

Abstract
Cerebral cavernous malformations are common vascular malformations with an unpredictable risk of hemorrhage which consequences range from headache to stroke or death. Three genes CCM1, 2, 3 have been linked to the disease. The encoded CCM proteins interact with each others within a large protein complex. Since less than two years, a plethora of new data has emerged on the signaling pathways in which they are involved. They regulate diverse aspects of endothelial cell morphogenesis and blood vessel stability such as cell-cell junctions, cell shape and polarity, or cell adhesion to extracellular matrix. Although fascinating, a global picture is hard yet to depict as little is known on how these pathways coordinate to orchestrate angiogenesis. We present here what is known on the structural domain organization of the CCM proteins, their association as a ternary complex and their subcellular localization. Numerous CCM partners have been identified by two-hybrid screens, genetic analyses or proteomic studies. We focus on the best characterized ones and we review data on the signaling pathways they regulate as a step toward better understanding the etiology of the disease.
Keywords: Cell-Matrix Junctions; physiology; Hemangioma, Cavernous, Central Nervous System; genetics; metabolism; physiopathology; Humans; Microfilament Proteins; genetics; metabolism; Microtubule-Associated Proteins; genetics; metabolism; Proto-Oncogene Proteins; genetics; metabolism; Signal Transduction; rap1 GTP-Binding Proteins; metabolism
Cerebral cavernous malformations (CCM) are common vascular malformations with a prevalence of 1 individual in 200–250. Leak of blood can be detected by magnetic resonance imaging around each lesion and individuals with these vascular lesions are subject to an unpredictable risk of hemorrhage in midlife. Although lesions have been described in a variety of vascular beds, clinical manifestations are most common in the central nervous system where consequences can be stroke, seizure or any kind of neurological disorder and can lead to death [1]. The lesions consist of densely packed, grossly-dilated, capillary-like sinusoids lined by a single layer of endothelium embedded in a thick collagen matrix. Importantly, these lesions lack components of organized mature vessels such as pericytes, astrocytic foot processes and intact endothelial cell-cell junctions [2]. Both sporadic and familial forms of CCM have been identified. The genetics of the disease is developed in a joined minireview by Riant et al. in this issue. Briefly, three loci have been mapped and the genes reponsible for the disease, CCM1 to 3, identified in these loci. Since less than two years, fascinating new data have emerged on the signaling pathways regulated by the products of these three genes. However a global picture is hard yet to depict since not much is known about how these signaling pathways coordinate. Many advances have been made in the description of the core complex formed by the association of these three proteins and in the identification of numerous CCM partners. In this review, we will focus on partners that open new avenues for CCM research and we will discuss recent insights into their role in cytoskeleton remodelling, regulation of cell matrix adhesion and cell-cell junction homeostasis.
I. Structural domain organization of CCM1/Krit1, CCM2/OSM/MGC4607 and CCM3/PDCD10 proteins
CCM1 gene encodes a protein also named Krit1 for K-Rev Interaction Trapped 1. Krit1 was first identified in 1997 as a partner of the small G-protein Krev-1/Rap1 from a yeast two-hybrid screen [3]. Two years later, the CCM1 locus was mapped to the gene encoding Krit1 [4] [5]. Krit1 is a 84 kDa scaffold protein with no catalytic activity which contains several distinct domains involved in protein-protein interaction (Figure 1). Remarkably, Krit1 possesses a C-terminal FERM (band Four.1 Ezrin Radixin Moesin) domain, a signature of membrane binding proteins like Talin, Ezrin, Radixin, or Moesin. FERM domains are composed of three subdomains F1 to F3 arranged in clover-shaped fashion. The F3 subdomain has a PTB (Phospho-Tyrosine Binding) fold. PTB domains recognize a canonical NPXY/F motif often found on the cytoplasmic tail of transmembrane receptors. Recruitment of PTB or FERM proteins to transmembrane receptors is a conserved mechanism used by cells to build intracellular signaling hubs. Remarkably, besides its FERM domain, Krit1 possesses three N-terminal NPXY/F motifs (figure 1). This peculiar structural organization allows the N and C-terminal halves of Krit1 to interact with each other in GST pull-down [6] or in a yeast two-hybrid interaction assay [7] suggesting that Krit1 could adopt a closed and opened conformation in vivo resulting from either intramolecular folding or dimerization. The first [6] or the third [7] NPXY/F motif could be involved in this interaction. A systematic mutagenesis of each of the three motifs should help to determine the contribution of each of them. Three ankyrin repeats are present between the NPXY/F motifs and the FERM domain (figure 1). Although ankyrin repeats are found in thousands of proteins and support interaction with many diverse proteins, no partner interacting with Krit1 ankyrin repeats has been found yet. Compared to Krit1, CCM2 and 3 are much simpler in their structural domain organization. CCM2 gene encodes a scaffold protein of 51 kDa containing also a PTB domain [8, 9], but no other known domain (figure 1). It was identified in a yeast two-hybrid screen using MEKK3 as bait to identify proteins involved in the response of the cell to hyperosmotic shock [10] and named Osmosensing Scaffold for MEKK3 (OSM). The last mutated gene CCM3 or PDCD10 (Programmed Cell Death 10) has been identified more recently [11] and is up-regulated in fibroblasts exposed to specific apoptosis inducers, such as staurosporine, cycloheximide, and TNF-α [12]. Apoptotic or on the contrary proliferative functions have been attributed to CCM3 [13, 14]. No homology with any known domain is found on CCM3 but it has been suggested that this small protein (25 kDa) folds as one stable domain [15] (figure 1).
Figure 1. Structural domains of CCM proteins.
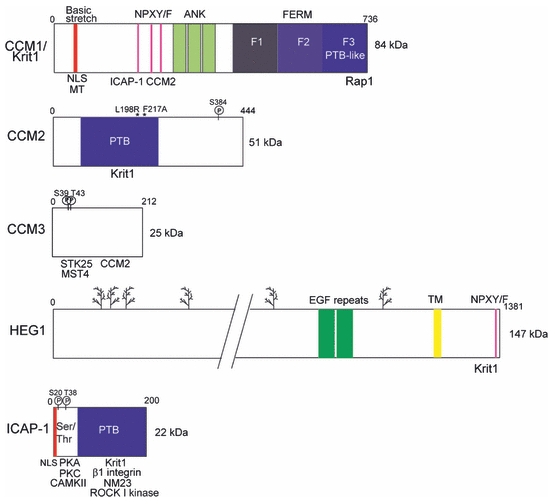
Krit1/CCM1 bears a C-terminal FERM (Band 4.1 Erzin Radixin Moesin) domain and three N-terminal NPXY/F motifs allowing either the folding of the protein on itself or its interaction with ICAP-1 and CCM2. ANK: Ankyrin domain, NLS: Nuclear Localization Signal, MT: microtubules. PTB (PhosphoTyrosine Binding) domain of CCM2/OSM interacts with a Krit1 NPXY/F motif. L198R and F217A mutations prevent CCM2 interaction with Krit1. CCM3 has no homology with any known domain. Its N-terminal fragment (L33 to K50) interacts with MST4, STK24, and STK25. Ser39 and Thr43 are substrate of phosphorylation by STK25. HEG1 is a heavily glycosylated ( )transmembrane protein carrying two extracellular EGF-like repeats and a C-terminal NPXY/F motif which interacts with Krit1. Its extracellular ligand is not known. ICAP-1 has a Ser/Thr riche N-terminus containing a NLS and sites of phosphorylation by Calmodulin-dependent kinase II, Protein kinase A and C. Reported interactions with β1 integrin, Krit1, ROCK I-kinase and NM23.
