

Abstract
Membrane receptor-activated signal transduction pathways are integral to cellular functions and disease mechanisms in humans. Identification of the full set of proteins interacting with membrane receptors by high throughput experimental means is difficult because methods to directly identify protein interactions are largely not applicable to membrane proteins. Unlike prior approaches that attempted to predict the global human interactome we used a computational strategy that only focused on discovering the interacting partners of human membrane receptors leading to improved results for these proteins. We predict specific interactions based on statistical integration of biological data containing highly informative direct and indirect evidences together with feedback from experts. The predicted membrane receptor interactome provides a system-wide view, and generates new biological hypotheses regarding interactions between membrane receptors and other proteins. We have experimentally validated a number of these interactions. The results suggest that a framework of systematically integrating computational predictions, global analyses, biological experimentation and expert feedback is a feasible strategy to study the human membrane receptor interactome.
Keywords: data integration, membrane proteins, protein-protein interaction network, receptor interactome, receptor crosstalk, signal transduction
1 Introduction
Membrane proteins are encoded by more than 25% of the genes in typical genomes and include structural proteins, channels and receptors [1]. Receptors in particular are attractive drug targets because they mediate the communication between the cell and its environment. There are two types of membrane receptors (Figure S6.1). Type I receptors are a broad group of diverse families of membrane receptors that directly or indirectly activate enzymatic activity, such as tyrosine kinase activity. Type II receptors refer to the large G protein coupled receptor (GPCR) family, to which 50% of current drugs are targeted [2]. A survey of the human genome has identified approximately 1000 membrane receptors equally divided between the two types [3]. Signaling mechanisms initiated by membrane receptors are complex and involve numerous proteins. For example, well over a hundred proteins have been shown to bind directly to the Epidermal Growth Factor Receptor (EGFR) [4–6]. Full receptor signalling pathways can include hundreds of proteins and on the order of a thousand interactions between them, as revealed in high-throughput experiments carried out for the TGF-β pathway [7, 8] and TNF-α pathway [9]. Furthermore, different receptor pathways cross-talk with each other. For example, binding of ligands to certain GPCRs can initiate transactivation of the EGFR [10–13]. Therefore multi-targeting several receptors can be more successful than targeting single receptors for treating complex diseases. Thus, in cancers where the EGFR is often overexpressed, significant effort is focused on identifying new targets that mediate or prevent signaling pathway crosstalk [14]. To fully understand signaling pathways and the crosstalk between them would require identification of the repertoire of all proteins that interact with membrane receptors (referred to as “the membrane receptor interactome” throughout this paper) and it is expected that such understanding would provide a useful resource in the study of complex diseases [15]. Protein interaction maps are also the cornerstones of phenome-interactome networks in human diseases [16].
The membrane receptor interactome is a subset of the human interactome and could therefore in principle be derived from large-scale human protein-protein interaction mapping studies. However, identification of a comprehensive human protein interactome, which is estimated to contain between 130,000 and 650,000 pairs [4, 17, 18], is largely out of reach despite phenomenal efforts [12, 19, 20]. The sum of all interactions for which there is some experimental evidence available is on the order of 30,000–40,000 [21], suggesting that at most 25% and perhaps as little as 5% of the interactions are known to date. Less than 10% of the known interactions involve membrane receptors. Data from small scale experiments identified approximately 2500 pairs of interacting proteins, where at least one of the proteins in the interacting pair is a receptor [4]. The high-throughput yeast two hybrid (Y2H) method is not well suited for identifying membrane protein interactions. This is because the interaction has to occur in the nucleus, a compartment inaccessible to proteins that normally reside in the plasma membrane. Alternative methods have been designed, for example the split ubiquitin system in which a transcription factor is only released when an interaction has taken place at the plasma membrane itself. While this approach has been used to identify membrane protein interactions in yeast [22], there are 10 times less receptors in yeast as compared to human cells. Thus, the yeast dataset only contains 12 proteins with homology to human membrane receptors (engaged in 47 interactions). Direct applications of methods to specifically target membrane protein interactions in human cells have not yet been reported. In principle, mass spectrometry-based approaches [12], or the luciferase reporter assay LUMIER [7] can identify membrane receptor interactions, but both approaches involve over-expression of proteins and an affinity-chromatography step, both of which can be non-trivial for membrane proteins due to the experimental difficulties arising from the need to maintain a hydrophobic environment for structural integrity of membrane proteins. Consequently, while the two available large-scale Y2H datasets contain no membrane receptors at all [19, 20], these interactions are also underrepresented in the human mass spectrometry and LUMIER based protein interaction screens. Using mass spectrometry, only 136 pairwise interactions involving 27 membrane receptors were identified, out of which the vast majority involves only 2 of the membrane receptors used as baits [12]. In the LUMIER screen of the TGF-β pathway, only 4 membrane receptor interactions out of 947 total interactions were reported [7]. Thus, direct experimental identification methods that will also work well for membrane receptors still require further developments.
In addition to direct experimental methods, computational approaches have proven useful in cataloguing the human protein interactome in a variety of ways [23]. These include probabilistic integration of several datasets [24, 25], learning from orthologous proteins in other model organisms [7, 26–28], text-mining algorithms applied to Medline abstracts [28] and quality assessments of existing interactome maps [29]. It has been clearly established that using direct and indirect data together as features in a supervised learning framework improves the success in predicting yeast protein interactions when compared to direct data alone [30–32]. Recent analysis of predicted interactions demonstrated that these approaches can be successfully applied to human data [21, 27, 33]. However, all of these studies have focused on the general protein interaction prediction task, covering the entire human proteome. This may in part contribute to disagreements between the computational predictions and the currently available high throughput results [32, 33].
In contrast to the general proteome approach, in this paper, we focused specifically on predicting membrane receptor interactions. An overview of the strategy taken here to identify the membrane receptor interactome is provided in Figure 1. First we extract features from diverse biological data sources, including sequence, structure, function and genomic information. We predict specific interactions involving receptors using the random forest classifier applied to the binary classification task of whether two proteins interact or not. Biological feedback is used to optimize feature extraction procedures. The predicted interactions for all human membrane receptors make up the human membrane receptor interactome. The predictions lend themselves to designing experimentally testable biological hypotheses. This could include for example global level properties of the network such as which proteins may serve as receptor hubs, or specific hypotheses on which pairs of proteins may interact.
Figure 1. Illustration of our combined computational-experimental approach to investigate the human membrane receptor interactome.

