotion velocity (Fig. 1B) [supplemental Movies 1 and 2 for wild-type and slo-1(gf), respectively, available at www.jneurosci.org as supplemental material]. We showed previously that a SLO-1::GFP fusion protein localizes similarly as wild-type SLO-1 in vivo (Wang et al., 2001). The subcellular localization of SLO-1(E350Q)::GFP was indistinguishable from that of SLO-1::GFP in neurons and body-wall muscle cells (Fig. 1C). Thus, the Pslo-1::SLO-1(E350Q) strain appeared to be appropriate for the intended genetic screen.
Figure 1.
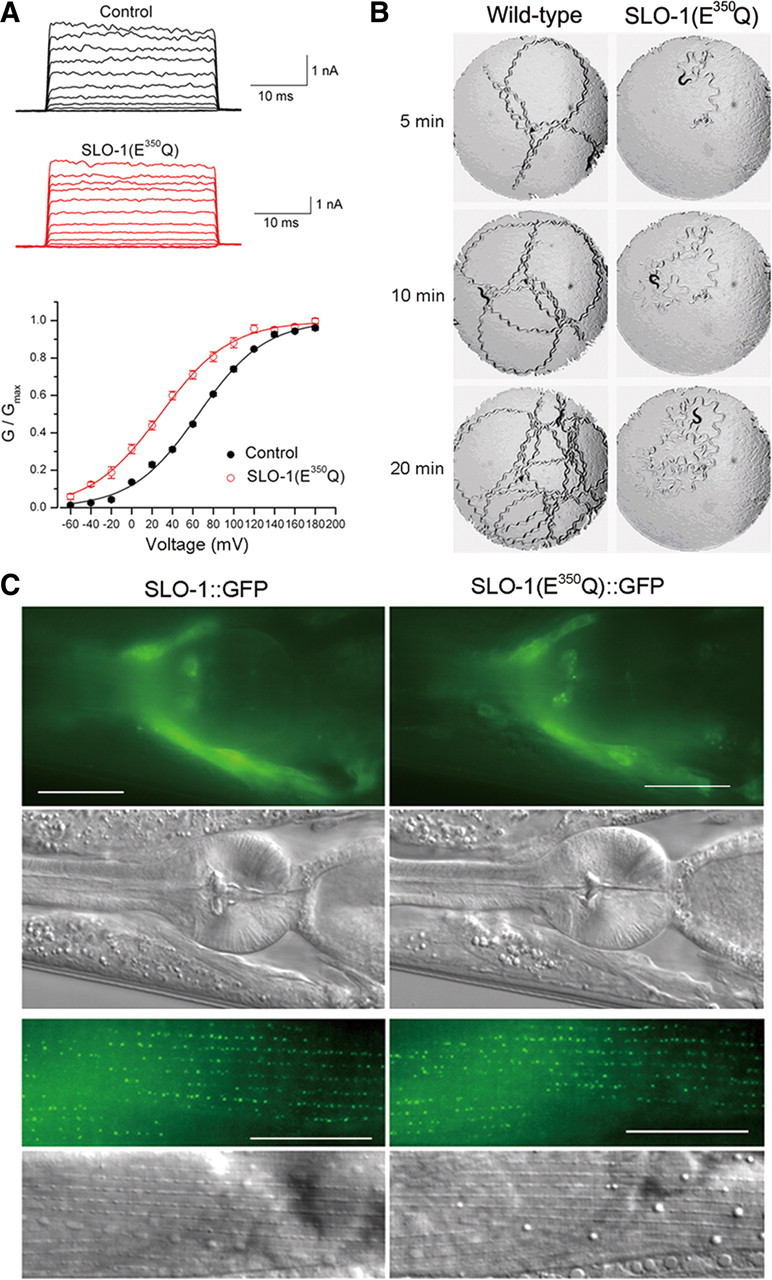
SLO-1(E350Q) mutation shifted channel G–V relationship and inhibited worm locomotion. A, Macroscopic currents of inside-out patches from Xenopus oocytes expressing wild-type SLO-1 (Control) or SLO-1(E350Q) and the G–V relationship. The G–V relationship was fitted to the Boltzmann function G = Gmax/[1 + exp(V50 − V)/k], where G is the conductance at voltage V, Gmax is the maximal conductance, V50 is the voltage at which G = 0.5 Gmax, and k is the slope factor. The V50 was 65.4 ± 1.4 mV for wild-type SLO-1 (n = 10) and 28.4 ± 3.2 mV for SLO-1(E350Q) (n = 6). B, Locomotion behavior of a wild-type worm and a transgenic worm expressing Pslo-1::SLO-1(E350Q). A piece of paper with a circular hole (7 mm in diameter) was laid on top of the bacterial lawn in the agar plate to restrict animal movements within the field of observation. A single adult hermaphrodite was placed in the center of the field to start the assay. Snapshots of the animal and its locomotion track were taken at 5, 10, and 20 min. The animal expressing SLO-1(E350Q) showed a distorted locomotion waveform and greatly reduced locomotion velocity. C, SLO-1(E350Q)::GFP localized similarly as SLO-1::GFP in both neurons and body-wall muscle cells. The top two rows show fluorescent and differential interference contrast (DIC) images of the head. Fluorescent signal was localized mainly in the nerve ring region. The bottom two rows show fluorescent and DIC images of body-wall muscle. Fluorescent signal was enriched at dense body areas. Scale bars, 20 μm.
From a screen of ∼24,000 haploid genomes, we isolated 25 mutants that suppressed the lethargic phenotype of slo-1(gf) worms. Six of the mutants belong to one gene, which was named bkip-1. bkip-1 was mapped to a 63 kb interval on the right arm of chromosome II (14,060k–14,123k) through single nucleotide polymorphism-based genetic mapping (Davis et al., 2005). To identify the candidate gene, we tested whether cosmids or PCR-amplified genomic DNA fragments within this interval could reinstate the lethargic phenotype of slo-1(gf) when expressed in slo-1(gf);bkip-1(zw10) double mutant, which has grossly normal mobility. We found that a genomic DNA fragment of 6 kb containing a predicted pseudogene (Y39G8C.3; www.wormbase.org) completely reinstated the lethargy of slo-1(gf) when expressed in the double mutant.