)transmembrane protein carrying two extracellular EGF-like repeats and a C-terminal NPXY/F motif which interacts with Krit1. Its extracellular ligand is not known. ICAP-1 has a Ser/Thr riche N-terminus containing a NLS and sites of phosphorylation by Calmodulin-dependent kinase II, Protein kinase A and C. Reported interactions with β1 integrin, Krit1, ROCK I-kinase and NM23.
II. CCM complexes and their sub-cellular localizations
Interactions within the CCM1, 2, 3 complex
Consistent with their involvement in the same pathology, Krit1/CCM1, CCM2 and CCM3 can interact altogether. Co-immunoprecipitations, GST pull-downs and mutagenesis have allowed identifying the interaction sites between the three proteins in this complex.
Endogenous or overexpressed Krit1 and CCM2 interact with each other [16, 17]. Mutations in the PTB domain of CCM2 on conserved residues critical for NPXY/F motif binding (figure 1) are deleterious for Krit1-CCM2 interaction. One, L198R, a single missense mutation found on CCM patient [9], the other, F217A, has been engineered based on homology structure with known PTB domain [16]. The N-terminus of CCM2 also takes part in this interaction as a lack of amino acid residues 11 to 68, an in frame-deletion observed in patients [18], prevents the interaction of CCM2 with Krit1 [19]. Conversely, the binding domain for CCM2 on Krit1 is still uncertain. Since their interaction involves CCM2 PTB domain, it is likely that the counterpart on Krit1 is one of its three NPXY/F motifs. Indeed, a yeast two-hybrid assay using small fragments of CCM2 centered on NPXY/F 2 and 3 have pinpointed these motifs as CCM2 interacting sites [17]. However, single amino acid substitution in each of these motifs has no effect on binding of Krit1 to CCM2 [16]. Additional mutagenesis on residues immediately N or C-terminal of the NPXY/F might be required to significantly reduce the affinity.
CCM2 interacts with CCM3 [15, 19] but their respective interaction sites are not known. None of the three CCM2 mutations cited above impairs CCM2 binding to CCM3 [19] showing that CCM2 binding domains to Krit1 and CCM3 are not redundant. Indeed, the three overexpressed proteins form a complex. CCM2 is the linker protein that brings together Krit1 and CCM3 which otherwise have no affinity for each other [15, 19, 20]. Remarkably, this ternary complex was detected by proteomic approaches [20, 21]. However, CCM3 was also identified by proteomic analysis as a component of an other large complex named STRIPAK (striatin-interacting phosphatase and kinase) which assembles phosphatases and kinases arranged around the protein phosphatase 2A (PP2A) core [21]. Interestingly, neither Krit1 nor CCM2 was detected in the STRIPAK complex, but small amounts of CCM3 could be pulled down along with Krit1 on CCM2 beads. This suggests that CCM3 associates with (at least) two different complexes; in substoichiometric amounts with the Krit1-CCM2 complex and in large amounts with the STRIPAK complex.
Shuttling of CCM proteins between the membrane and nucleus
These in vitro data suggest that the three CCM associate in a ternary complex in vivo but that they are also very likely engaged in several other complexes having different localizations (figure 2). As such, Krit1 associates with the β1 integrin regulator ICAP-1 (as discussed below) and this complex can shuttle between the cytosol and the nucleus. Both Krit1 and ICAP-1 have a NLS motif in their N-terminus and both localize in a NLS dependent manner to the nucleus of transfected cells [16] [7]. Interestingly, it has been shown that during cell spreading, ICAP-1 shuttles from the plasma membrane to the nucleus where it stimulates transcription and cellular proliferation [22]. However, binding of CCM2 to Krit1 inhibits the nuclear translocation of Krit1/ICAP-1 complex. Indeed, co-transfection of CCM2 together with Krit1 and ICAP-1 induces the formation of a ternary complex between the three proteins that sequesters Krit1/ICAP-1 in the cytosol [16] [7]. The association of CCM2 with Krit1/ICAP-1 could therefore be a key event and the target of upstream signaling pathways to control Krit1/ICAP-1 transcriptional regulatory functions.
Figure 2. Emerging signaling pathways and vascular processes controlled by the CCM proteins.
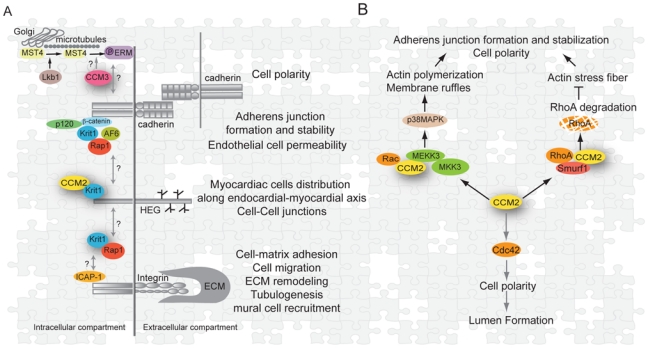
(A)Cadherins, HEG1 and integrins are three transmembrane receptors connected to CCM proteins or functions. All three receptors are already known to play in different steps of vessel morphogenesis. Possible cross-talks between their dependent signaling pathways through CCM proteins are represented by arrows.
(B)CCM2 is a scaffold for small GTPases of the Rho family and for p38MAPK kinase. It is involved in actin cytoskeleton remodeling through scaffolfing of Rac, activation of the p38 MAPK kinase pathway and proteosomal degradation of RhoA. CCM2 could also be involved directly or indirectly in Cdc42 activation. As a result, cell-cell junctions, cell polarity and lumen formation are likely dependent on CCM2 signaling.
Transport along microtubules could be a way for Krit1 and its partners to shuttle between the cytoplasm and the nucleus. Interestingly, α and β tubulins have been identified by proteomic analysis of proteins co-immunoprecipitating with flagged CCM2 in stably transfected macrophages [20]. The presence of tubulin subunits in the pulled down complex depended on CCM2-Krit1 interaction since a functional PTB domain was required on CCM2 suggesting that Krit1 is the direct partner of tubulin. In fact, Krit1 has been shown to co-sediment with in vitro polymerized microtubules [6], and to co-localize with microtubules in BAEC [23]. Two binding sites for microtubules have been mapped on Krit1: the one that contributes the most to the binding overlaps with the NLS sequence, the other lies in its last 50 amino acid residues.