Step 1. Feature Extraction. Features were collected from diverse data sources, including sequence information, gene expression, functional annotation, tissue location, homologous interactions, and domain based association evidence. Step 2. Prediction for all receptors. Evidence was integrated using a random forest classifier for protein-protein interaction prediction. The random forest is an ensemble classifier and uses a collection of decision trees to model the relationship between multiple evidence and protein interactions. Step 3. Receptor interactome identification. Receptor interactome was defined as described in ‘Methods’. Visualizations were done with Cytoscape [54]. Nodes are drawn in different colors, with green representing type I receptors, blue for GPCR, pink for ligands and red for other human gene products. Step 4. Interaction validations. Specific pairs with high likelihood of interaction based on random forest score were validated experimentally.
2 Methods
Evidence Integration with a Random Forest Classifier
Combining evidence from many different sources as features in a supervised learning framework has proven a successful strategy in predicting protein interactions in yeast [24, 30, 31, 34] and in human [24, 25]. Here, we employ the random forest binary classification approach [35] for integrating multiple data sets to predict interactions for human membrane receptors de novo. In this approach, the evidence sources for interactions are treated as features describing examples of known positives and negatives (“gold-standards”). The derived feature distribution is used to yield predictions for unknown examples with a statistical reliability.
Features
Feature attributes for each protein pair are extracted from data sets that may be related to interactions. These include sequence information, gene expression, functional annotation, tissue location, homologous interactions, and domain based association evidence (Figure 1, Step 1, and Figure 2A). There are many possible ways to encode evidence sources into feature attributes and it is an important factor for the reliability of the computational predictions [36]. For instance, detailed encoding of available features leads to 130 attributes for each pair. While the overall performance of a prediction system based on these features was reasonable (data not shown), biological insight was used to improve the predictions (Figure 1, back arrow). For example, in manual inspection of specific predictions, it appeared that functional similarity dominated the selected binding partners and we therefore reduced the number of feature items derived from Gene Ontology (GO) [37] functional similarity. Biological feedback was also used in optimizing the feature similarity measures. We finally settled on 27 feature attributes for each protein pair. These are listed in Figure 2A. The first three features are similarity measures derived from GO. The fourth attribute describes two proteins’ tissue positions. The next sixteen features are Pearson’s correlation between two genes in sixteen gene expression sets. The next four attributes describe how likely theses two proteins interact in other species. The last feature is the interaction probability from the domain point of view. A summary of how each feature was encoded is provided in details in Supplement S1.
Figure 2. Statistical comparison of performance in the human membrane receptor protein interaction prediction task.

A. Diverse biological data sets are collected and used as evidence to predict PPIs for receptors via evidence integration with the random forest (RF) classifier. 27 feature attributes for each protein pair are extracted from data sets that may be related to interactions. Each feature’s name and index number are listed in the upper-left table. To generate the RF, we select for each tree a bootstrap sample of the training data. Next, for every node in these trees a random subset of the attributes is chosen and the attribute achieving the best division is selected. Once model trees are grown, protein pairs are propagated down and the ‘votes’ from all trees are used to compute interaction scores. B. The RF classifier was compared to three other classifiers: Support Vector Machine, Naïve Bayes, and Logistic Regression. Precision (Prediction Accuracy) refers to the fraction of predictions that are known to be correct. Recall (Sensitivity) refers to the fraction of known interactions that are correctly predicted. The prediction accuracy versus sensitivity curve is then plotted for different cutoffs on the predicted score cutoffs. At recall rates of 10%~20% RF outperforms all other methods. A global analysis based on AUC scores also indicates that RF outperforms the other methods (see Figure S2.1). C. Evaluation of two different negative gold standards. The negative pairs were either sampled entirely at random or using random receptor-protein pairs with similar functions. D. Performance comparison between receptor interactome identification task and general human PPI prediction task. Classifiers trained only using membrane receptors outperform those trained on the global interaction data. E. Performance comparison between using the full feature attributes and using just the top 10 ranked features based on the Gini criterion. RF classifier is used for subfigures C-E.
Gold standard
The gold-standard data set to train the classifier should ideally be (a) generated independently from the evidence sources, (b) sufficiently large for reliable statistics, and (c) free of systematic bias [30]. The gold-standard positives were extracted from the Human Protein Reference Database (HPRD) [4]. This data set contained 2522 high-confidence pair-wise protein interactions, where at least one of the interacting proteins is a receptor. The interactions downloaded from the HPRD included only those that were detected by low-throughput approaches revealing physical binding, excluding high-throughput derived results. A list of 904 human receptor proteins from the Human Plasma Membrane Receptome (HPMR) database [3] was used to filter the HPRD for these positive interactions. There exists the concern that homologous protein pairs might cause bias in the training, as well as over-estimation of the performance of the method. We investigated this issue and found no evidence to substantiate this concern (see Supplementary S2).
Identification of gold-standard negatives is less straight-forward. Because of the nature of laboratory experiments, it is very difficult to prove that two proteins do not interact and a negative dataset is therefore not available. One strategy that has been proposed is to sample interactions from disparate localization [30], decreasing the chance that the negative dataset is contaminated with positive interactions. We have tested this idea and determined that such a strategy reduced the accuracy of our classifier (see Supplement S2). This observation may be attributable to the fact that this strategy introduces bias into the dataset [25, 34]. Another strategy is to randomly sample pairs for which an interaction is not reported in the HPRD (recommended in [18]). Considering the small fraction of interacting pairs in the total set of potential protein pairs (estimated to be less than 0.1%), the error for contamination is expected to be very low [36]. We thus used a random set of receptor-protein pairs excluding all known HPRD pairs as our negative training set. The drawback of the random negative set is that random proteins with different biological functions may be very easily distinguished from interacting proteins leading to a biased classifier. This may hurt the prediction performance because the classifier cannot learn the fine distinctions between interacting protein pairs and functionally related non-interacting pairs. We therefore constructed another negative set consisting of random pairs with similar molecular functions that are not in the HPRD. We compared the success of predicting membrane receptor interactions using these two negative gold-standard datasets (Results).
Random forest classifier
Classification algorithms use the features and the gold standard labels to learn differences between positive (‘interaction’) and negative (‘noninteraction’) examples of protein pairs (Figure 1, Step 2 and Figure 2A). We chose the random forest (RF) classifier [35] based on its success in yeast protein interaction prediction [36] and a performance comparison was carried out for human membrane receptors (Results). This classifier utilizes a collection of decision trees to determine if a protein pair interacts or not (Figure 2A). Within each decision tree, the non-leaf nodes are labeled with feature attributes, the arcs out of a node are labeled with possible value ranges of the attribute, and the leaves of the tree are labeled with classification decisions (interacting or not). In order to classify a protein pair, the pair is propagated down each tree based on its feature values and a decision is made based on the terminal node that is reached. Then the RF makes the final prediction based on the majority vote over all the trees in the model. Among the many possible machine learning approaches that could be applied, RF is particularly suitable to address the difficulties of this task because it can easily combine different types of data (including discrete, continuous and categorical data), does not assume feature independence (many biological datasets are expected to be correlated), and is particularly robust against noise and missing values.