The initiation and termination sites of this gene were then determined through 5′ and 3′ RACE. The full-length cDNA is predicted to encode a peptide of 60 aa residues (Fig. 2A). Expression of this cDNA under the control of bkip-1 promoter (Pbkip-1) in slo-1(gf);bkip-1(zw10) also completely reinstated the lethargic phenotype, indicating that the cDNA encodes a functional protein. Kyte–Doolittle hydrophilicity plot of the predicted translational product reveals a stretch of hydrophobic residues (21–46 aa) that potentially serves as a transmembrane domain (Fig. 2B). Molecular lesions were identified in all the six bkip-1 alleles through sequencing genomic DNA (Fig. 2A). bkip-1(zw10), which is a putative null resulting from a mutation in the splice acceptor site of the first intron, was used for all subsequent analyses. Blast search of genome databases revealed BKIP-1 homologs in several nematode species (Fig. 2C) but not in mammals.
Figure 2.
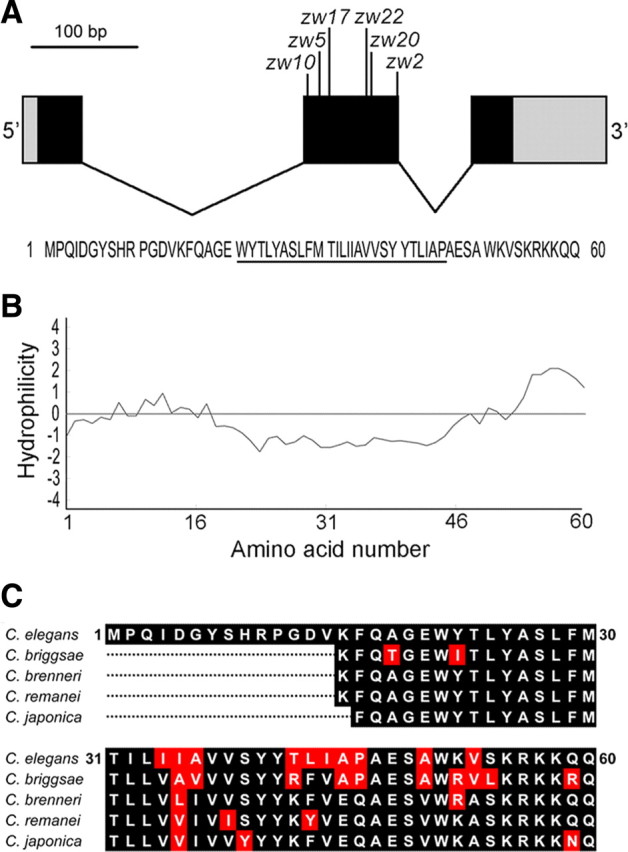
bkip-1 encodes a novel protein with homologs in nematodes. A, A diagram showing bkip-1 exon–intron organization and locations of the mutations identified in bkip-1 alleles. Coding exons are shown in black, 5′ and 3′ untranslated regions are in gray, and introns are as black lines. Molecular lesions of the six bkip-1 alleles are as follows: zw2 (P46L); zw5 (G19S), zw10 (splice acceptor site mutation leading to a frame shift after K15 and then a stop codon, LNQHGKYQSEKSSSEFASRPYK STOP); zw17 (W21 to STOP), zw20 (V37D), and zw22 (single nucleotide deletion in exon 2 leading to a frame shift after A36 and then a stop codon, QSSHIIHLLLLLNQHGKYQSEKSSSEFASRPYK STOP). Bottom, Deduced amino acid sequence of BKIP-1. The putative transmembrane domain is underlined. B, Kyte–Doolittle hydrophilicity plot of BKIP-1 showing that a stretch of hydrophobic amino acid residues that could potentially serve as a membrane-spanning domain. C, Alignment of BKIP-1 with putative homologs in several nematode species. The putative homologs were identified through blast search of the respective nematode genomes. The N-terminal portions of the putative homologs were excluded from the alignment because they cannot be readily predicted.
bkip-1(lf) suppressed the inhibitory effect of slo-1(gf) on locomotion speed, which is determined mainly by the functions of neurons and body-wall muscle cells. To determine whether BKIP-1 functions in neurons or body-wall muscle cells, we analyzed the effects of expressing wild-type BKIP-1 in neurons and body-wall muscle cells in slo-1(gf);bkip-1(lf) separately. We found that the effect of bkip-1(lf) on the slo-1(gf) phenotype could be reversed by expressing wild-type BKIP-1 under the control of either the pan-neuronal rab-3 promoter (Prab-3) (Nonet et al., 1997) or the muscle-specific myo-3 promoter (Pmyo-3) (Okkema et al., 1993) (Fig. 3A) (supplemental Movies 3–6, available at www.jneurosci.org as supplemental material). These observations suggest that BKIP-1 functions in both neurons and muscle cells.
Figure 3.

BIKP-1 functioned together with SLO-1 in controlling worm behavior. A, bkip-1(lf) mutant suppressed the locomotion defects caused by slo-1(gf), which could be reversed by expressing wild-type (WT) BKIP-1 either in neurons under the control of Prab-3 or in body-wall muscle cells under the control of Pmyo-3. Twenty to 30 worms were analyzed in each group. B, bkip-1(lf) mutant suppressed the egg-laying defect caused by slo-1(gf), which could be reversed by expressing wild-type BKIP-1 in muscle cells under the control of Pmyo-3. The number of eggs retained in the uterus was counted 24 h after late L4 stage at room temperature. Ten worms were analyzed in each group. C, D, bkip-1(lf) increased the head-bending angle, which could be reversed by expressing wild-type BKIP-1 in muscle cells under the control of Pmyo-3 but not in neurons under the control of Prab-3. Both the mean full-amplitude dorsal/ventral bending angle (C), as depicted in the diagram, and the root mean square of bending activities (D) were analyzed. The number of worms analyzed for the head-bending angle was 10 for each group. For all panels, data are shown as mean ± SE, and *indicates a statistically significant difference between the indicated groups (A) or compared with the wild-type (B–D) (p < 0.01, one-way ANOVA with Bonferroni's post hoc test).
slo-1(gf) mutants are known to be associated with an Egl phenotype (egg-laying defect) (Davies et al., 2003; Kim et al., 2009). Consistent with the previous reports, we observed that a significantly larger number of eggs were retained in the uterus of the slo-1(gf) strain compared with the wild type. This Egl phenotype of slo-1(gf) was suppressed by bkip-1(lf). It appears that the function of BKIP-1 in body-wall and/or vulval muscle cells was required for causing the Egl phenotype of slo-1(gf) because the phenotype could be reinstated by expressing wild-type BKIP-1 selectively in muscle cells of the slo-1(gf);bkip-1(lf) double mutant (Fig. 3B). Intriguingly, rescuing bkip-1(lf) selectively in neurons of the double mutant significantly reduced the number of retained eggs compared with the wild type (Fig. 3B). The underlying mechanism for this unexpected neuronal effect of BKIP-1 in the slo-1(gf);bkip-1(lf) background remains to be determined.