PTB and FERM domains have structural features enabling their interaction with phosphoinositides in membranes. As such, Krit1, CCM2 and CCM3 bind to phosphoinositides [6, 20]. Purified Krit1 binds to liposomes only when supplemented with phosphoinositides [6]. Modelisation of Krit1 FERM domain using known structures has highlighted a basic cleft between the F1 and F3 subdomains which could likely interact with the negative charges of phosphate groups. CCM2 and CCM3 also directly bind to phospholipids as shown by overlay experiments on phosphatidylinositol phosphate arrays [20]. CCM2 most likely interacts via its PTB domain. CCM3 lipid interacting domain is not yet known. CCM2 binds preferentially to mono- over bi-phosphorylated phosphatidylinositols, a result we also observed for Krit1 (our unpublished data). Conversely, CCM3 has a higher affinity for bi-and tri-phosphorylated phosphatidylinositols an additional argument suggesting that Krit1 together with CCM2 might localize to different membrane compartments than CCM3.
III. New partners for the CCM proteins: what they tell us on putative regulated signaling pathways
The CCM proteins are expressed in many different cell types. Thus, a crucial and intriguing question about the etiology of the cavernous malformations in blood vessels is to ask what is unique to endothelial cells. Indeed, depletion of CCM2 targeted to the endothelium and not to the surrounding tissue results in vascular defects in mouse embryos [24, 25] (see Chan et al, this issue). One possibility is that specific subsets of interactions occur in endothelial cells. Even though many studies have not been conducted in endothelial cells, they have been very helpful to identify new partners for CCM proteins. As such, proteomic studies performed in macrophages and astrocytes cell lines have helped identifying not less than 114 proteins interacting with CCM2 [20]. We will restrict in this review to the best characterized partners which can give clues on the function of the CCM proteins in vascular integrity.
An increasing amount of data indicates that the CCM proteins are connected to the plasma membrane and regulate cell-cell adhesion, cell shape and polarity and most likely cell adhesion to extracellular matrix (figure 2). This makes very much sense with regard to the phenotype of the CCM lesions where endothelial cells are loosely joined to each others, mural cells (i.e. pericytes and astrocytes) are absent, and the basal lamina surrounding the endothelium is abnormal [2]. Both cell-cell adhesion and cell polarity require the assembly of two specialized intercellular adhesion structures that regulate vascular permeability. Adherens Junctions (AJ) initiate and maintain strong contacts between endothelial cells and promote Tight Junctions (TJ) assembly. TJ are specialized in the passage of ions and solutes through the paracellular route. They may also act as a physical barrier along the cell surface allowing the asymmetrical distribution of proteins and lipids between apical and basolateral domains, a phenomenon known as cell polarization. Cell adhesion to extracellular matrix requires integrins clustered in highly dynamic adhesive structures which regulate cytoskeleton rigidity, extracellular matrix remodeling and probably cell-cell junctions.
1. Rap1, the master regulator of cell-cell and cell-extracellular matrix adhesion
Over the past years, it has been established that the Ras family small G protein Rap1 stimulates cell adhesion to extracellular matrix by activating integrins and cell-cell adhesion by stimulating the formation and maintenance of adherens junctions. It does so by activating a large number of effectors involved for most of them in regulating actin dynamics [26, 27]. Rap1 was the first reported Krit1 partner since it was used as the bait to clone Krit1 in a yeast two-hybrid screen [3]. This interaction has been questioned until 2007 when two groups confirmed by biochemical in vitro assays [6] and functional studies [28] that Krit1 is a Rap1 effector. However, Rap1 is not found in the CCM complex defined by proteomic analysis suggesting that Rap1-Krit1 could form an independent complex. Interestingly, Rap1a and 1b knock-out mice show defective angiogenesis, characterized by delayed perinatal retinal vascularization, reduced microvessel sprouting from aortic rings in response to angiogenic factors or reduced neovascularization of ischemic hind limbs [29] [30, 31]. Reduction of the function of Rap1b using morpholinos in zebrafish embryos disrupts endothelial junctions and provokes intracranial hemorrhage. Importantly, a minor reduction of Rap1b in combination with similar reduction in Krit1 results in high incidence of intracranial hemorrhage, whereas injection of each morpholinos independently has almost no effect [32]. This indicates that Rap1 and Krit1 act in a common molecular pathway. Indeed, Ginsberg’s group showed that small interfering RNA depletion of Krit1 blocks the ability of Rap1 to stabilize endothelial cell-cell junctions in culture cells [28].
2. CCM partners in cell-cell junctions
Proteins of Adherens Junctions
Endogenous Krit1 localizes to cell-cell junctions on a BAEC confluent monolayer and co-immunoprecipitates with the Rap1 effector AF-6/afadin, β-catenin and p120-catenin. This localization requires Krit1 FERM domain and is dependent upon activation of Rap1 [28]. Consistently, it was shown that in vitro Rap1 binding to Krit1 FERM domain enhances Krit1 association with liposomes most likely by inducing a conformational change of its basic pocket that gives Krit1 a better affinity for phosphoinositides [6]. Depletion of Krit1 by siRNA leads to the disruption of β-catenin localization to AJs and increases the permeability of the monolayer barrier [28], a phenotype reminiscent of what is observed in human lesions. Therefore Krit1 by localizing β-catenin to AJ is likely involved in the formation and the maintenance of the endothelial barrier (figure 2a). However it is not yet known whether Krit1- β-catenin interaction is direct.
The transmembrane glycosylated protein HEG1
HEG1 is a transmembrane protein of unknown function bearing a large extracellular domain with two EGF-like domains, a transmembrane segment and a short cytoplasmic tail (100 amino acids) with a conserved C-terminal NPXY/F motif (figure 1). Its extracellular domain is predicted to be highly glycosylated. It is expressed specifically in the endothelium and the endocardium. No extracellular ligand is known yet. HEG1 is the mammalian homolog of the zebrafish heart of glass. Zebrafish Heart of glass mutants show enlarged cardiac chambers resulting from unproper distribution of myocardiac cells along the endocardial-to-myocardial axis [33]. Two other genes, santa and valentine, functioning in the same molecular pathways were identified and turned out to be Krit1 and CCM2 respectively. They display the same phenotype as heart of glass when disrupted in zebrafish or when a combination of low doses morpholinos against the three proteins is injected [34]. Recently, HEG1 and CCM2 were also shown to genetically interact in mouse [35]. Indeed, Heg1−/−; Ccm2lacZ/+ [35] like Ccm2−/− mice [24, 25] have severe cardiovascular defects and die early in development owing to a failure of nascent endothelial cells to form patent vessels. Both heg1−/− and ccm2−/− mice displayed shortened endothelial junctions compared to control littermates [35]. More details can be found in a joined mini-review on animal models of the CCM disease (Chan et al., this issue). In addition, the ternary complex between HEG1, Krit1 and CCM2 has been biochemically demonstrated [35] (figure 2a). A CCM2 mutant unable to bind Krit1 is not recruited in the HEG1-Krit1 complex suggesting that Krit1 is the adaptor which connects CCM2 to the transmembrane receptor. It is very likely that the association of HEG1 with Krit1 requires HEG1 NPXY/F motif and Krit1 FERM domain but this remains to be tested.