Experimental procedures
We experimentally validated predicted interactions for the EGFR. We chose this receptor because it is one of the best known, and with more than 100 confirmed binding partners, it is the receptor with the currently largest set of known interactions for any receptor in the HPRD [27]. Three novel interactions, EGFR-Hck, EGFR-dynamin-2 and EGFR-TGF-β1 were chosen for validation. The corresponding experimental procedures are described here, while most sources of reagents and cell lines used are provided in Supplement S3.
Interactions studies with the EGFR were carried out with full-length EGFR as well as truncated versions lacking the extracellular and transmembrane domains. In one construct, the entire cytoplasmic domain including the juxtamembrane domain was retained (referred to as C+J-EGFR) and in a second construct, the juxtamembrane domain was deleted (referred to as C-EGFR). For details on the construction of the respective plasmids, refer to Supplement S3. Other details including the sources of chemicals, antibodies and other purified proteins, and cell lines are described in Supplement S3.
EGFR-Hck pair
To study the interaction of EGFR with Hck, COS-1 cells were transiently transfected with a plasmid carrying the gene corresponding to C-EGFR or C+J-EGFR, tagged with a 9-mer sequence corresponding to the 1D4 antibody epitope to facilitate immunoprecipitation. The soluble COS-1 cell extract was incubated with 1D4-sepharose beads and washed. For detection of the Hck interaction, Sf9 cells infected with a baculovirus carrying Histidine-tagged Hck were extracted and added to the 1D4-EGFR beads. The beads were washed with large volumes of buffer. In control experiments, 1D4-beads were incubated with cell extracts of untransfected COS-1 cells. C-EGFR or C+J-EGFR was eluted from the 1D4-columns with nonapeptide and the samples were probed with the 1D4 antibody for EGFR and with the anti-Histidine tag antibody for Hck.
For co-immunoprecipitation experiments in cancer cell lines, UM-22A and 1483 cells were plated at a density of 2 × 105 cells/ml in 10 cm plates and incubated at 37°C for 48 hours in regular growth media (DMEM plus 10% fetal bovine serum). Lysates were obtained and prepared for an immunoprecipitation assay. 500μg of lysate was mixed with 2 μg of Hck antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and incubated overnight at 4°C with rotation. 40 μl of Protein G agarose beads were added to the lysate/antibody solution and incubated for 2 hours at 4°C with rotation. Immunoprecipitates were resolved on an 8% SDS PAGE gel, transferred to a nitrocellulose membrane and probed with an anti-EGFR antibody (BD Transduction, CA). The membrane was developed with Luminol reagent by autoradiography. The membrane was stripped in Restore Western Blot Stripping Buffer and probed with an anti-Hck antibody.
EGFR-Dynamin 2 pair
For detection of the interaction between EGFR and the large GTPase dynamin-2, GFP-tagged dynamin-2 was expressed by transient transfection in COS-1 cells. The washed C-EGFR or C+J-EGFR and control beads were incubated with soluble GFP-dynamin-2 cell extract, washed and eluted. An anti-GFP antibody Western blot was used to probe for GFP-dynamin-2 in the EGFR elution fractions.
EGFR-TGF-β1 pair
To study the interaction of EGFR with TGF-β1, full-length EGFR expressing Hep3B human hepatoma cell lysate proteins were incubated with TGF-β1. EGFR and TGF-β1 were immunoprecipitated with anti-EGFR antibody and anti-TGF-β1 antibody, respectively, and probed on Western blots with anti-TGF-β1 and anti- EGFR antibodies. In a separate experiment, purified GST-EGFR and TGF-β1 proteins were mixed and incubated. The complex was immunoprecipitated using an anti-GST antibody and protein-A-sepharose. TGF-β1 protein, which was co-immunoprecipitated with GST-EGFR, was detected on Western blots of the immunoprecipitate with anti-TGF-β1 antibody.
3 Results and Discussion
Random Forest Classifier Performance
Relying on the gold standard data, we statistically evaluated the performance of our approach using “Prediction accuracy vs. Sensitivity” curves and the results are shown in Figure 2B-E. Here prediction accuracy (or Precision) refers to the fraction of interacting pairs predicted by the classifier that are truly interacting. Sensitivity (or Recall) measures how many of the known pairs of interacting proteins have been identified by the learning model. Both measures can take values from 0.0 to 1.0 (the larger the better). Often, there is an inverse relationship between Prediction accuracy and Sensitivity, where it is possible to increase one at the cost of reducing the other. A “Prediction accuracy vs. Sensitivity” curve always goes from the top left to the bottom right of the graph. For an ideal system, the graph drops steeply on the right side. Examining the entire curve is very informative, but since in PPI networks the number of non-interacting pairs far exceeds the number of interacting pairs, we are interested in the performance of our models under conditions when the “Sensitivity” is small (which means the false positive (FP) rate is very low). Note that even FP=0.1 is not meaningful for this task, see explanations in Supplementary S1 for details. First, we compared the RF classifier to several other popular classification algorithms, Naïve Bayes [24, 30], Logistic Regression [29] and Support Vector Machine classifiers [38], which have been applied to PPI prediction tasks in similar cases before. As can be seen in Figure 2B, the RF performs best for this task, since its curve is mostly above others when the “sensitivity” is small (for instance less than 0.5). This conclusion is supported by Figure S2.1, where the random forest method performs best on partial AUC score criteria, where partial AUC score summarizes the above performance curve using one number for a certain “sensitivity” rate (see details in Supplement S1). In addition, we could see that when the rate of “sensitivity-recall” is extremely small (smaller than 0.05), RF achieves lower precisions compared to Naïve Bayes (averaged over 12 repeats). This means that for a test set with about 80 positive interactions, when RF finds four true positive interactions by a score cutoff, it predicts more interaction examples as compared to Naïve Bayes. We argue that this might not mean that the RF performs worse than Naïve Bayes since the negative examples in the test corpus are noisy random instances and RF might be able to identify those highly likely interacting pairs that have not been covered by the “gold standard” labels.
Next, we investigated the two settings of gold standard negatives: the random strategy (random pairs not in HPRD) and the co-functional random strategy (co-functional random pairs not present in HPRD). Figure 2C shows the result of this comparison. We found that the first negative set (random pairs) led to better results compared to the more constrained negative set (requiring different functional categories). We have thus used the fully random negative set for the remainder of our analysis.