bkip-1(lf) mutants appeared jerky and hyperactive, which grossly resembled slo-1(lf) mutants (Wang et al., 2001). slo-1(lf) is characterized by an increased head-bending angle in quantitative analyses of the locomotion behavior (Kim et al., 2009; Chen et al., 2010). To determine whether BKIP-1 functions together with SLO-1 in regulating head bending, we analyzed the head-bending angle using an automated worm tracking and analysis system (Chen et al., 2010). We observed that the head-bending angle was similarly increased in the bkip-1(lf) mutant as in the slo-1(lf) mutant; furthermore, the severity of this phenotype was not additive in the slo-1(lf);bkip-1(lf) double mutant (Fig. 3C,D). The head-bending phenotype of bkip-1(lf) could be rescued by expressing wild-type BKIP-1 in muscle cells but not in neurons (Fig. 3C,D). These observations collectively suggest that BKIP-1 functions together with SLO-1 in body-wall muscle cells to affect the head-bending angle.
BKIP-1 was coexpressed and colocalized with SLO-1 in neurons and muscle cells
The expression pattern of bkip-1 was similar to that of slo-1 when analyzed by expressing GFP under the control of their native promoters. Both genes were expressed in most, if not all, neurons as well as several types of muscle cells (Fig. 4A). The only obvious difference in the expression patterns between the two genes was that bkip-1, but not slo-1, was expressed in the head mesodermal cell (Fig. 4A). To determine the subcellular localization of BKIP-1, we fused GFP to the C terminus of BKIP-1 and expressed the fusion protein under the control of Pbkip-1. Similar to SLO-1 (Wang et al., 2001), BKIP-1 was enriched in the nerve ring and in body-wall muscle dense bodies (Fig. 4B).
Figure 4.

Expression and subcellular localization patterns of bkip-1 were similar to those of slo-1. A, GFP showed similar expression patterns with Pslo-1 and Pbkip-1. Strong expression was observed in head neurons (not labeled), nerve ring (NR), ventral cord (VC), tail neurons (not labeled), body-wall muscle (BM), head muscle (HM), vulval muscle (VM), anal depressor muscle (ADM), and stomatointestinal muscle (SIM). Note that bkip-1, but not slo-1, was also expressed in head mesodermal cell (HMC). Because of the mosaic expression of the transgenes, fluorescence intensity was somewhat variable from cell to cell. B, Subcellular localization of BKIP-1::GFP and SLO-1::GFP fusion proteins in the nerve ring and body-wall muscle cells were similar. The green puncta in body-wall muscle cells correspond to locations of dense bodies. Scale bars, 20 μm.
BKIP-1 and SLO-1 appeared to physically interact both in vitro and in vivo
The similar expression and subcellular localization patterns of SLO-1 and BKIP-1 suggest that these two proteins might interact physically. To examine this possibility, we transiently expressed Myc-tagged SLO-1 (SLO-1::Myc) and HA-tagged BKIP-1 (BKIP-1::HA) in HEK293 cells and performed coimmunoprecipitation experiments. SLO-1 has two major structural components: the N-terminal portion (1–352 aa) containing seven membrane-spanning domains including the pore domain, and the cytoplasmic C-terminal portion (353–1140) containing two RCK domains (Jiang et al., 2002; Salkoff et al., 2006) (Fig. 5A). We found that BKIP-1 coimmunoprecipitated with either full-length SLO-1 or SLO-1 N terminal (SLO-1Δ371–1140) but not SLO-1 C terminal (SLO-1Δ1–370) (Fig. 5B), suggesting that SLO-1 N terminal is required for association with BKIP-1. The interaction between BKIP-1 and SLO-1 appeared to be Ca2+ independent (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). We also observed that both the putative transmembrane domain of BKIP-1 and its C terminal were necessary for its interaction with SLO-1 in coimmunoprecipitation, whereas the N terminal was dispensable for the interaction (Fig. 5C). These observations suggest that SLO-1 and BKIP-1 physically interact with each other and that the N-terminal portion of SLO-1 and the transmembrane as well as the C-terminal domains of BKIP-1 are important in mediating the interaction.
Figure 5.

BKIP-1 and SLO-1 coimmunoprecipitated from transfected HEK293 cells. A, Diagrams of putative membrane topologies of SLO-1 and BKIP-1. The numbers indicate the positions in which truncation or deletion was made in the experiments shown in B and C. B, BKIP-1 coimmunoprecipitated with full-length SLO-1 or SLO-1Δ371–1140 but not SLO-1Δ1–370. C, BKIP-1Δ1–13 but not BKIP-1Δ30–42 or BKIP-1Δ51–60 coimmunoprecipitated with SLO-1. In both B and C, BKIP-1 and its variants were tagged with HA, whereas SLO-1 and its variants were tagged with Myc. Immunoprecipitation (IP) and immunoblot (IB) were performed with Myc and HA antibodies. All specific bands are indicated by their relative molecular sizes, and nonspecific bands are indicated by arrows.
We then asked whether BKIP-1 and SLO-1 might interact in vivo by performing the bimolecular fluorescence complementation (BiFC) assay (Chen et al., 2007; Shyu et al., 2008). In this assay, the nonfluorescent N- and C-terminal portions of yellow fluorescent protein (YFPa and YFPc) are fused separately to a pair of proteins of interest. The fluorophore of YFP may be reconstituted if these two proteins are physically very close (Shyu et al., 2008). Specifically, we inserted YFPa into the linker region between the two RCK domains of SLO-1 (Jiang et al., 2002; Yusifov et al., 2008; Wu et al., 2010; Yuan et al., 2010) and fused YFPc to the C terminus of BKIP-1 and three variants with different deletions (Δ1–13, Δ30–42, and Δ51–60). The SLO-1::YFPa fusion protein was coexpressed with each of the BKIP-1 fusions in neurons and body-wall muscle cells. In transgenic animals, BiFC was observed in both neurons and body-wall muscle cells (Fig. 6). Deletion of the transmembrane domain of BKIP-1 abolished the BiFC (Fig. 6), which is consistent with the coimmunoprecipitation data (Fig. 5C). However, unlike the coimmunoprecipitation data (Fig. 5C), BiFC still occurred after deleting the C terminal of BKIP-1 (Fig. 6). Together, the coimmunoprecipitation and BiFC analyses suggest that the putative membrane-spanning domain of BKIP-1 allows BKIP-1 to be physically close to SLO-1, whereas both the membrane-spanning domain and the C terminal are needed for BKIP-1 to bind to SLO-1.
