

Abstract
Pseudoxanthoma elasticum (PXE), an autosomal recessive multisystem disorder, is caused by mutations in the ABCC6 gene, and approximately 300 distinct mutations representing >1,000 mutant alleles have been disclosed thus far. Few population‐based studies have reported mutational hotspots in some geographic areas. In this study, we attempted to correlate recurring mutations with the individuals’ ethnic origin. Specifically, we plotted our international database of 70 families from distinct or mixed ethnic backgrounds against their mutations. The frequent p.R1141X mutation was distributed widely across Europe, while deletion of exons 23–29 (del23–29) was encountered in Northern Europe and in Northern Mediterranean countries. p.R1138W may be a marker for French descent, evidenced by its presence also in French Canadians. The splice site transition mutation 3736–1G→A was seen in the neighboring countries Greece and Turkey, whereas 2542 delG occurs only in the Japanese. Two mutations seem to be present worldwide without evidence of a founder effect, p.Q378X and p.R1339C, suggesting the presence of mutational hotspots. Knowledge of this distribution will allow us to streamline mutation screening through a targeted, stepwise approach when the ethnicity of a patient is known. This will facilitate the identification of individuals at risk, improving their care to prevent ophthalmological and vascular disease. Clin Trans Sci 2010; Volume 3: 295–298
Keywords: genes, cardiovascular diseases, genetics
Introduction
Pseudoxanthoma elasticum (PXE; Online Mendelian Inheritance of Man no. 264800 for the autosomal recessive variant) is a heritable connective tissue disorder characterized by progressive calcification of elastic fibers in the skin, retina, and the cardiovascular system. 1 , 2 , 3 In general, the earliest clinical manifestations are dermal lesions that present as small yellowish papules primarily at flexural sites, such as the axillae, antecubital fossae, neck, and groin. These papules tend to coalesce into larger plaques rendering the skin inelastic and redundant. Laxity of the affected skin causes the appearance of premature aging and may have an extensive cosmetic impact. The diagnosis of PXE is confirmed by histopathological evaluation of dermal lesions using von Kossa and Verhoeff‐van Gieson stains that demonstrate mineralization of elastic fibers. The characteristic ophthalmological finding in PXE is the presence of angioid streaks in the retina resulting from breaks in the calcified elastic lamina of the Bruch’s membrane. Consequently, these fractures result in neovascularization and bleeding, which may cause a loss of visual acuity and lead to primarily central blindness. 4 Cardiovascular complications arise from the progressive calcification of the elastin‐rich arterial blood vessel. Clinical sequelae include intermittent claudication, arteriosclerosis that may lead to hypertension, internal bleeding from the gastric arterial blood vessels, and myocardial infarction at a relatively young age. 5 , 6 , 7
PXE is inherited in an autosomal recessive pattern, and mutations in the ATP‐binding cassette (ABC) subfamily C member 6 (ABCC6) gene are the cause of PXE. 8 , 9 , 10 , 11 The ABCC6 gene is comprised of 31 exons spanning approximately 73 kb of genomic DNA on the chromosomal region 16p13.1. The gene codes for the ABCC6 protein of 1,503 amino acids, also known as multidrug resistance‐associated protein 6 (MRP6). ABCC6 is predicted to embody three transmembrane‐spanning domains comprising a total of 17 transmembrane helices. ABCC6 also contains two evolutionarily conserved intracellular nucleotide binding folds (NBF1 and NBF2) that are critical for ATP hydrolysis and for the function of the protein as a transmembrane efflux transporter. 12 ABCC6 is expressed predominantly in the liver and kidneys, and to a lesser extent, if at all, in tissues clinically affected by PXE. 13 , 14 , 15 The precise physiological function of ABCC6 and what molecule(s) this transmembrane protein transports in vivo are currently unknown. 2 , 3
To date close to 300 distinct ABCC6 mutations associated with PXE have been disclosed in various populations (for reviews see Refs. 16, 17). The mutations identified include deletions, insertions, single base pair substitutions, and putative splice site mutations. Examination of the mutation database 17 reveals that premature termination codon‐causing mutations are distributed fairly evenly along the entire gene. In case of missense mutations resulting in substitution of a critical amino acid, the vast majority of mutations has been found toward the carboxy‐terminal end of the protein, corresponding to exons 24 and 30 of the gene. The frequency of mutations in these exons encoding the NBFs and in the seventh and eighth cytoplasmic loop between the 15th and 16th transmembrane regions, respectively, would suggest that these protein domains are of critical importance for normal ABCC6 function.