An alternative way to predict partners of human membrane receptors is to predict the general human interactome first, and then extract membrane receptor interactions from this general set. This is a viable option since the features used in the training of our approach are not membrane specific. However, as Figure 2D clearly shows, the precision and recall of the receptor interactome is higher when training on the receptor-only gold standard as opposed to training on the entire human gold standard. This is probably due to the ability of the classifier to highlight features that are uniquely important for classifying membrane receptors interactions. The performance was better for all of the different classifiers used, when they were trained on receptors only (Supplement S2). In both scenarios, the RF classifier performed best (Supplement S2). Thus, focusing on a subset (here, membrane receptors) of the human interactome allows us to generate better predictions. This is particularly encouraging given the fact that the membrane receptors are a group of proteins that are experimentally difficult to study.
Finally, we investigated which features are informative for the membrane receptor interaction prediction task. Figure 2E shows the performance when only the top ten most discriminative features were used to train and test the RF. The top-ranked features were selected using the Gini criterion (see Supplement S1), which has been proven useful in investigating feature importance for protein interaction predictions in yeast [36]. Among the top ten Gini ranked features, five of them are similarities of gene expressions. The other features ranked in the list of the top ten most informative features include sequence alignment score, domain-domain interaction features, the homology derived interactions from yeast, co-tissue positions and the co-biological-process feature from GO. While the top ten features alone achieve reasonable predictions (for details, see Supplement S2), the performance is still significantly less than when using all features, suggesting that despite overlap, all features used contain highly complementary information that can be successfully used for the classification.
Experimental Studies
To demonstrate how the predictions can be used to design experiments, we conducted validation experiments for the EGFR. The predicted EGFR interactome is shown in Figure 3A. Many previously known as well as novel interactions are highly ranked and it depends on the interest of the investigator how to choose amongst them. We chose three novel interactions, EGFR-Hck, EGFR-dynamin-2 and EGFR-TGF-β1, all highly ranked with scores 2.7, 3.1 and 3.4, respectively.
Figure 3. EGFR related interactions.

A. EGFR related interaction network in the predicted interactome. EGFR is colored yellow, other type I receptors are green, ligands are pink, and other soluble proteins are red. Ligand assignments were extracted from GO [37]. B. Experimental validation of the predicted interaction between EGFR and dynamin-2. EGFR (either the entire cytoplasmic domain, C+J-EGFR (see Methods), or the cytoplasmic domain lacking the juxtamembrane domain (C-EGFR, see Methods) were bound to an antibody column and dynamin-2 was bound and co-eluted with epitope peptide. Only the results for C+J-EGFR are shown in the figure as the results with C-EGFR were very similar. The panel is an anti-GFP blot to detect dynamin-2. The lanes are labeled as follows: S, standard (dynamin-2 transfected COS-1 cell lysate); FT, flow through; W, last wash fraction; E1-E5, elution fractions 1 through 5. C. Experimental validation of the predicted interaction between EGFR and Hck. Panel I. C-EGFR or C+J-EGFR was bound to an antibody column and Hck was bound and co-eluted with epitope peptide. Only the results for C-EGFR are shown in the figure, as C+J-EGFR gave very similar results. The panel represents an anti-histidine blot to detect Hck. E1-E6 denotes elution fractions 1 through 6. Panel II. EGFR and HCK also interact in 2 HNSCC cell line models. UM-22A and 1483 cells were plated in regular growth media for 48 hours and lysates were obtained. 500 μg of protein was immunoprecipitated with anti-Hck antibody, resolved by SDS-PAGE and transferred to nitrocellulose membrane. The membrane was probed with anti-EGFR antibody (left panel) and stripped and reprobed for Hck using an anti-Hck antibody (right panel). Panel III. Hck and EGFR interaction is not dependent on EGFR activation. UM-22A cells were serum starved for 72 hours and treated with EGF for 10 minutes. Lysates were collected and immunoprecipitated with 2μg of Hck, resolved by SDS-PAGE and immunoblotted with EGFR and Hck using specific antibodies. Panel IV. Downmodulation of Hck decreases survival of HNSCC. UM-22A cells were transiently transfected with Hck siRNA for 72 hours. The percentage cell survival was determined by MTT assay. D. Interaction of EGFR with TGF-β1. Panel I. Co-immunoprecipitation of GST-EGFR and TGF-β1. Lane 1, TGF-β1 only. Lane 2, no GST-EGFR. Lanes 2, 3, 4, 5, 6 represent immunoprecipitation experiments when GST-EGFR and TGF-β1 were incubated at ratios 0:1, 1:50, 1:25, 1:5 and 1:1, respectively. Panels II, III. Signal transduction results. Western Blots of PCI-37A cells treated with no ligand i.e. NoTx (“no treatment”, lane 1 in Panel II and III), EGF, TGF-β1 (lanes 2 and 3 in Panel II and III) and cells preincubated with C225 (lanes 4–6 in Panel II) and ALK4 inhibitor (lanes 4–6 in Panel III). The EGF concentration was 10ng/ml and the TGF-β1 concentration was 5pM. The blots were detected with anti-MAPK and anti-phospho-MAPK antibodies. The phospho-MAPK and MAPK antibodies recognize the p44/42 MAPK (Erk1/2) proteins, so there are two bands at 44kd and 42kd, respectively. As a control, we have included β-tubulin for each blot as an indicator for equal loading.
EGFR-Dynamin 2 pair
Dynamin-2 is a protein regulating vesicle formation on lipid membranes. A functional link between EGFR and dynamin-2 was already known because catalytically inactive dynamin-2 is no longer able to internalize the EGFR [39, 40] but it was not known that the two proteins physically interact. To validate this prediction, we carried out co-immunoprecipitation experiments with proteins expressed in COS-1 cells, which confirmed that the EGFR cytoplasmic domain interacts with dynamin-2 (Figure 3B). This suggests that the EGFR might be mechanistically involved in its internalization at the molecular level and not only at the regulatory level.
EGFR-Hck pair
The Src-homology kinase Hck is a signaling protein with a role in HIV-1 pathogenesis [41] and oncogenesis [42]. While several other Src-homology kinases are known to interact with receptors, via their SH2 domains, a previous large screen of the binding of SH2 domains in the human genome has revealed that the presence of such a domain in itself is not a proof that proteins containing these domains interact with the EGFR [5]. The experiments carried out to validate the prediction that Hck and the EGFR interact, are shown in Figure 3C. First, we carried out co-purification experiments with the cytoplasmic domain of EGFR over-expressed in COS-1 cells, and Hck over-expressed in insect cells. The experiment provides evidence that the EGFR cytoplasmic domain interacts with Hck (Figure 3C, Panel I).