Figure 6.

BKIP-1 and SLO-1 appeared to be physically very close in vivo. Bimolecular fluorescence complementation assays were performed by fusing the N-terminal portion of YFP (YFPa) to full-length SLO-1 (SLO-1::YFPa) and fusing the C-terminal portion of YFP (YFPc) to either full-length BKIP-1 (BKIP-1::YFPc) or BKIP-1 with deletions (BKIP-1Δ1–13::YFPc, BKIP-1Δ51–60::YFPc, and BKIP-1Δ30–42::YFPc). In both neurons and body-wall muscle cells, all the BKIP-1 fusions except BKIP-1Δ30–42::YFPc were able to reconstitute YFP fluorophore with the coexpressed SLO-1::YFPa. Scale bars, 20 μm.
SLO-1 required BKIP-1 to regulate neurotransmitter release
A well established function of SLO-1 is to regulate neurotransmitter release at the C. elegans NMJ (Wang et al., 2001; Liu et al., 2007). Decreased neurotransmitter release at the NMJ could be a major cause for the locomotion defects observed in slo-1(gf) worms. To determine whether BKIP-1 is needed for this function of SLO-1, we analyzed the effect of bkip-1(lf) on ePSCs recorded from body-wall muscle cells at two different extracellular Ca2+ concentrations (5 mm and 500 μm). The higher [Ca2+] is more suitable for determining whether slo-1(gf) reduces ePSC amplitude and whether this effect may be reversed by bkip-1(lf), whereas the lower [Ca2+] is more suitable for testing whether bkip-1(lf) could increase ePSC amplitude as slo-1(lf) does (Liu et al., 2007). Indeed, at 5 mm [Ca2+]o (Fig. 7A), the ePSC amplitude was greatly reduced in slo-1(gf) animals compared with the wild type, and this effect was reversed by bkip-1(lf). bkip-1(lf) alone did not increase ePSC amplitude, which resembles slo-1(lf) and is probably attributable to limited capacity of the readily releasable pool of synaptic vesicles (Wang et al., 2001). At 500 μm [Ca2+]o (Fig. 7B), ePSC amplitude was increased to a similar degree in bkip-1(lf) and slo-1(lf) as well as in the slo-1(lf);bkip-1(lf) double mutant. The effects of bkip-1(lf) on neuromuscular transmission at both Ca2+ concentrations could be eliminated by expressing wild-type BKIP-1 in neurons but not in muscle cells (Fig. 7A,B). These observations suggest that SLO-1 requires BKIP-1 to regulate neurotransmitter release at the presynaptic site.
Figure 7.
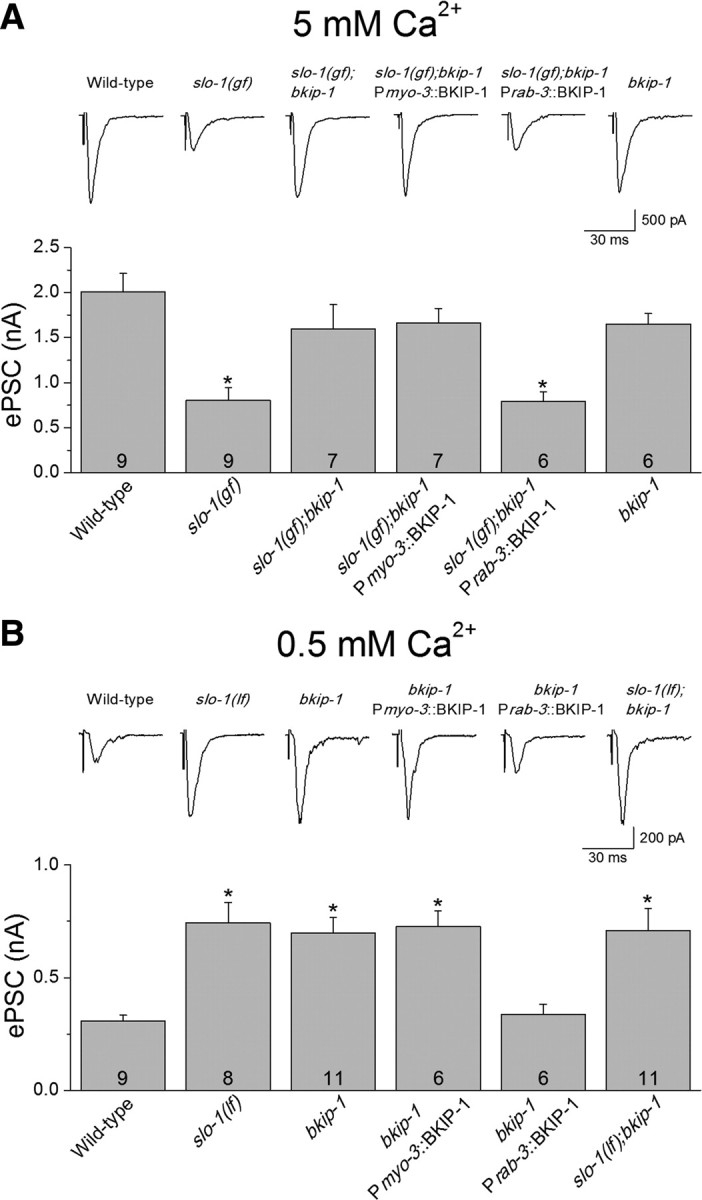
Effects of bkip-1 mutation on neurotransmitter release. A, At 5 mm [Ca2+]o, the amplitude of ePSCs at the neuromuscular junction was significantly smaller in worms expressing SLO-1(gf), which could be reversed by bkip-1(lf) (zw10 allele). ePSC amplitude in bkip-1(lf) alone was comparable with that in wild-type animal. B, At 500 μm [Ca2+]o, ePSC amplitude of bkip-1(lf) was significantly larger than that of the wild type but similar to that of slo-1(md1745) [a putative null (Wang et al., 2001)] or the double-mutant slo-1(md1745);bkip-1(zw10). In both A and B, the neuromuscular transmission phenotype of bkip-1(lf) was rescued by neuronal but not muscle expression of wild-type BKIP-1. The holding potential was −60 mV. * indicates a statistically significant difference compared with the wild-type (p < 0.01, one-way ANOVA with Bonferroni's post hoc test). Error bars represent SE. The number of samples analyzed is indicated inside each column.