As a hint toward a function, HEG1 is evolutionary related to mucin 13 [36]. Mucins are either secreted or inserted as transmembrane glycoproteins in polarized epithelia. Transmembrane mucin 1 can associate with FGFR3 (Fibroblast Growth Factor receptor 3) [37], and with β-catenin to activate β-catenin-driven transcription of Wnt target genes [38, 39]. Interestingly, an emerging idea concerning mucins function is that loss of polarity through a breach in the cell layer could enable growth factor receptors and mucins to associate and engage in signaling to activate gene transcription designed to repair the breach and reestablish cell polarity [40]. This signaling pathway would make sense with regard to the loss of the integrity of the endothelial barrier and a putative dysfunction of repair mechanisms in CCM lesions. Consistent with this line, an elegant work from E. Dejana’s group has shown that Wnt/β-catenin signaling is required for endothelial cell expression of proteins necessary to the development of the blood brain barrier [41]. Therefore, under the control of HEG1, Krit1 could be involved along with β-catenin in the dual role of stabilization of cell-cell junctions and regulation of the expression of blood brain barrier specific players.
3. Partners in cell shape remodeling and polarity
Along with a role of Krit1 in cell-cell adhesion, a network of data identifies the CCM complex as a scaffold for the Rho family GTPases RhoA, Rac and Cdc42, and for the MAPK (mitogen activated protein kinases) and STK (Ser/Thr kinases) kinases. These proteins regulate endothelial cell shape and polarity. How RhoA, Rac and Cdc42 interplay to orchestrate cell-cell junction formation and polarity is still under active investigation and finely reviewed in [42]. Nevertheless, emerging data suggest that CCM proteins are involved in the spatio-temporal tuning of these small GTPases and consequently are able to remodel actin cytoskeleton (figure 2b).
CCM2 as a scaffold of actin cytoskeleton machinery
CCM2/OSM has been first identified by two-hybrid screening as a scaffold for the MEKK3/MKK3 complex [10] which is necessary to restore cell volume and shape in response to hyperosmotic shock. p38 MAPK is a downstream substrate of MEKK3 (mitogen-activated protein kinase kinase kinase 3). MAPKs are ubiquitously expressed and contribute to a wide variety of cell responses to very diverse stimuli. MAPKs are the terminal kinases in a three kinase phosphorelay module, in which MAPKs are phosphorylated and activated by mitogen-activated protein kinase kinase MKKs, which themselves are phosphorylated and activated by mitogen-activated protein kinase kinase kinase (MKKKs) [43].
It is a critical kinase for long-term cellular adaptation to prolonged hyperosmotic exposure. It regulates gene transcription and actin remodeling. This pathway is conserved from yeast to mammals and in multiple tissues suggesting its importance in cellular physiology beyond that of hyperosmolarity responses. Indeed, the p38 MAPK pathway has also been shown to play an important role in angiogenesis. Deletion of MEKK3 causes severe vascular defects [44] and defective angiogenesis in Rap1b-deficient mice is associated with an impaired p38 MAPK signaling pathway [30]. Moreover, P38 MAPK is required for vEGF (vascular Epidermal Growth Factor) effect on actin remodeling in HUVEC [45]. P38 MAPK signaling pathway leads to the activation of HSP27 (Heat Shock Protein 27), an F-actin cap binding protein which in turn activates actin polymerization and stabilization. It is proposed that CCM2 exists in a stable complex with MEKK3. Upon a hyperosmotic stress, CCM2 and MEKK3 are recruited to membrane ruffles through a direct interaction of CCM2 with Rac, where they co-localize with F-actin [10]. Therefore, CCM2 would serve as a scaffold for the actin polymerization machinery (figure 2b). A link between CCM2, Rac and MEKK3 has been confirmed by proteomic analysis of the CCM complex [20].
Control of RhoA degradation and actin stress fibers formation by CCM2
More recently, the effects of the depletion of CCM2 on endothelial cell cytoskeletal architecture and signaling have been studied [25]. CCM2 depletion by small interfering RNA leads to an increased number of actin stress fibers and enhanced permeability of the endothelial layer, a phenotype also observed upon depletion of Krit1 [28]. In addition to Rac1, CCM2 co-immunoprecipitates with RhoA. CCM2 depletion leads to increased activated RhoA, whereas it has no effect on activation of Rac1 [25]. Contrary to what was observed in hyperosmotic shock, CCM2 depletion does not affect p38 MAPK signaling but rather an other MAPK module, i.e. the JNK (c-Jun N-terminal kinase), MKK4, MKK7 pathway [25]. JNK activation is blocked by the ROCK (Rho-associated kinase) inhibitor Y-27632 suggesting that CCM2 loss activates the JNK pathway through RhoA. Therefore, a physiological function of CCM2 may be to limit RhoA activation. Johnson’s group recently gave a molecular explanation of this inhibitory effect by identifying the E3 ubiquitin ligase Smurf1 as a new CCM2 partner. They show by co-immunoprecipitation on overexpressed proteins that Smurf1 interacts with CCM2 through a PTB/NPXY interaction and that this interaction leads to loss of RhoA [46] (figure 2b). Proteosomal degradation is one of the modes used by cells to spatially restrict small G protein signaling. In particular, localized degradation of RhoA has already been involved in the control of cell polarity or migration [47, 48].
Importantly, HEG1, only expressed in endothelial cells, could very well be a long sought piece of the puzzle which gives the CCM pathway its endothelial specific nature. Interaction of Krit1 with HEG1 and VE-Cadherin in endothelial monolayer might create a physical link between these receptors to negatively control RhoA-dependent stress fibers formation and to promote Rac-dependent cell-cell junction.
Putative regulation of lumenogenesis by CCM2 via Cdc42 activation
Contrary to Rac and RhoA, no interaction has been observed between CCM2 and Cdc42. However, depletion in CCM2 leads to less basal activated Cdc42 implying that CCM2 is somehow involved in activating Cdc42 [25]. Besides its role on actin filament bundling during filopodia formation and cell migration, Cdc42 has a conserved role in regulating cell polarity in many eukaryotic cells mainly through interaction with the polarity complex PAR (PAR6-PAR3-aPKC). Cdc42 affects cell-cell junction formation and the polarized trafficking of proteins to the apical and basal domains [49].
Concomitantly with a decrease in the level of activated Cdc42 [25], knock-down of CCM2 in HUVEC has been reported to decrease lumen formation in 3D in vitro culture [25, 35] (figure 2b). This is consistent with the already described role of Cdc42 in lumenogenesis. During capillary formation, endothelial cells assemble into chains, polarize and generate apical membrane vesicles via pinocytosis. The intracellular vesicles then coalesce into an elongate vacuole-like structure spanning the length of the cell which fuses with the plasma membrane to open to the exterior and establish luminal continuity with the next cell in the chain [50]. Cdc42 and Rac1 are both required for lumenogenesis by involving Pak2, Pak4 and the PAR complex [51]. Consistently, in CCM2 depleted mice or zebrafish, endothelial cells failed to organize in lumenized vessels. However, endothelial vacuole-like structures form normally in the intersegmental vessels of zebrafish embryos lacking CCM2 as visualized using GFP-Cdc42 to label these vacuoles [35]. On the contrary, CCM2 deficient HUVEC presented a strong decrease in vacuoles and lumen formation in a 3D in vitro culture [25]. Whereas it is proposed in [35] that steps downstream of the formation of the vacuoles might be affected by the loss of CCM2 and lead to the absence of a lumen, quantification of intracellular vacuoles in [25] pinpoints to a default at the level of vacuoles formation. Further experiments will be needed to solve the discrepancy between these results.