EGFR is a direct target for treatment of head and neck cancer with the FDA approved drug cetuximab. Inhibitions of cancer growth in head and neck cancer cells, which over-express the EGFR, are augmented by co-inhibition of src family kinases (Grandis laboratory unpublished observations). Xi et al also reported that phosphorylation of src family kinases requires EGFR kinase activity [43]. Src is known to bind to different phosphotyrosine sites on the intracellular kinase domain of EGFR, further mediating the activation of EGFR and its downstream signaling events [44]. To demonstrate whether Hck, as a member of the Src family kinases, interacts with EGFR in the head and neck cancer cells, a co-immunoprecipitation assay was performed. Figure 3C, Panel II illustrates that under normal growth conditions EGFR interacts with Hck in both UM-22A and 1483 HNSCC cell lines. EGF treatment does not increase the intensity of the co-immunoprecipitated band (Figuer 3C, Panel III). Interestingly, the src family kinase inhibitor, dasatinib, also has high affinity for Hck [45] suggesting that in cancer cell lines Hck may interact with EGFR and contribute to tumor progression pathways. To test this hypothesis, HNSCC cells were transfected with Hck siRNA (Supplement 3, Figure S3.3). There was a 20% decrease in proliferation in transfected cells (Figure 3C, Panel IV). However, there was no effect on HNSCC invasive ability (data not shown). Thus, the contribution of Hck interaction with the EGFR appears to be less important than that of other src-family kinases, such as c-Src.
EGFR-TGF-β1 pair
Transforming growth factor beta 1 (TGF-β1) is an extracellular ligand that is functionally linked to the EGFR because TGF-β1 binding to its normal receptor, the TGF receptor, is believed to transactivate EGFR [43]. While we did not observe co-immunoprecipitation of TGF-β1 with a full-length EGFR expressing Hep3B human hepatoma cell lysate (data not shown), it was found to co-immunoprecipitate with purified GST-EGFR (Figure 3D, Panel I), suggesting the possibility of a weak physical interaction between the two proteins. This indicates that activation may not only be via transactivation but also via direct binding and activation of the EGFR by TGF-β1. We were able to confirm the functional interaction between EGFR and TGF-β1 by measuring the ligand-induced phosphorylation level increases in MAPK. PCI-37A squamous cell carcinoma of the head and neck cells were incubated with EGF and TGF-β1 and expression levels of MAPK and phospho-MAPK were detected on a western blot (Figure 3D, Panels II and III). TGF-β1 is able to stimulate MAPK to similar levels as EGF (Figure 3D, Panel II), consistent with a strong functional link between the two pathways [43]. However, it appears that this may not be mediated by direct interaction between the EGFR and TGF-β1 because the TGF-β1 receptor inhibitor Alk4 abrogated TGF-β1 activation of MAPK (Figure 3D, Panel III).
A database of human membrane receptor interactions
While experimental researchers may investigate individual membrane receptors in detail and choose the random forest cut-off score according to their preferences, it is also of interest to generate a global set of interactions, especially when comparing predictions to other existing databases.
To generate the database we trained the final RF classifier using a positive set containing all known receptor interaction pairs (2522). The negative training set contains 250,000 random pairs that do not have overlap with any of the HPRD pairs. T-test was used to measure the statistical significance of predicted scores, based on training with multiple random negative sets (see Supplementary S2). To estimate what RF cut-off we should use to generate a reliable membrane receptor interactome network graph, we investigated the distribution of predicted scores for known HPRD pairs and the remaining random receptor-protein pairs in testing sets. As seen in Figure 4, a cut-off of 2.0 is stringent in the sense that it is well able to separate the two classes. This cut-off resulted in a recall range of around 20% in the performance evaluation experiments (more details in Supplementary S2). We therefore generated the membrane receptor interactome using this cut-off†. The derived network contains 9100 edges, and includes 559 membrane receptors and 1750 non-receptors (Figure 5). Figure 5 shows the graphical overview of the predicted interactions. Receptors are colored as green (type I) or blue (GPCRs) nodes. Selected subnetworks in this interactome were visualized in Supplement S4 as well as their matched subgraphs in HPRD. Figure 3A also draws the interaction subgraph connected to EGFR in this predicted interactome.
Figure 4. Histogram of the distributions of predicted scores.
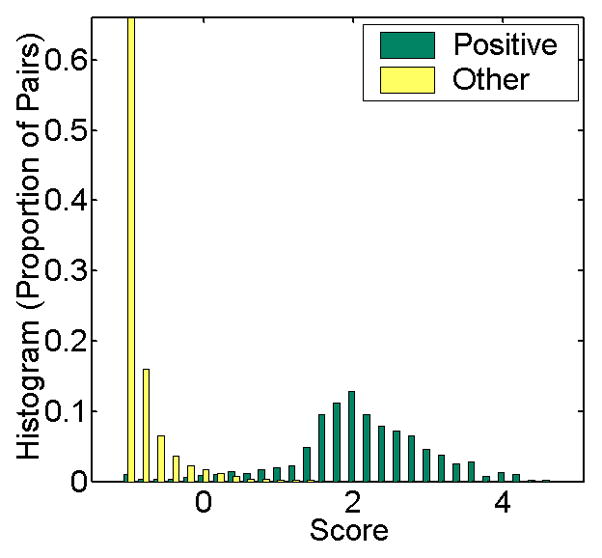
The figure presents a histogram of the distributions of predicted scores for known and random receptor-protein pairs. Yellow bars are for positive pairs (labeled in HPRD) and green bars represent the remaining (random) pairs. Y-axis is the fraction of examples within each class receiving the score. We found that a cut-off of 2.0 is stringent and separates the two classes well. Note however that since the set of non-interacting pairs is much larger than the set of interacting pairs this score will still lead to many false positives (or, a precision of close to 20%) as indicated in Figure 2.
Figure 5. The predicted human membrane receptor interactome.

Graphical overview of the entire network of interactions. Receptors in the GPCR family are colored blue, type I receptors are green (except EGFR, which is highlighted in yellow), ligands are pink, and other soluble proteins are red. Ligand assignments were extracted from GO [37]. Visualizations were performed using Cytoscape [54].
Of the 9100 edges, each representing a pairwise interaction, 1462 edges are already in the HPRD [4]. In addition, 257 edges overlap with those determined by a previous probabilistic integration effort which targeted the general “human interactome” [24]. 220 edges are also included in the STRING database [32]. Three additional edges were validated by the recent TAP-MS screen of human proteins [12], and 2 out of 4 receptor interactions identified in the LUMIER system [7] are members of our receptor interactome. We also considered the homology of the human receptors to membrane proteins studied in the yeast membrane protein interactome screen [22]. Of the 12 human homologs for these membrane receptors only two are type II receptors (STE3 and GRP1) and the majority of the remaining proteins are putative but not confirmed type I receptors. No overlap with our human membrane receptor interactome was observed. In contrast, we observed a higher overlap with a human membrane receptor specific experimental study of the four ERBB receptors [5]: 50 of the 181 interactions discovered in that study are also included in our predicted interaction set. These results and a detailed comparison of our RF score distributions in the receptor-related interactions included in these datasets are provided in Supplementary S2. In addition, all receptor pairs in our predicted interactome with the RF cutoff 1.0 are shared in Supplementary S6. Both their RF scores and the related p-values are included in this shared EXCEL sheet.