BKIP-1 regulated SLO-1 channel properties in Xenopus oocytes
To determine whether BKIP-1 modulates SLO-1 functional properties, we analyzed macroscopic currents in inside-out patches from Xenopus oocytes expressing either SLO-1 alone or SLO-1 plus BKIP-1 at three different cytoplasmic [Ca2+]. BKIP-1 showed two obvious effects on SLO-1 functional properties. First, BKIP-1 significantly affected the G–V relationship of the channel, increasing V50 by ∼20 mV at 10 μm [Ca2+] but decreasing V50 by ∼20 mV at 100 μm and 1 mm [Ca2+] (Fig. 8B,C). Second, BKIP-1 greatly decreased SLO-1 activation rate at the lower (10 μm) but not the higher (100 μm and 1 mm) [Ca2+] (Fig. 8A,D). In addition, patches from oocytes coexpressing BKIP-1 generally showed much larger current amplitudes compared with the control (Fig. 8A, not quantified). These observations suggest that BKIP-1 regulates SLO-1 functional properties in a Ca2+-dependent manner.
Figure 8.
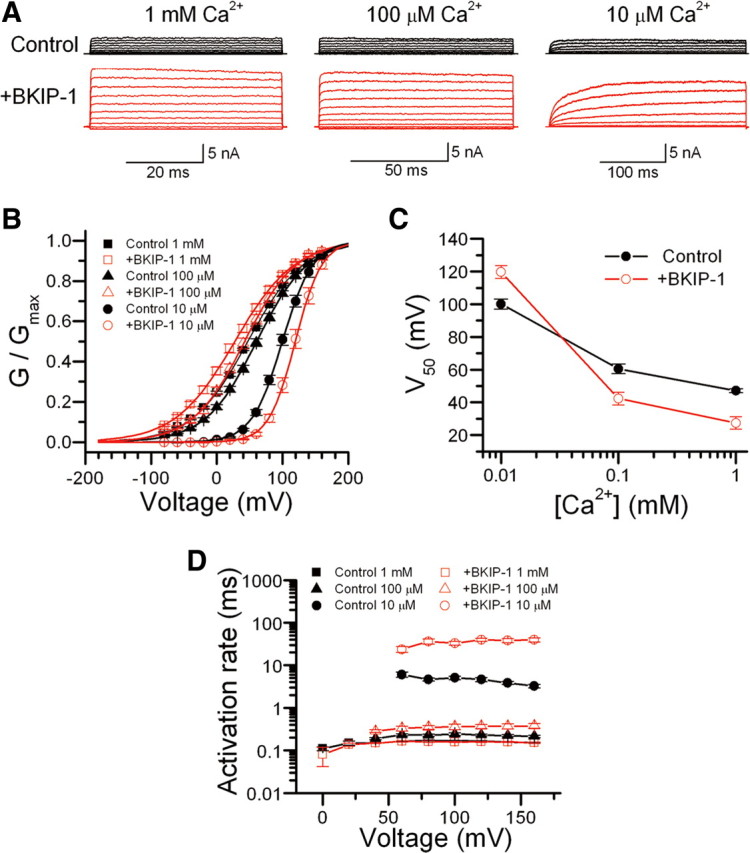
Effects of BKIP-1 on SLO-1 channel functional properties in Xenopus oocytes. A, Representative macroscopic current traces in inside-out patches from oocytes expressing SLO-1 alone (Control) or SLO-1 plus BKIP-1 (+BKIP-1) in response to voltage steps (−80 to +160 mV at 20 mV intervals and 50, 100, or 300 ms pulse duration) at different [Ca2+] in the bath solution. B, The G–V relationships of Control and +BKIP-1 at different [Ca2+] were fitted to the Boltzmann function G = Gmax/[1 + exp(V50 − V)/k], where G is the conductance at voltage V, Gmax is the maximal conductance, V50 is the voltage at which G = 0.5 Gmax, and k is the slope factor. C, Effects of BKIP-1 on SLO-1 V50 at different [Ca2+]. BKIP-1 increased V50 at 10 μm [Ca2+] but decreased V50 at 100 μm and 1 mm [Ca2+]. The difference was statistically significant at all [Ca2+] (p < 0.01, unpaired t test). D, Effect of BKIP-1 on SLO-1 activation kinetics. Activation time constant was determined by a single-exponential fit to the current trace at each voltage step. BKIP-1 significantly slowed SLO-1 activation at 10 μm [Ca2+] but not at the higher [Ca2+], and this effect of BKIP-1 was independent of membrane potential (two-way ANOVA). Data are shown as mean ± SE. The numbers of patches (n) analyzed were 10 in all cases except for Control (n = 12) and +BKIP-1 (n = 13) at 1 mm [Ca2+].
The effects of BKIP-1 on SLO-1 functional properties are reminiscent of those of human β4-subunit (hβ4) on human Slo1 (hSlo1) (Brenner et al., 2000). Therefore, we tested whether hβ4 could substitute for BKIP-1 in regulating SLO-1 function by expressing hβ4 in the slo-1(gf);bkip-1(lf) double mutant under the control of Pbkip-1. We found that expression of hβ4 failed to reinstate either the Egl (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) or the lethargic phenotype of slo-1(gf) (data not shown), suggesting that BKIP-1 and hβ4 may be interacting with distinct domains or sequences of the BK channel.
BKIP-1 increased SLO-1 surface expression
SLO-1 is enriched in the nerve ring in which the density of synapses is the highest in the animal as well as in dense body regions of body-wall muscle cells (Wang et al., 2001). To determine whether BKIP-1 regulates SLO-1 expression or subcellular localization, we created a strain expressing integrated Pslo-1::SLO-1::GFP. This transgene was then crossed into bkip-1(lf). Compared with the wild type, GFP fluorescent puncta in muscle dense bodies were dimmer (data not quantified) and GFP epifluorescence in the nerve ring was significantly reduced in bkip-1(lf) mutant (Fig. 9A,B).