Putative control of cell polarization by CCM3 through GCKIII kinases
Using yeast two-hybrid screen and proteomic analysis, STK24, STK25 and MST4 were identified as partners of CCM3 [14, 15, 21]. These ser/thr kinases belong to the germinal center kinases III (GCKIII) subfamily, and are related to the yeast protein kinase sterile 20 (Ste20). STK25 (Serine/threonine Kinase 25) and MST4 (Mammalian sterile twenty-like 4) bind at the N-terminus of CCM3 between leucine 33 and Lysine 50 [52], a region deleted by an in frame deletion of exon 5 in a family of patients [11]. CCM3 is phosphorylated by STK25 at ser 39 and thr 43 [52] but the role of this phosphorylation is not known yet. Both STK25 and MST4 localize to the Golgi apparatus in unpolarized cells and regulate cell migration and polarity [53]. Interestingly, MST1, a GCK kinase which interacts with the Rap1 effector RAPL, translocates from the Golgi on vesicles moving along microtubules aimed at assembling specialized plasma membrane domains such as leading edge during T cell polarization [54].
The recent connection of MST4 with Lkb1 function in cell polarity might help understanding CCM3 role. Lkb1 is a tumor suppressor gene responsible for Peutz-Jeghers syndrome, a cancer predisposition disorder characterized by gastrointestinal polyps. Lkb1 regulates cell polarity in epithelial cells in a cell autonomous fashion. Ten Klooster and collaborators recently showed that upon Lkb1 activation, MST4 translocates from the Golgi to the sub-apical domain of the epithelial cell near the brush border where it phosphorylates ezrin [55], a membrane-actin microfilaments linker necessary for normal microvilli. Whereas Lkb1 seems to control MST4 subcellular localization, CCM3 might regulate MST4 kinase activity (figure 2a). Indeed, it has been shown that CCM3 enhances MST4 activity in vitro [14]. It would therefore be very interesting to place CCM3 in the newly described Lkb1 pathway and to check whether it also applies to endothelial polarization by regulating function of ERM proteins such as ezrin or moesin. Interestingly, phosphorylated ezrin is localized to cell-cell junction in endothelial cells and regulates cell-cell junction formation and stability [56]. Importantly, conditional Lkb1 deletion targeted to endothelial cells leads to embryonic death with loss of vascular smooth muscle cells (vSMCs) around the vessels and vascular disruption [57], a phenotype also observed in CCM lesions. This phenotype is attributed to loss of TGFβ production by endothelial cells and to blocking of its subsequent signaling to adjacent differenciating vSMCs.
4. Partners in Cell-ExtraCellular Matrix adhesion
The last published articles on animal CCM models strongly emphasize the role of CCM proteins on the control of cell-cell junctions. However, we think that a control of the interaction of endothelial cells with their surrounding environment should not be ruled out. Indeed, ultrastructural analyses of CCM lesions clearly demonstrated the absence of perivascular ensheating cells or astrocytic foot processes around the vessel and the presence of a thicker and multilayered collagenous matrix [2]. Moreover and strikingly, no defects in cell junctioning between endothelial cells was observed in zebrafish CCM1 and CCM2 mutants but instead increased spreading of endothelial cell around dilated vessels [58]. Finally, the first chronologically identified CCM partner, ICAP-1 (Integrin Cytoplasmic Associated Protein-1) is involved in regulating cell adhesion to extracellular matrix. ICAP-1 has been identified as a Krit1 partner in a yeast two-hybrid screen and their interaction was confirmed by co-immunoprecipitation [59, 60]. ICAP-1 is present in the CCM complex identified by proteomic analysis [20]. Like CCM2, ICAP-1 has a C-terminal PTB domain linked to a short N-terminal moiety (60 a.a) containing several consensus sites for kinases (figure 1). Its PTB domain interacts with the first NPXY/F motif of Krit1. Importantly, a ternary complex can form between ICAP-1, Krit1 and CCM2 [16], suggesting that Krit1 could act as a scaffold for ICAP-1 and CCM2 dependent signaling pathways.
ICAP-1 inhibits β1 integrin activation and focal adhesion assembly
Whereas its role in the CCM complex is not known yet, ICAP-1 has been well characterized as inhibitor of β1 integrin activation by talin. ICAP-1 binds specifically to β1 integrin cytoplasmic tail [61]. Its overexpression in cells leads to disruption of β1 integrin focal adhesions, subsequently decreased cell adhesion to fibronectin and increased cell migration [62, 63]. ICAP-1 competes in vitro with talin for binding to β1 integrin. Consistently, live cell imaging performed in Icap-1 deficient MEF confirmed that ICAP-1 inhibits β1 integrin high affinity state favored by talin, slows down the rate of focal adhesion assembly and controls matrix sensing [64]. In addition, ICAP-1 interacts with ROCK and recruits it to β1 integrin in the lamellipodia [65]. The most evident phenotype of ICAP-1 deficient mice is their smaller size and weight, their cranio-facial abnormalities and a general skeletal defect due to reduced proliferation and differentiation defect in osteoblast cells [66]. In addition, C57Bl6 ICAP-1 deficient mice display a high rate of perinatal mortality (D. Bouvard & R. Fässler, personal communication). Whether ICAP-1 deficient mice suffer from vascular defects is not known yet. Importantly, depletion of Krit1 by small interfering RNA leads to the depletion of ICAP-1 in HeLa or HUVEC [67]. This reduced level of ICAP-1 is not due to a down-regulation of its mRNA [67] therefore implying that ICAP-1 is stabilized upon its association with Krit1. This observation suggests that ICAP-1 might also be reduced in patients mutated on CCM1 gene.
β1 integrin regulates vascular morphogenesis: a target for CCM proteins?
Ligand-activated integrins are essential to control intracellular actin cytoskeleton organization [68] and extracellular matrix (ECM) remodeling [69]. Mouse models were very valuable to highlight the role of β1 integrin in blood vessel morphogenesis. Indeed, conditional deletion of β1 integrin in endothelial cells induces general vascular defects including reduced branching and sprouting and is embryonic lethal [70–72]. Interestingly, blood vessels are frequently discontinuous [71], cranial vessels are dilated [71, 72], and sporadic large cerebral hemagiomas can be seen [72]. Moreover, the staining of fibronectin (FN), a ligand of α5β1 integrin, is reduced and more diffused in mutant embryo basement membranes around the vessels [71].