Webserver and Database Interface
To enable biomedical researchers to benefit from our human membrane receptor interactome, we have implemented a web server “HMRI” (http://flan.blm.cs.cmu.edu/HMRI/). An interactive interface allows users the retrieval of interactions. Researchers interested in specific membrane receptors can enter individual or sets of proteins and retrieve their rank-ordered interactions, along with the evidence supporting these predictions. A visual representation of the network graph of queried interactions is also provided. Supplement S5 describes the interface in more details.
4 Discussion
It is becoming increasingly clear that blockage of one signaling pathway as a disease treatment strategy may not be sufficient in many cases [11]. Accelerating the identification of communication points between pathways is therefore expected to have major impact for biomedical research. Signal transduction pathways often involve and in fact are frequently initiated by membrane receptors. It is therefore important to enhance our understanding of the scope and details of interactions involving this class of proteins.
In this paper, we described a multi-layered approach to predict and validate protein-protein interactions relating to human membrane receptors systematically. Due to the experimental challenges present for this type of proteins we relied on biological datasets providing indirect evidence about protein interaction relationships. Since each of these datasets only provides partial information we developed and applied a classification strategy to integrate evidence from different data sources for predictions of receptor-protein interactions.
Evaluation of the performance of our classification method showed that the precision and recall of interactions were best when using only receptor related protein pairs as the positive set and random pairs (not in HPRD) as the negative set, incorporating the full set of optimally encoded features. Under these conditions, the RF achieves the best accuracy when compared to Naïve Bayes, Logistive Regression and SVM classifiers. We believe the reason for the random forest (RF) classifier performing better than the other classifiers is because this classifier makes decisions based on relationships between features. While Naïve Bayes and regression classifiers assume that each feature is an independent measurement and produce a combined weighted vote from each of the features, decision trees (and RF which is based on them) follow paths down the tree, testing for correlations between different features. Thus, decision tree classifiers are much more appropriate when we expect features to be highly dependent as is the case in the current problem. We expect the RF to perform better than other decision tree classifiers, because the currently available direct and indirect protein interaction data is inherently noisy and contains many missing values. The randomization and ensemble strategies within the RF make it more robust to noise when compared to others.
The prediction accuracy of RF (the fraction of predictions that are known to be correct) is 20% at a sensitivity (the fraction of known interactions that are correctly predicted) of 16%. This performance is comparable to large-scale experimental PPI data sets in general [32, 33] and superior to the receptor related pairs extracted from previous general human interactomes, both predicted [24, 25] and experimentally determined [12]. A detailed comparison of our predictions with previous related data sets is provided in Supplement S2. To appreciate the utility of a 20% accuracy for experimentalists, consider that only 0.1% (1 in 1000) of random protein pairs are estimated to actually interact [17]. Thus, without any useful predictions biologists would need to test more than 1000 potential pairs for one true interaction. In contrast, when using our list we expect 1 in 5 experiments to lead to an interacting pair. In practice, the ratio of success can be even higher because expert biologists will utilize prior information and knowledge when designing experiments.
It has been suggested previously, that focusing on specific subnetworks may provide more reliable information [12, 46]. Here we show that focusing on predicting membrane receptors generates better predictions than selecting interactions related to membrane receptors from a general human interactome. We expect that many other protein families would equally be able to benefit from this approach, for example kinase or phosphate networks, or proteins involved in transcriptional regulation.
One goal of developing a method to predict receptor interactions reliably is to provide specific experimentally testable hypotheses. Given the expense and effort in conducting wet-lab experiments, these will likely be conducted on a small scale, involving one or few membrane receptors. To provide example scenarios, we chose the EGFR as a representative. We conducted pull-down experiments for interactions with Hck, dynamin-2 and TGF-β1. All three proteins could be co-eluted with the EGFR specifically. While pull-down experiments are not the ultimate proof for a direct physical interaction as the same result will also be obtained with complexes, where the interaction can be mediated by another protein, these results qualitatively support a linkage between the EGFR and these proteins. In the case of the TGF-β1 previous experiments had already established such a linkage [43], but in the case of Hck and dynamin-2 these are novel linkages. For example, while dynamin-2 is known to be required for EGFR endocytosis because of dynamin-2’s role in pinching off membrane vesicles, the stability of the interaction throughout the detergent solubilisation and immunoaffinity chromatography suggests that these two proteins are more directly linked. An inverted scenario is provided by the Hck-EGFR interaction. Here, it was neither known whether the two proteins interact nor if they are functionally linked. Given the importance of src family kinases for EGFR mediated cancer progression, we hypothesized based on the prediction and the experimental support from pull-down experiments that Hck may play a role also. However, anti-Hck siRNA knock-out experiments revealed that other src family kinases are probably better targets: while a 20% decrease in proliferation of cancer cells transfected with Hck siRNA was observed, there was no effect in a tumor invasion assay.
Discovery of a general relationship between interaction and biological function is an important, unresolved challenge. It is thus not trivial to address the question when is an interaction “real”. An obvious criterion is affinity, but it is increasingly becoming clear that even very weak and transient interactions with affinities in the μM to mM range can have biological relevance. For example, the functionally important but ultra-weak interaction between the adaptors PINCH-1 and Nck-2, results – if disrupted – in severe impairment of focal adhesion formation [47]. This interaction was undetectable by pull-down experiments [47], while it was observed in a yeast-2-hybrid screen [48] and by NMR [36]. The need for washing solid-support bound complexes extensively in pull-down experiments results in a bias of this assay to detect high-affinity, stable interactions. This dependence on the experimental setup may also be the reason why we did not observe a TGF-β1/EGFR interaction by co-immunoprecipitation in cell lysates, while we do confirm the interaction with purified components. This indicates that the affinity between these two proteins is probably low. Detection of weak interactions such as this or the PINCH-1/Nck-2 example on a proteome-wide scale has recently become possible by the revolutionary shift towards quantitative proteomics. Incorporation of isotope labels [49, 50] and other approaches [51] to differentiate between unspecific background binders and specific interactors reduces manipulation and time of the experiment, making it more likely to also detect weak interactions [52]. Therefore, these approaches promise to shift the current bias of PPI databases in containing stable interactions to a more realistic representation of the diverse types of interactions observed in vivo ranging in affinities over 12 orders of magnitude [53]. Correlating random forest score with affinities will require affinity information to be deposited together with the interaction. To date, most interactions in PPI databases are reported as binary values, without specification of affinity, and computational models focus on binary predictions accordingly. The RF score thus reflects our confidence in an interaction being “real” but does not directly relate to affinity.