Sequencing of all 31 exons and flanking intronic sequences of the ABCC6 gene in each new patient makes genotyping notably arduous and time consuming. Knowledge of recurring mutations can facilitate an efficient and time saving screening strategy that will promote methods in identifying individuals at risk. In this study we have identified recurrent mutations in our international database of 70 patients with biopsy‐proven PXE from over 30 diverse cultural or cross‐native descents. This study was specifically designed to identify hotspot mutations in the ABCC6 gene in different ethnic groups, implement a stepwise mutation screening approach when the ethnicity of a patient is known, and provide a methodical and cost‐effective diagnostic screening strategy.
Methods
Patients
DNA samples from our international database of 70 affected patients were subjected to study. Among the study cohort, 60 patients were unrelated and 5 patients shared kinship with one first degree relative affected with PXE. Thus, a total of 60 unrelated and 10 related patients were examined. The patients were either of Asian (Chinese, Japanese), French Canadian, Mediterranean (Greek, Italian, Kosovoan, Spanish, Turkish), Northern European (British, German, Irish, Polish, Russian), or Scandinavian descent (Finnish, Norwegian, Swedish). Informed consent was obtained from all subjects, and the study was approved by the Institutional Review Board of Thomas Jefferson University. The diagnosis of PXE was based on dermatological, ophthalmologic, and/or cardiovascular evaluations. In each proband, the dermal lesions were histologically confirmed to be consistent with the diagnosis of PXE by the observation of calcified elastic fibers in biopsy specimens upon Verhoeff‐van Gieson and/or von Kossa staining.
Mutation analysis
Genomic DNA was isolated from peripheral blood leukocytes of patients affected with PXE, from their unaffected relatives, and from 50 unaffected and unrelated controls of each ethnic group, according to standard procedures. Mutation detection comprised polymerase chain reaction (PCR) amplification of each of the 31 exons in the ABCC6 gene using primer pairs placed on the flanking intronic sequences. Primer sequences for PCR amplification were obtained from the BAC clone A‐962B4 (GenBank accession no. U91318), and the intron–exon junctions of ABCC6 were analyzed by comparison with the published cDNA sequence (Gen Bank accession no. AF076622). The PCR products were analyzed on 1% or 2% agarose gels. PCR products were then examined by heteroduplex scanning by conformation‐sensitive gel electrophoresis or denaturing high‐performance liquid chromatography. The PCR products exhibiting heteroduplex bands were subjected to direct automated nucleotide sequencing (Applied Biosystems model 377 sequencing system; Carlsbad, CA, USA). Restriction enzyme digestions were utilized for the confirmation of nucleotide changes in PXE patients and for the affirmation of their absence in 50 unrelated and unaffected controls. The restriction enzyme reactions were performed according to the manufacturers’ recommendations, and the digestion products were separated on 2% or 3% agarose gels.
Results
ABCC6 mutation analysis was carried out in our international cohort of 70 PXE patients, among which 60 were unrelated and 5 shared one first degree relative. The patients with one first degree relative affected with PXE had different mutations on each allele. Therefore, these patients were considered separately for mutation frequency analysis. PXE patients were from Asian (8.6%), French Canadian (9.3%), Northern Mediterranean (18.6%), Northern European (55.7%), and Scandinavian (7.9%) ethnic groups. Two unrelated patients were of mixed descent. One patient was Northern European and French Canadian, and the second was Northern European and Scandinavian. Both patients were homozygous for the disease‐causing mutation and the alleles were divided amongst both ethnic origins for mutation frequency analysis.
Mutations were identified in 129 of the putative 140 mutant alleles examined, yielding a mutation detection rate of 92.1%. In our database, mutant alleles occurred in compound heterozygous and homozygous forms, the majority of mutant alleles being compound heterozygous. A total of eight recurrent mutations were distinguished in the cohort, including nonsense (p.Q378X and p.R1141X), missense (p.R1138W and p.R1339C), splice site (3736–1G/A, 2787 + 1G/T), frameshift (2542 delG), and multiexon deletion (del exon 23–29).
In our PXE cohort, the recurring nonsense mutation p.R1141X was present in all five ethnic groups, with the exception of Asian, representing 15.4% of French Canadian, 19.2% of Mediterranean, 20.5% of Northern European, and 36.4% of Scandinavian mutations; overall it occurred in 22.9% (32/140) of all the alleles examined. The high frequency of this mutation in the Mediterranean and Northern European population may be explained by a founder effect. 18 The allele p.R1141X was found to share a common haplotype identical by descent in French 19 and Italian patients. 20 In three different studies examining German, 21 Italian, 20 and French PXE patients, 19 R1141X was found to be the most frequent mutation. p.R1141X segregated in a compound heterozygous mode in 50% of French Canadians, 80% in Mediterranean, 75% in Northern Europeans, and in 33.3% of Scandinavians. Homozygous allocation occurred in 50% of French Canadians, 18.7% of Northern Europeans, and 66.7% of Scandinavians whereas heterozygous distribution appeared only in Mediterraneans (20%) and Northern Europeans (6.3%). The p.R1141X mutation can be detected by BsiYI restriction enzyme digestion and agarose gel electrophoresis, and confirmation can be attained by sequencing.