β1 integrin regulates several processes involved in vascular morphogenesis such as extracellular matrix remodeling and growth factor delivery, lumen formation and recruitment of mural cells [73–75]. Three-dimensional in vitro culture experiments and chorioallantoic membrane assays in chicken embryos have shown that FN fibrillogenesis is required for endothelial cell tubulogenesis [76]. In vivo, FN fibrillogenesis is likely a α5β1 integrin-driven process resulting in extracellular FN organization in fibrils [69, 77] which modulates environment rigidity. Remarkably, at identical substrate densities, plating endothelial cells on rigid surfaces promotes cell-ECM interactions and endothelial cell dispersion whereas plating endothelial cells on softer surfaces promotes cell-cell interactions and network formation [78]. Additionally, FN fibrillogenesis organizes the deposition of collagen [79]. This regulates cell contractility and migration and might be crucial for proper tubulogenesis. Moreover, organized matrix can tether soluble growth factors like vEGF or TGFβ and generate gradients that elicit endothelial chemotactic responses. It has been shown that matrix-bound vEGF induces capillary sprouting with small lumen whereas soluble vEGF induces capillary hyperplasia and lumen enlargement [80]. The major dilation observed in CCM lesions in humans could be a consequence of incorrect growth factors gradient. Lumenogenesis per se is another process possibly involving β1 family integrin. It is proposed that integrins signal to Rac and Cdc42 to activate vacuolization [74, 81]. Finally, β1 integrin promotes blood vessel maturation by stimulating the adhesion of mural cells to endothelial cells. For instance, α4β1 integrin on endothelial cells can interact with VCAM-1 (Vascular Cell Adhesion Molecule - 1), a transmembrane adhesion receptor present on mural cells to mediate apposition of the two cell types [82]. Conversely, β1 integrin in pericytes is necessary for their correct spreading along the vessels [83, 84]. The defect in coverage with mural cells in CCM lesions might be a consequence of β1 integrin dysfunction either in endothelial or mural cells.
Since ICAP-1 regulates β1 integrin function, CCM proteins may consequently regulate processes involving β1 integrin (figure 2a). Interestingly, it has been reported using yeast two-hybrid assays that Krit1 can compete with β1 integrin for binding to ICAP-1 [60] suggesting that Krit1 could regulate ICAP-1 inhibitory effect on β1 integrin. Conversely, β1 integrin and ICAP-1 could regulate Krit1 functions on cell-cell adhesion. These intriguing hypotheses need further work to be tested.
What about HEG1?
The numerous HEG1 glycosylated moieties might bind to galactoside-binding lectins, named galectins, as mucins do. Upon binding to galectin-3, epithelial cell MUC1 clusters on the cell surface possibly unraveling adhesion sites and this leads to binding of the epithelial cell to endothelial cell [85]. Moreover, galectin-3 was reported to regulate α2β1 binding to collagen-I and IV [86]. Consistently, early adhesion events of cells to ECM involving other kind of receptors than integrins like proteoglycan or hyaluronan receptors were reported to precede the formation of adhesive structures driven by integrins [87]. Therefore, HEG1 together with integrins could participate in a temporally regulated adhesion process to either ECM or mural cells.
V. Perspectives
The last two years have been extraordinarily rewarding by opening new avenues for the comprehension of the physiology of the CCM proteins. While many hints about various signaling pathways have been collected, numerous holes persist in the jigsaw puzzle making it difficult to catch sight of the entire painting. In the future, efforts will have to be put on describing cross-talks between these different pathways. What stands out for now is that HEG1 could ignite endothelial specific pathways involving CCM proteins that are necessary for morphogenesis of blood vessels. Putative molecular links between HEG1 and Adherens junctions on one side and integrins on the other side deserve to be thoroughly explored. If molecular links between the two types of cell adhesion turn out to involve CCM partners, they may lift the veil on the long known but poorly understood cross-talk between integrins and cadherins.
Acknowledgments
We thank Olivier Destaing, Daniel Bouvard, Sophie Béraud Dufour, Mireille Faurobert and Reinhard Fässler for helpful discussions and comments on the manuscript. This work was supported by the CNRS, INSERM, the Région Rhône-Alpes and the association pour la recherche contre le cancer (ARC).
List of abbreviations
- AJ
Adherens Junction
- BAEC
Bovine Aortic Endothelial Cell
- CCM
Cerebral Cavernous Malformation
- ECM
ExtraCellular Matrix
- FERM
Band 4.1 Ezrin Radixin Moesin
- FN
Fibronectin
- GCK
Germinal Center Kinase
- ICAP-1
Integrin Cytoplasmic Adaptor Protein-1
- HEG1
Heart of Glass 1
- HUVEC
Human Vein Umbilical Endothelial Cells
- Krit1
K-Rev Interacting 1
- MAPK
Mitogen Activated Protein Kinase
- MEKK3
Mitogen Activated Protein Kinase Kinase Kinase 3
- MKK
Mitogen Activated Protein Kinase Kinase
- NLS
Nuclear Localization Signal
- OSM
Osmosensing scaffold for MEKK3
- PTB
PhosphoTyrosine Binding
- STRIPAK
striatin-interacting phosphatase and kinase
- TJ
Tight Junction
References
- 1.Marchuk DA, et al. Vascular morphogenesis: tales of two syndromes. Hum Mol Genet. 2003;12(1):R97–112. doi: 10.1093/hmg/ddg103. [DOI] [PubMed] [Google Scholar]
- 2.Clatterbuck RE, et al. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J Neurol Neurosurg Psychiatry. 2001;71(2):188–92. doi: 10.1136/jnnp.71.2.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serebriiskii I, et al. Association of Krev-1/rap1a with Krit1, a novel ankyrin repeatcontaining protein encoded by a gene mapping to 7q21-22. Oncogene. 1997;15(9):1043–9. doi: 10.1038/sj.onc.1201268. [DOI] [PubMed] [Google Scholar]
- 4.Sahoo T, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum Mol Genet. 1999;8(12):2325–33. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- 5.Laberge-le Couteulx S, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23(2):189–93. doi: 10.1038/13815. [DOI] [PubMed] [Google Scholar]
- 6.Beraud-Dufour S, et al. Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. Febs J. 2007;274(21):5518–32. doi: 10.1111/j.1742-4658.2007.06068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Francalanci F, et al. Structural and functional differences between KRIT1A and KRIT1B isoforms: a framework for understanding CCM pathogenesis. Exp Cell Res. 2009;315(2):285–303. doi: 10.1016/j.yexcr.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Liquori CL, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73(6):1459–64. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denier C, et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004;74(2):326–37. doi: 10.1086/381718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uhlik MT, et al. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol. 2003;5(12):1104–10. doi: 10.1038/ncb1071. [DOI] [PubMed] [Google Scholar]