Figure 9.

BKIP-1 increased SLO-1 expression in C. elegans and transfected HEK293 cells. A, SLO-1::GFP expression in body-wall muscle cells. SLO-1::GFP puncta at dense bodies appeared weaker in bkip-1(zw10) mutant compared with the wild type. The bright signal in the middle was of the ventral cord and motor neuron somas. B, SLO-1::GFP epifluorescence in the nerve ring was decreased in bkip-1(zw10) mutant. The mean fluorescence intensity of the nerve ring region (surrounded by the white line) was measured with NIH Image J software and compared between the two groups (n = 15 for both groups). * indicates a statistically significant difference compared with the wild type (WT) (p < 0.01, paired t test). C, slo-1 transcription was not altered by bkip-1(lf). GFP was expressed under the control of slo-1 promoter. GFP expression in neurons and muscle cells was indistinguishable between the wild type and bkip-1(zw10). NR, Nerve ring; VC, ventral cord; BM, body-wall muscle; HM, head muscle. D, Effects of BKIP-1 on SLO-1 total and surface protein levels. HEK293 cells were cotransfected with either SLO-1 and BKIP-1 or SLO-1 and the empty vector for BKIP-1. The amount of SLO-1 protein was quantified by densitometry and normalized first by α-tubulin and then by SLO-1 control (without BKIP-1). * indicates a statistically significant difference (p < 0.05, paired t test). Data are shown as mean ± SE and were from three experiments. E, BKIP-1 expression was not altered in slo-1 mutant. BKIP-1::GFP fusion was expressed in the wild-type and slo-1(md1745) mutant under the control of bkip-1 promoter. GFP epifluorescence in both neurons and body-wall muscle cells was indistinguishable between the wild-type and the slo-1 mutant. Scale bars, 20 μm.
Decreased SLO-1::GFP signal in the nerve ring and muscle dense bodies of bkip-1(lf) mutant could be attributable to decreased gene transcription or decreased protein synthesis/trafficking. To determine whether BKIP-1 regulates slo-1 transcription, we compared the expression of a Pslo-1::GFP transcriptional fusion between the wild-type and bkip-1(lf) mutant. GFP expression in both neurons and body-wall muscle cells was indistinguishable between the two groups (Fig. 9C), suggesting that BKIP-1 does not regulate slo-1 transcription. To determine whether BKIP-1 regulates SLO-1 surface expression, we examined the total and surface SLO-1 protein levels in HEK293 cells transfected with either Myc-tagged SLO-1 alone or together with HA-tagged BKIP-1. Cotransfection with BKIP-1 significantly increased the amount of SLO-1 protein in the plasma membrane but did not show a significant effect on SLO-1 total protein (Fig. 9D). These observations suggest that BKIP-1 does not affect SLO-1 protein synthesis; instead, it increases SLO-1 expression on the plasma membrane. We also examined whether SLO-1 has an effect on BKIP-1 expression by expressing Pbkip-1::BKIP-1::GFP in the wild-type and slo-1(lf) mutant. We found that BKIP-1::GFP expression in both neurons and body-wall muscle cells was indistinguishable between these two groups (Fig. 9E).
Discussion
In the present study, we identified a novel auxiliary subunit of the BK channel through a genetic approach. Several lines of evidence suggest that BKIP-1 is critical to SLO-1 function in vivo. First, synaptic and behavioral phenotypes of bkip-1(lf) and slo-1(lf) mutants were indistinguishable; both mutants showed increased neurotransmitter release at the NMJ and increased head-bending angle. Second, bkip-1(lf) essentially eliminated the lethargy, the egg-laying defect, and defective neurotransmitter release caused by slo-1(gf). Third, BIKP-1 facilitated SLO-1 subcellular localization. This is the first time showing that knock-out of an auxiliary subunit phenocopies knock-out of BK channel α-subunit.
Hydrophilicity analyses suggest that BKIP-1 may be a single-pass membrane protein. This structural feature is reminiscent of KCNEs (also known as MinK and MinK-related peptides), which indiscriminately regulate voltage-gated potassium channels in heterologous expression systems (McCrossan and Abbott, 2004). Sequence alignment analyses using the Clustal method showed that the level of sequence identity between BKIP-1 and KCNEs is as low as that between BKIP-1 and randomly picked unrelated membrane proteins (e.g., C. elegans innexins). BKIP-1 is also distinct from KCNE/MiRP homologs in C. elegans (Park et al., 2005) and CALF-1, which is a recently identified single-pass membrane protein and facilitates membrane trafficking of voltage-gated Ca2+ channels in C. elegans (Saheki and Bargmann, 2009). Because BKIP-1 does not have significant homology to any previously characterized molecules but has homologs at least in some nematode species, it represents a new class of proteins.
The existence of BKIP-1 homologs in different nematode species suggests that BKIP-1 had evolved at least tens of millions of years ago and that its presence may be essential to maintaining the species during evolution. Nevertheless, BKIP-1 homologs were not readily identified in human or mouse. Given that there are regions of significant sequence divergence between C. elegans SLO-1 and mammalian Slo1 (Wei et al., 1996) and that it is unknown which specific region of SLO-1 interacts with BKIP-1, it is possible that BKIP-1 analogs exist in mammals but were missed in the blast search because of its small size and sequence divergence. It has been proved many times that proteins with similar structures and functions exist in mammals but were not immediately obvious during the discovery of a novel protein in invertebrates. For example, C. elegans SOL-1 was initially identified as a CUB-domain protein required for glutamate receptor function without mammalian homologs (Zheng et al., 2004). However, structurally and functionally related proteins (e.g., NETO2) were later identified in mammals (Zhang et al., 2009). Therefore, more analyses are needed before we know whether or not there are proteins like BKIP-1 in mammals.
The effects of BKIP-1 on SLO-1 activation and G–V relationship were Ca2+ dependent. Specifically, BKIP-1 increased V50 and slowed channel activation rate at the lower cytoplasmic [Ca2+] (10 μm) but decreased V50 and showed no obvious effect on channel activation rate at the higher [Ca2+] (100 μm and 1 mm). The effects of BKIP-1 on SLO-1 functional properties are reminiscent of those of hβ4 on hSlo1. hβ4 also slows BK channel activation at 10 μm [Ca2+]i, increases V50 at lower [Ca2+]i (≤10 μm), but deceases V50 at higher [Ca2+]i (≥50 μm) (Brenner et al., 2000). Presumably, these Ca2+-dependent properties of BK channel auxiliary proteins would make the channel to have low activity at rest but enable it to be more active in response to action potentials.