In conclusion, we describe a computational method that uses a random forest classifier to integrate diverse direct and indirect features for predicting protein interactions involving human membrane receptors. Early computational methods for predicting human protein interactions have focused primarily on exploiting homology to other organisms [7, 26–28], but because of the uncertainty in choosing the best homolog in human for a protein found in fly, worm, mouse or yeast, this can lead to an overestimation of the true equivalent interactions in human [21]. Integrative approaches have therefore been developed [24, 25], but these included little expert feedback for feature extraction or computational predictions. An iterative integration of computational and experimental expertise is necessary to make the whole prediction system more robust and accurate. Furthermore, we found that an approach which focused on a specific protein family yields more reliable predictions. The predictions can be viewed as hypothesis-generating tools that should help researchers prioritize experiments for identifying interaction partners of specific membrane receptors. To illustrate this we presented a number of experimental studies exemplifying the different scenarios of biological hypotheses involving protein interactions that can be tested. Since we predict thousands of previously unknown interactions, these experiments serve to demonstrate the potential of our membrane receptor interactome in generating novel and experimentally testable hypotheses. To allow other biologists to utilize these predictions, we developed a web-interface that allows access to all of our predictions along with the evidence supporting them.
Several aspects of our interaction classification step could be further revised to improve the overall performance. For example, the feature sets employed currently are general. Membrane receptor specific evidence, such as structural clues given by the membrane topology of these proteins or membrane receptor family specific information such as the knowledge of G protein coupling specificity in the case of GPCR’s, might be able to better capture the properties of membrane receptor related interactions.
Supplementary Material
Acknowledgments
This work was supported in part by National Science Foundation Grants ITR 0225656, CAREER 0448453 and CAREER CC044917, National Institutes of Health Grants NLM108730, R01 CA098372 and AI060422 and the Sofya Kovalvskaya Award (to JKS) from the Humboldt Foundation.
Abbreviations
- PPI
protein-protein interaction
- EGFR
Epidermal Growth Factor Receptor
- GPCR
G protein coupled receptors
- FP
false positive
- RF
Random Forest
- GO
Gene Ontology
Footnotes
RF cut-off can be chosen less stringent depending on the task, e.g. when filtering potential interactions to be tested experimentally for specific membrane receptors.
Contributor Information
Yanjun Qi, Email: qyj@cs.cmu.edu.
Harpreet K. Dhiman, Email: dimpledhiman@yahoo.com.
Neil Bhola, Email: neb17@pitt.edu.
Ivan Budyak, Email: budyak@biochem.umass.edu.
Siddhartha Kar, Email: skar@pitt.edu.
David Man, Email: sdm14@pitt.edu.
Arpana Dutta, Email: ard36@pitt.edu.
Kalyan Tirupula, Email: kalyan@ccbb.pitt.edu.
Brian I. Carr, Email: Brian.Carr@kimmelcancercenter.org.
Jennifer Grandis, Email: grandisjr@upmc.edu.
Ziv Bar-Joseph, Email: zivbj@cs.cmu.edu.
Judith Klein-Seetharaman, Email: jks33@pitt.edu.
References
- 1.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muller G. Towards 3D structures of G protein-coupled receptors: a multidisciplinary approach. Curr Med Chem. 2000;7:861–888. doi: 10.2174/0929867003374534. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Shlomo I, Yu Hsu S, Rauch R, Kowalski HW, Hsueh AJ. Signaling receptome: a genomic and evolutionary perspective of plasma membrane receptors involved in signal transduction. Sci STKE. 2003:17. doi: 10.1126/stke.2003.187.re9. [DOI] [PubMed] [Google Scholar]
- 4.Mishra GR, Suresh M, Kumaran K, Kannabiran N, et al. Human protein reference database--2006 update. Nucleic Acids Res. 2006;34:D411–414. doi: 10.1093/nar/gkj141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones RB, Gordus A, Krall JA, MacBeath G. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 6.Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Molecular Systems Biology. 2005;10:1038. doi: 10.1038/msb4100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
- 8.Colland F, Jacq X, Trouplin V, Mougin C, et al. Functional proteomics mapping of a human signaling pathway. Genome Res. 2004;14:1324–1332. doi: 10.1101/gr.2334104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouwmeester T, Bauch A, Ruffner H, Angrand PO, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 10.Vivian WY, Thomas SM, Zhang Q, Wentzel AL, et al. Mitogenic effects of gastrin-releasing peptide in head and neck squamous cancer cells are mediated by activation of the epidermal growth factor receptor. Oncogene. 2003;22:6183–6193. doi: 10.1038/sj.onc.1206720. [DOI] [PubMed] [Google Scholar]
- 11.Thomas SM, Bhola NE, Zhang Q, Contrucci SC, et al. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006;66:11831–11839. doi: 10.1158/0008-5472.CAN-06-2876. [DOI] [PubMed] [Google Scholar]
- 12.Ewing RM, Chu P, Elisma F, Li H, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gschwind A, Prenzel N, Ullrich A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002;62:6329–6336. [PubMed] [Google Scholar]
- 14.Zhang Q, Thomas SM, Lui VW, Xi S, et al. Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc Natl Acad Sci. 2006;103:6901–6906. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White AW, Westwell AD, Brahemi G. Protein-protein interactions as targets for small-molecule therapeutics in cancer. Expert Rev Mol Med. 2008;10:e8. doi: 10.1017/S1462399408000641. [DOI] [PubMed] [Google Scholar]
- 16.Lage K, Karlberg EO, Storling ZM, Olason PI, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 17.Stumpf MP, Thorne T, de Silva E, Stewart R, et al. Estimating the size of the human interactome. Proc Natl Acad Sci U S A. 2008;105:6959–6964. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venkatesan K, Rual JF, Vazquez A, Stelzl U, et al. An empirical framework for binary interactome mapping. Nat Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 20.Stelzl U, Worm U, Lalowski M, Haenig C, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 21.Ramirez F, Schlicker A, Assenov Y, Lengauer T, Albrecht M. Computational analysis of human protein interaction networks. Proteomics. 2007;7:2541–2552. doi: 10.1002/pmic.200600924. [DOI] [PubMed] [Google Scholar]