- 11.Bergametti F, et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76(1):42–51. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busch CR, Heath DD, Hubberstey A. Sensitive genetic biomarkers for determining apoptosis in the brown bullhead (Ameiurus nebulosus) Gene. 2004;329:1–10. doi: 10.1016/j.gene.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, et al. Apoptotic Functions of PDCD10/CCM3, the Gene Mutated in Cerebral Cavernous Malformation 3. Stroke. 2009 doi: 10.1161/STROKEAHA.108.527135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma X, et al. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol Biol Cell. 2007;18(6):1965–78. doi: 10.1091/mbc.E06-07-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voss K, et al. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics. 2007;8(4):249–56. doi: 10.1007/s10048-007-0098-9. [DOI] [PubMed] [Google Scholar]
- 16.Zawistowski JS, et al. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 2005;14(17):2521–31. doi: 10.1093/hmg/ddi256. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, et al. Interaction between krit1 and malcavernin: implications for the pathogenesis of cerebral cavernous malformations. Neurosurgery. 2007;60(2):353–9. doi: 10.1227/01.NEU.0000249268.11074.83. discussion 359. [DOI] [PubMed] [Google Scholar]
- 18.Liquori CL, et al. Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am J Hum Genet. 2007;80(1):69–75. doi: 10.1086/510439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stahl S, et al. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum Mutat. 2008;29(5):709–17. doi: 10.1002/humu.20712. [DOI] [PubMed] [Google Scholar]
- 20.Hilder TL, et al. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res. 2007;6(11):4343–55. doi: 10.1021/pr0704276. [DOI] [PubMed] [Google Scholar]
- 21.Goudreault M, et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol Cell Proteomics. 2009;8(1):157–71. doi: 10.1074/mcp.M800266-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fournier HN, et al. Nuclear translocation of integrin cytoplasmic domain-associated protein 1 stimulates cellular proliferation. Mol Biol Cell. 2005;16(4):1859–71. doi: 10.1091/mbc.E04-08-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunel M, et al. KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc Natl Acad Sci U S A. 2002;99(16):10677–82. doi: 10.1073/pnas.122354499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulday G, et al. Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations. Dis Model Mech. 2009;2(3–4):168–77. doi: 10.1242/dmm.001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitehead KJ, et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med. 2009;15(2):177–84. doi: 10.1038/nm.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bos JL. Linking Rap to cell adhesion. Current Opinion in Cell Biology. 2005;17(2):123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Ponsioen B, et al. Direct spatial control of Epac1 by cAMP. Mol Cell Biol. 2009 doi: 10.1128/MCB.01630-08. MCB.01630–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glading A, et al. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol. 2007;179(2):247–54. doi: 10.1083/jcb.200705175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carmona G, et al. Role of the small GTPase Rap1 for integrin activity regulation in endothelial cells and angiogenesis. Blood. 2009;113(2):488–97. doi: 10.1182/blood-2008-02-138438. [DOI] [PubMed] [Google Scholar]
- 30.Chrzanowska-Wodnicka M, et al. Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice. Blood. 2008;111(5):2647–56. doi: 10.1182/blood-2007-08-109710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan J, et al. Rap1a is a key regulator of fibroblast growth factor 2-induced angiogenesis and together with Rap1b controls human endothelial cell functions. Mol Cell Biol. 2008;28(18):5803–10. doi: 10.1128/MCB.00393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gore AV, et al. Combinatorial interaction between CCM pathway genes precipitates hemorrhagic stroke. Dis Model Mech. 2008;1(4–5):275–81. doi: 10.1242/dmm.000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mably JD, et al. heart of glass regulates the concentric growth of the heart in zebrafish. Curr Biol. 2003;13(24):2138–47. doi: 10.1016/j.cub.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 34.Mably JD, et al. santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development. 2006;133(16):3139–46. doi: 10.1242/dev.02469. [DOI] [PubMed] [Google Scholar]
- 35.Kleaveland B, et al. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat Med. 2009;15(2):169–76. doi: 10.1038/nm.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lang T, Hansson GC, Samuelsson T. An inventory of mucin genes in the chicken genome shows that the mucin domain of Muc13 is encoded by multiple exons and that ovomucin is part of a locus of related gel-forming mucins. BMC Genomics. 2006;7:197. doi: 10.1186/1471-2164-7-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren J, et al. MUC1 Oncoprotein Functions in Activation of Fibroblast Growth Factor Receptor Signaling. Mol Cancer Res. 2006;4(11):873–883. doi: 10.1158/1541-7786.MCR-06-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gopal U, et al. Interaction of MUC1 with beta-catenin modulates the Wnt target Gene cyclinD1 in <I>H</I>. <I>pylori</I>-induced gastric cancer. Molecular Carcinogenesis. 2007;46(9):807–817. doi: 10.1002/mc.20311. [DOI] [PubMed] [Google Scholar]
- 39.Huang L, et al. MUC1 cytoplasmic domain coactivates Wnt target gene transcription and confers transformation. Cancer Biol Ther. 2003;2(6):702–6. [PubMed] [Google Scholar]
- 40.Singh PK, Hollingsworth MA. Cell surface-associated mucins in signal transduction. Trends Cell Biol. 2006;16(9):467–76. doi: 10.1016/j.tcb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 41.Liebner S, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183(3):409–17. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iden S, Collard JG. Crosstalk between small GTPases and polarity proteins in cell polarization. Nat Rev Mol Cell Biol. 2008;9(11):846–59. doi: 10.1038/nrm2521. [DOI] [PubMed] [Google Scholar]
- 43.Cuevas BD, Abell AN, Johnson GL. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26(22):3159–71. doi: 10.1038/sj.onc.1210409. [DOI] [PubMed] [Google Scholar]
- 44.Yang J, et al. Mekk3 is essential for early embryonic cardiovascular development. Nat Genet. 2000;24(3):309–13. doi: 10.1038/73550. [DOI] [PubMed] [Google Scholar]
- 45.Rousseau S, et al. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15(18):2169–77. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 46.Crose LE, et al. Cerebral cavernous malformation 2 protein promotes Smad ubiquitin regulatory factor 1-mediated RhoA degradation in endothelial cells. J Biol Chem. 2009 doi: 10.1074/jbc.C900009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozdamar B, et al. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307(5715):1603–9. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 48.Sahai E, et al. Smurf1 regulates tumor cell plasticity and motility through degradation of RhoA leading to localized inhibition of contractility. J Cell Biol. 2007;176(1):35–42. doi: 10.1083/jcb.200605135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 50.Kamei M, et al. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature. 2006;442(7101):453–6. doi: 10.1038/nature04923. [DOI] [PubMed] [Google Scholar]
- 51.Koh W, Mahan RD, Davis GE. Cdc42- and Rac1-mediated endothelial lumen formation requires Pak2, Pak4 and Par3, and PKC-dependent signaling. J Cell Sci. 2008;121(Pt 7):989–1001. doi: 10.1242/jcs.020693. [DOI] [PubMed] [Google Scholar]