BK channels perform many important physiological functions in vivo. In the nervous system, BK channels colocalize with voltage-sensitive Ca2+ channels at the presynaptic terminal (Roberts et al., 1990; Robitaille et al., 1993; Issa and Hudspeth, 1994; Yazejian et al., 2000) and serve as an important regulator of neurotransmitter release (Robitaille et al., 1993; Hu et al., 2001; Wang et al., 2001; Raffaelli et al., 2004; Liu et al., 2007). Thus, BKIP-1 potentially plays important roles in the nervous system by modulating the BK channel and possibly in other tissues as well.
Footnotes
This work was supported by National Institute of Health Grants 1R01MH085927 and 5R01GM083049 (Z.W.W.) and National Science Foundation Grant 0619427 (Z.W.W.). We thank Aguan Wei for suggestions about this manuscript, Yanping Wang for suggestions on biochemical experiments, and Audrey Fraser (Sanger Institute, Cambridge, UK) for providing cosmids.
References
- Behrens R, Nolting A, Reimann F, Schwarz M, Waldschütz R, Pongs O. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel β subunit family. FEBS Lett. 2000;474:99–106. doi: 10.1016/s0014-5793(00)01584-2. [DOI] [PubMed] [Google Scholar]
- Brelidze TI, Niu X, Magleby KL. A ring of eight conserved negatively charged amino acids doubles the conductance of BK channels and prevents inward rectification. Proc Natl Acad Sci U S A. 2003;100:9017–9022. doi: 10.1073/pnas.1532257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel β subunits, hKCNMB3 and hKCNMB4. J Biol Chem. 2000;275:6453–6461. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8:1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- Chen B, Liu Q, Ge Q, Xie J, Wang ZW. UNC-1 regulates gap junctions important to locomotion in C. elegans. Curr Biol. 2007;17:1334–1339. doi: 10.1016/j.cub.2007.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Liu P, Wang SJ, Ge Q, Zhan H, Mohler WA, Wang ZW. α-catulin CTN-1 is required for BK channel subcellular localization in C. elegans body-wall muscle cells. EMBO J. 2010;29:3184–3195. doi: 10.1038/emboj.2010.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AG, Pierce-Shimomura JT, Kim H, VanHoven MK, Thiele TR, Bonci A, Bargmann CI, McIntire SL. A central role of the BK potassium channel in behavioral responses to ethanol in C. elegans. Cell. 2003;115:655–666. doi: 10.1016/s0092-8674(03)00979-6. [DOI] [PubMed] [Google Scholar]
- Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics. 2005;6:118. doi: 10.1186/1471-2164-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiatt SM, Shyu YJ, Duren HM, Hu CD. Bimolecular fluorescence complementation (BiFC) analysis of protein interactions in Caenorhabditis elegans. Methods. 2008;45:185–191. doi: 10.1016/j.ymeth.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R, Laake P, Pongs O, Knaus HG, Ottersen OP, Storm JF. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21:9585–9597. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa NP, Hudspeth AJ. Clustering of Ca2+ channels and Ca2+-activated K+ channels at fluorescently labeled presynaptic active zones of hair cells. Proc Natl Acad Sci U S A. 1994;91:7578–7582. doi: 10.1073/pnas.91.16.7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- Kim EY, Zou S, Ridgway LD, Dryer SE. β1-subunits increase surface expression of a large-conductance Ca2+-activated K+ channel isoform. J Neurophysiol. 2007;97:3508–3516. doi: 10.1152/jn.00009.2007. [DOI] [PubMed] [Google Scholar]
- Kim H, Pierce-Shimomura JT, Oh HJ, Johnson BE, Goodman MB, McIntire SL. The dystrophin complex controls bk channel localization and muscle activity in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000780. doi: 10.1371/journal.pgen.1000780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus HG, Folander K, Garcia-Calvo M, Garcia ML, Kaczorowski GJ, Smith M, Swanson R. Primary sequence and immunological characterization of β-subunit of high conductance Ca2+-activated K+ channel from smooth muscle. J Biol Chem. 1994;269:17274–17278. [PubMed] [Google Scholar]
- Levy DI, Wanderling S, Biemesderfer D, Goldstein SA. MiRP3 acts as an accessory subunit with the BK potassium channel. Am J Physiol Renal Physiol. 2008;295:F380–F387. doi: 10.1152/ajprenal.00598.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen B, Yankova M, Morest DK, Maryon E, Hand AR, Nonet ML, Wang ZW. Presynaptic ryanodine receptors are required for normal quantal size at the Caenorhabditis elegans neuromuscular junction. J Neurosci. 2005;25:6745–6754. doi: 10.1523/JNEUROSCI.1730-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen B, Ge Q, Wang ZW. Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J Neurosci. 2007;27:10404–10413. doi: 10.1523/JNEUROSCI.5634-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology. 2004;47:787–821. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- McManus OB, Helms LM, Pallanck L, Ganetzky B, Swanson R, Leonard RJ. Functional role of the β subunit of high conductance calcium-activated potassium channels. Neuron. 1995;14:645–650. doi: 10.1016/0896-6273(95)90321-6. [DOI] [PubMed] [Google Scholar]
- Meera P, Wallner M, Toro L. A neuronal beta subunit (KCNMB4) makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proc Natl Acad Sci U S A. 2000;97:5562–5567. doi: 10.1073/pnas.100118597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ. Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci. 1997;17:8061–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A. Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics. 1993;135:385–404. doi: 10.1093/genetics/135.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KH, Hernandez L, Cai SQ, Wang Y, Sesti F. A family of K+ channel ancillary subunits regulate taste sensitivity in Caenorhabditis elegans. J Biol Chem. 2005;280:21893–21899. doi: 10.1074/jbc.M502732200. [DOI] [PubMed] [Google Scholar]
- Plüger S, Faulhaber J, Fürstenau M, Löhn M, Waldschütz R, Gollasch M, Haller H, Luft FC, Ehmke H, Pongs O. Mice with disrupted BK channel β1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circ Res. 2000;87:E53–E60. doi: 10.1161/01.res.87.11.e53. [DOI] [PubMed] [Google Scholar]