- 22.Miller JP, Lo RS, Ben-Hur A, Desmarais C, et al. Large-scale identification of yeast integral membrane protein interactions. Proc Natl Acad Sci U S A. 2005;102:12123–12128. doi: 10.1073/pnas.0505482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. Part II. Computational methods to predict protein and domain interaction partners. PLoS Comput Biol. 2007;3:e43. doi: 10.1371/journal.pcbi.0030043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhodes DR, Tomlins SA, Varambally S, Mahavisno V, et al. Probabilistic model of the human protein-protein interaction network. Nat Biotechnol. 2005;8:951–959. doi: 10.1038/nbt1103. [DOI] [PubMed] [Google Scholar]
- 25.Scott MS, Barton GJ. Probabilistic prediction and ranking of human protein-protein interactions. BMC Bioinformatics. 2007;8:239. doi: 10.1186/1471-2105-8-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehner B, Fraser AG. A first-draft human protein-interaction map. Genome Biol. 2004:5. doi: 10.1186/gb-2004-5-9-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathivanan S, Periaswamy B, Gandhi TK, Kandasamy K, et al. An evaluation of human protein-protein interaction data in the public domain. BMC Bioinformatics. 2006;7(Suppl 5):S19. doi: 10.1186/1471-2105-7-S5-S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramani AK, Li Z, Hart GT, Carlson MW, et al. A map of human protein interactions derived from co-expression of human mRNAs and their orthologs. Mol Syst Biol. 2008;4:180. doi: 10.1038/msb.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braun P, Tasan M, Dreze M, Barrios-Rodiles M, et al. An experimentally derived confidence score for binary protein-protein interactions. Nat Methods. 2009;6:91–97. doi: 10.1038/nmeth.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jansen R, Yu H, Greenbaum D, Kluger Y, et al. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302:449–453. doi: 10.1126/science.1087361. [DOI] [PubMed] [Google Scholar]
- 31.Lee I, Date SV, Adai AT, Marcotte EM. A probabilistic functional network of yeast genes. Science. 2004;306:1555–1558. doi: 10.1126/science.1099511. [DOI] [PubMed] [Google Scholar]
- 32.von Mering C, Jensen LJ, Kuhn M, Chaffron S, et al. STRING 7--recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007;35:D358–362. doi: 10.1093/nar/gkl825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Futschik ME, Chaurasia G, Herzel H. Comparison of human protein-protein interaction maps. Bioinformatics. 2007;23:605–611. doi: 10.1093/bioinformatics/btl683. [DOI] [PubMed] [Google Scholar]
- 34.Qi Y, Klein-Seetharaman J, Bar-Joseph Z. Random Forest Similarity for Protein-Protein Interaction Prediction from Multiple Sources. Pacific Symposium on Biocomputing. 2005;10:531–542. [PubMed] [Google Scholar]
- 35.Breiman L. Random Forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 36.Vaynberg J, Qin J. Weak protein-protein interactions as probed by NMR spectroscopy. Trends Biotechnol. 2006;24:22–27. doi: 10.1016/j.tibtech.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 37.Ashburner M, Ball CA, Blake JA, Botstein D, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ben-Hur A, Noble WS. Kernel methods for predicting protein-protein interactions. Bioinformatics. 2005;21(Suppl 1):i38–46. doi: 10.1093/bioinformatics/bti1016. [DOI] [PubMed] [Google Scholar]
- 39.Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 1994;27:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- 41.Trible RP, Emert-Sedlak L, Smithgall TE. HIV-1 nef selectively activates Src family kinases Hck, Lyn, and c-Src through direct SH3 domain interaction. J Biol Chem. 2006;281:27029–27038. doi: 10.1074/jbc.M601128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pecquet C, Nyga R, Penard-Lacronique V, Smithgall TE, Murakami H, Régnier A, Lassoued K, Gouilleux F. The Src tyrosine kinase Hck is required for Tel-Abl- but not for Tel-Jak2-induced cell transformation. Oncogene. 2007;26:1577–1585. doi: 10.1038/sj.onc.1209949. [DOI] [PubMed] [Google Scholar]
- 43.Xi S, Zhang Q, Dyer KF, Lerner EC, et al. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J Biol Chem. 2003;278:31574–31583. doi: 10.1074/jbc.M303499200. [DOI] [PubMed] [Google Scholar]
- 44.Stover DR, Becker M, Liebetanz J, Lydon NB. Src phosphorylation of the epidermal growth factor receptor at novel sites mediates receptor interaction with Src and P85 alpha. J Biol Chem. 1995;270:15591–15597. doi: 10.1074/jbc.270.26.15591. [DOI] [PubMed] [Google Scholar]
- 45.Lombardo LJ, Lee FY, Chen P, Norris D, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 46.Xia Y, Lu L, Gerstein M. Integrated prediction of the helical membrane protein interatome in yeast. J Mol Biol. 2006;357:339–349. doi: 10.1016/j.jmb.2005.12.067. [DOI] [PubMed] [Google Scholar]
- 47.Velyvis A, Vaynberg J, Yang Y, Vinogradova O, et al. Structural and functional insights into PINCH LIM4 domain-mediated integrin signaling. Nat Struct Biol. 2003;10:558–564. doi: 10.1038/nsb938. [DOI] [PubMed] [Google Scholar]
- 48.Tu Y, Li F, Wu C. Nck-2, a novel Src homology2/3-containing adaptor protein that interacts with the LIM-only protein PINCH and components of growth factor receptor kinase-signaling pathways. Mol Biol Cell. 1998;9:3367–3382. doi: 10.1091/mbc.9.12.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blagoev B, Kratchmarova I, Ong SE, Nielsen M, et al. A proteomics strategy to elucidate functional protein-protein interactions applied to EGF signaling. Nat Biotechnol. 2003;21:315–318. doi: 10.1038/nbt790. [DOI] [PubMed] [Google Scholar]
- 50.Ranish JA, Yi EC, Leslie DM, Purvine SO, et al. The study of macromolecular complexes by quantitative proteomics. Nat Genet. 2003;33:349–355. doi: 10.1038/ng1101. [DOI] [PubMed] [Google Scholar]
- 51.Rinner O, Mueller LN, Hubalek M, Muller M, et al. An integrated mass spectrometric and computational framework for the analysis of protein interaction networks. Nat Biotechnol. 2007;25:345–352. doi: 10.1038/nbt1289. [DOI] [PubMed] [Google Scholar]
- 52.Vermeulen M, Hubner NC, Mann M. High confidence determination of specific protein-protein interactions using quantitative mass spectrometry. Curr Opin Biotechnol. 2008;19:331–337. doi: 10.1016/j.copbio.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 53.Nooren IM, Thornton JM. Diversity of protein-protein interactions. Embo J. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shannon P, Markiel A, Ozier O, Baliga NS, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.