- 52.Katrin V, et al. Functional analyses of human and zebrafish 18-amino acid in-frame deletion pave the way for domain mapping of the cerebral cavernous malformation 3 protein. Hum Mutat. 2009;30(6):1003–1011. doi: 10.1002/humu.20996. [DOI] [PubMed] [Google Scholar]
- 53.Preisinger C, et al. YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3{zeta} J Cell Biol. 2004;164(7):1009–1020. doi: 10.1083/jcb.200310061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katagiri K, Imamura M, Kinashi T. Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nat Immunol. 2006;7(9):919–28. doi: 10.1038/ni1374. [DOI] [PubMed] [Google Scholar]
- 55.ten Klooster JP, et al. Mst4 and Ezrin induce brush borders downstream of the Lkb1/Strad/Mo25 polarization complex. Dev Cell. 2009;16(4):551–62. doi: 10.1016/j.devcel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 56.Pujuguet P, et al. Ezrin Regulates E-Cadherin-dependent Adherens Junction Assembly through Rac1 Activation. Mol Biol Cell. 2003;14(5):2181–2191. doi: 10.1091/mbc.E02-07-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Londesborough A, et al. LKB1 in endothelial cells is required for angiogenesis and TGFbetamediated vascular smooth muscle cell recruitment. Development. 2008;135(13):2331–8. doi: 10.1242/dev.017038. [DOI] [PubMed] [Google Scholar]
- 58.Hogan BM, et al. ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum Mol Genet. 2008;17(16):2424–32. doi: 10.1093/hmg/ddn142. [DOI] [PubMed] [Google Scholar]
- 59.Zhang J, et al. Interaction between krit1 and icap1alpha infers perturbation of integrin beta1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum Mol Genet. 2001;10(25):2953–60. doi: 10.1093/hmg/10.25.2953. [DOI] [PubMed] [Google Scholar]
- 60.Zawistowski JS, et al. KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum Mol Genet. 2002;11(4):389–96. doi: 10.1093/hmg/11.4.389. [DOI] [PubMed] [Google Scholar]
- 61.Chang DD, et al. ICAP-1, a novel beta1 integrin cytoplasmic domain-associated protein, binds to a conserved and functionally important NPXY sequence motif of beta1 integrin. J Cell Biol. 1997;138(5):1149–57. doi: 10.1083/jcb.138.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bouvard D, et al. Disruption of focal adhesions by integrin cytoplasmic domain-associated protein-1 alpha. J Biol Chem. 2003;278(8):6567–74. doi: 10.1074/jbc.M211258200. [DOI] [PubMed] [Google Scholar]
- 63.Zhang XA, Hemler ME. Interaction of the integrin beta1 cytoplasmic domain with ICAP- 1 protein. J Biol Chem. 1999;274(1):11–9. doi: 10.1074/jbc.274.1.11. [DOI] [PubMed] [Google Scholar]
- 64.Millon-Fremillon A, et al. Cell adaptive response to extracellular matrix density is controlled by ICAP-1-dependent beta1-integrin affinity. J Cell Biol. 2008;180(2):427–41. doi: 10.1083/jcb.200707142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peter JMS, et al. Integrin cytoplasmic domain-associated protein-1 (ICAP-1) interacts with the ROCK-I kinase at the plasma membrane. J Cell Physiol. 2006;208(3):620–628. doi: 10.1002/jcp.20699. [DOI] [PubMed] [Google Scholar]
- 66.Bouvard D, et al. Defective osteoblast function in ICAP-1-deficient mice. Development. 2007;134(14):2615–25. doi: 10.1242/dev.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, et al. krit1 modulates beta1-integrin-mediated endothelial cell proliferation. Neurosurgery. 2008;63(3):571–8. doi: 10.1227/01.NEU.0000325255.30268.B0. discussion 578. [DOI] [PubMed] [Google Scholar]
- 68.Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10(1):21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 69.Leiss M, et al. The role of integrin binding sites in fibronectin matrix assembly in vivo. Current Opinion in Cell Biology. 2008;20(5):502–507. doi: 10.1016/j.ceb.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 70.Tanjore H, et al. Beta1 integrin expression on endothelial cells is required for angiogenesis but not for vasculogenesis. Dev Dyn. 2008;237(1):75–82. doi: 10.1002/dvdy.21385. [DOI] [PubMed] [Google Scholar]
- 71.Carlson TR, et al. Cell-autonomous requirement for {beta}1 integrin in endothelial cell adhesion, migration and survival during angiogenesis in mice. Development. 2008;135(12):2193–2202. doi: 10.1242/dev.016378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lei L, et al. Endothelial Expression of 1 Integrin Is Required for Embryonic Vascular Patterning and Postnatal Vascular Remodeling. Mol Cell Biol. 2008;28(2):794–802. doi: 10.1128/MCB.00443-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97(11):1093–107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 74.Iruela-Arispe ML, Davis GE. Cellular and Molecular Mechanisms of Vascular Lumen Formation. Developmental Cell. 2009;16(2):222–231. doi: 10.1016/j.devcel.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Astrof S, Hynes RO. Fibronectins in vascular morphogenesis. Angiogenesis. 2009 doi: 10.1007/s10456-009-9136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou X, et al. Fibronectin fibrillogenesis regulates three-dimensional neovessel formation. Genes Dev. 2008;22(9):1231–43. doi: 10.1101/gad.1643308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biology. 2005;24(6):389–399. doi: 10.1016/j.matbio.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 78.Deroanne CF, Lapiere CM, Nusgens BV. In vitro tubulogenesis of endothelial cells by relaxation of the coupling extracellular matrix-cytoskeleton. Cardiovasc Res. 2001;49(3):647–658. doi: 10.1016/s0008-6363(00)00233-9. [DOI] [PubMed] [Google Scholar]
- 79.Sottile J, et al. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am J Physiol Cell Physiol. 2007;293(6):C1934–46. doi: 10.1152/ajpcell.00130.2007. [DOI] [PubMed] [Google Scholar]
- 80.Lee S, et al. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol. 2005;169(4):681–91. doi: 10.1083/jcb.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bayless KJ, Davis GE. The Cdc42 and Rac1 GTPases are required for capillary lumen formation in three-dimensional extracellular matrices. J Cell Sci. 2002;115(Pt 6):1123–36. doi: 10.1242/jcs.115.6.1123. [DOI] [PubMed] [Google Scholar]
- 82.Garmy-Susini B, et al. Integrin alpha4beta1-VCAM-1-mediated adhesion between endothelial and mural cells is required for blood vessel maturation. J Clin Invest. 2005;115(6):1542–51. doi: 10.1172/JCI23445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abraham S, et al. Integrin beta1 subunit controls mural cell adhesion, spreading, and blood vessel wall stability. Circ Res. 2008;102(5):562–70. doi: 10.1161/CIRCRESAHA.107.167908. [DOI] [PubMed] [Google Scholar]
- 84.Grazioli A, et al. Defective blood vessel development and pericyte/pvSMC distribution in alpha 4 integrin-deficient mouse embryos. Dev Biol. 2006;293(1):165–77. doi: 10.1016/j.ydbio.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 85.Yu LG, et al. Galectin-3 interaction with Thomsen-Friedenreich disaccharide on cancerassociated MUC1 causes increased cancer cell endothelial adhesion. J Biol Chem. 2007;282(1):773–81. doi: 10.1074/jbc.M606862200. [DOI] [PubMed] [Google Scholar]
- 86.Friedrichs J, et al. Galectin-3 regulates integrin alpha2beta1-mediated adhesion to collagen-I and -IV. J Biol Chem. 2008;283(47):32264–72. doi: 10.1074/jbc.M803634200. [DOI] [PubMed] [Google Scholar]
- 87.Cohen M, et al. Dynamic study of the transition from hyaluronan- to integrin-mediated adhesion in chondrocytes. EMBO J. 2006;25(2):302–11. doi: 10.1038/sj.emboj.7600960. [DOI] [PMC free article] [PubMed] [Google Scholar]