- Raffaelli G, Saviane C, Mohajerani MH, Pedarzani P, Cherubini E. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J Physiol. 2004;557:147–157. doi: 10.1113/jphysiol.2004.062661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Saheki Y, Bargmann CI. Presynaptic CaV2 calcium channel traffic requires CALF-1 and the α2δ subunit UNC-36. Nat Neurosci. 2009;12:1257–1265. doi: 10.1038/nn.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salkoff L, Butler A, Ferreira G, Santi C, Wei A. High-conductance potassium channels of the SLO family. Nat Rev Neurosci. 2006;7:921–931. doi: 10.1038/nrn1992. [DOI] [PubMed] [Google Scholar]
- Schopperle WM, Holmqvist MH, Zhou Y, Wang J, Wang Z, Griffith LC, Keselman I, Kusinitz F, Dagan D, Levitan IB. Slob, a novel protein that interacts with the Slowpoke calcium-dependent potassium channel. Neuron. 1998;20:565–573. doi: 10.1016/s0896-6273(00)80995-2. [DOI] [PubMed] [Google Scholar]
- Shahidullah M, Reddy S, Fei H, Levitan IB. In vivo role of a potassium channel-binding protein in regulating neuronal excitability and behavior. J Neurosci. 2009;29:13328–13337. doi: 10.1523/JNEUROSCI.3024-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu YJ, Hiatt SM, Duren HM, Ellis RE, Kerppola TK, Hu CD. Visualization of protein interactions in living Caenorhabditis elegans using bimolecular fluorescence complementation analysis. Nat Protoc. 2008;3:588–596. doi: 10.1038/nprot.2008.16. [DOI] [PubMed] [Google Scholar]
- Toro B, Cox N, Wilson RJ, Garrido-Sanabria E, Stefani E, Toro L, Zarei MM. KCNMB1 regulates surface expression of a voltage and Ca2+-activated K+ channel via endocytic trafficking signals. Neuroscience. 2006;142:661–669. doi: 10.1016/j.neuroscience.2006.06.061. [DOI] [PubMed] [Google Scholar]
- Uebele VN, Lagrutta A, Wade T, Figueroa DJ, Liu Y, McKenna E, Austin CP, Bennett PB, Swanson R. Cloning and functional expression of two families of β-subunits of the large conductance calcium-activated K+ channel. J Biol Chem. 2000;275:23211–23218. doi: 10.1074/jbc.M910187199. [DOI] [PubMed] [Google Scholar]
- Wallner M, Meera P, Toro L. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane β-subunit homolog. Proc Natl Acad Sci U S A. 1999;96:4137–4142. doi: 10.1073/pnas.96.7.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZW, Saifee O, Nonet ML, Salkoff L. SLO-1 potassium channels control quantal content of neurotransmitter release at the C. elegans neuromuscular junction. Neuron. 2001;32:867–881. doi: 10.1016/s0896-6273(01)00522-0. [DOI] [PubMed] [Google Scholar]
- Wei A, Jegla T, Salkoff L. Eight potassium channel families revealed by the C. elegans genome project. Neuropharmacology. 1996;35:805–829. doi: 10.1016/0028-3908(96)00126-8. [DOI] [PubMed] [Google Scholar]
- Weiger TM, Holmqvist MH, Levitan IB, Clark FT, Sprague S, Huang WJ, Ge P, Wang C, Lawson D, Jurman ME, Glucksmann MA, Silos-Santiago I, DiStefano PS, Curtis R. A novel nervous system β subunit that downregulates human large conductance calcium-dependent potassium channels. J Neurosci. 2000;20:3563–3570. doi: 10.1523/JNEUROSCI.20-10-03563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Yang Y, Ye S, Jiang Y. Structure of the gating ring from the human large-conductance Ca2+-gated K+ channel. Nature. 2010;466:393–397. doi: 10.1038/nature09252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Hirschberg B, Smolik S, Forte M, Adelman JP. dSLo interacting protein 1, a novel protein that interacts with large-conductance calcium-activated potassium channels. J Neurosci. 1998;18:2360–2369. doi: 10.1523/JNEUROSCI.18-07-02360.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Ding JP, Lingle CJ. Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J Neurosci. 1999;19:5255–5264. doi: 10.1523/JNEUROSCI.19-13-05255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Ding JP, Zeng XH, Duan KL, Lingle CJ. Rectification and rapid activation at low Ca2+ of Ca2+-activated, voltage-dependent BK currents: consequences of rapid inactivation by a novel β subunit. J Neurosci. 2000;20:4890–4903. doi: 10.1523/JNEUROSCI.20-13-04890.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazejian B, Sun XP, Grinnell AD. Tracking presynaptic Ca2+ dynamics during neurotransmitter release with Ca2+-activated K+ channels. Nat Neurosci. 2000;3:566–571. doi: 10.1038/75737. [DOI] [PubMed] [Google Scholar]
- Yuan P, Leonetti MD, Pico AR, Hsiung Y, MacKinnon R. Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science. 2010;329:182–186. doi: 10.1126/science.1190414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusifov T, Savalli N, Gandhi CS, Ottolia M, Olcese R. The RCK2 domain of the human BKCa channel is a calcium sensor. Proc Natl Acad Sci U S A. 2008;105:376–381. doi: 10.1073/pnas.0705261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarei MM, Song M, Wilson RJ, Cox N, Colom LV, Knaus HG, Stefani E, Toro L. Endocytic trafficking signals in KCNMB2 regulate surface expression of a large conductance voltage and Ca2+-activated K+ channel. Neuroscience. 2007;147:80–89. doi: 10.1016/j.neuroscience.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Zhang W, St-Gelais F, Grabner CP, Trinidad JC, Sumioka A, Morimoto-Tomita M, Kim KS, Straub C, Burlingame AL, Howe JR, Tomita S. A transmembrane accessory subunit that modulates kainate-type glutamate receptors. Neuron. 2009;61:385–396. doi: 10.1016/j.neuron.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Mellem JE, Brockie PJ, Madsen DM, Maricq AV. SOL-1 is a CUB-domain protein required for GLR-1 glutamate receptor function in C. elegans. Nature. 2004;427:451–457. doi: 10.1038/nature02244. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Schopperle WM, Murrey H, Jaramillo A, Dagan D, Griffith LC, Levitan IB. A dynamically regulated 14–3-3, Slob, and Slowpoke potassium channel complex in Drosophila presynaptic nerve terminals. Neuron. 1999;22:809–818. doi: 10.1016/s0896-6273(00)80739-4. [DOI] [PubMed] [Google Scholar]