The second most frequent mutation was deletion of exons 23–29, occurring in 12.9% (18/140) of the alleles, and was encountered predominantly in Northern European (18.3%) and Mediterranean (11.5%) descendants. This mutation was not identified in Asian, French Canadian or Scandinavian populations. Haplotype analysis of two French families sharing the mutation (del exons 23–29) carried out by Chassaing et al. 19 suggested that there was not a founder origin for this recurrent mutation.
Exon 24 of the ABCC6 gene was the most affected in our cohort of patients. In this exon a recurrent mutation, p.R1138W, occurred exclusively in 46.1% of French Canadians. The mutation was homozygous in 66.7% of the patients and compound heterozygous in 33.3%. Exon 24 also harbored other mutations (p.R1138Q, p.R1164X, and p.R1164Q) that did not appear to have a predilection for specific ethnicities.
Two additional core mutations established in Mediterraneans and Scandinavians were p.Q378X and p.R1339C. p.R1339C segregated exclusively in 11.5% of Mediterraneans and 9.1% of Scandinavians. p.Q378X distributed in 3.8% of Mediterraneans and 9.1% of Scandinavians. We did not identify p.Q378X in the French Canadian population, but it did appear in Northern Europeans (3.8%) and Asians (0.8%).
There have been few studies to date that analyze mutations in the ABCC6 gene in PXE patients of Asian origin. We screened five Japanese and one Chinese patient with biopsy‐proven PXE for mutations. The mutation 2542 delG appears to be a marker for Asian descent, because this mutation did not surface in other ethnic origins and was observed in two alleles (16.7%) in two unrelated Japanese patients. This mutation was also encountered by Noji et al. 22 in a Japanese patient who was homozygous for the deletion 2542_2543 del G in exon 19 of ABCC6 and heterozygous for a 6 kb deletion (FH‐Tonami‐1) in the LDL receptor gene. The second son of the Japanese patient examined by Noji et al. 22 was heterozygous for 2542_2543 del G in ABCC6 and heterozygous for the deletion FH‐Tonami‐1. Noji et al. also identified two novel missense mutations p.R1221C and p.R1357W in a Japanese patient with PXE. We did not encounter these mutations in our study. Analysis of mutations found in Japanese patients by Noji et al., 22 combined with our study, suggested that neither the nonsense mutation p.R1141X nor the large deletion ABCC6 del 23–29 are frequent in this ethnicity group.
In Mediterraneans, the splice‐site mutation 3736–1G/A was primarily seen in descendants of Greece and Turkey. In two unrelated Greek and Turkish patients this mutation was observed in compound heterozygous and homozygous forms, respectively. In two patients of Greek and Turkish origin Chassaing et al. 19 discovered two novel mutations, p.A766D in exon 18 and p.E1400K in exon 29. These mutations were not identified in our Greek or Turkish patients.
In French Canadians and Northern Europeans we discovered in exon 21 a recurring novel mutation, 2787 + 1G/T. This mutation occurred in 7.7% of French Canadians and 5.1% of Northern Europeans. We did not encounter this mutation in other ethnicities.
Discussion
In the present study we have performed mutational analysis for all 31 exons and flanking intronic sequences of the ABCC6 gene in 70 PXE patients. We screened all 140 alleles of patients from Asian, French Canadian, Northern Mediterranean, Northern European, and Scandinavian ethnic groups. Utilizing PCR, followed by heteroduplex scanning and direct sequencing, our mutation detection rate was 92.1%. In this study, we attempted to correlate recurring mutations with the individuals’ ethnic origin. Altogether, we identified eight recurrent mutations and recognized distinct trends. In general, our results revealed a distribution of mutations along the ABCC6 gene comparable to those published for other European countries. The majority of mutations are located in the carboxy‐terminal part of the ABCC6 protein and approximately 50% of these mutations occur within the two intracellular loops connecting the 13th and 14th, as well as the 15th and 16th transmembrane‐spanning segments. The clustering of these core mutations suggests that these two intracellular loops are essential for the function of ABCC6.
The recurring p.R1141X mutation was distributed widely across Europe, however, deletion of exons 23–29 was encountered primarily in Northern European and Mediterranean descendants. p.R1138W may be a marker for French descent, evidenced by its presence also in French Canadians. The splice‐site mutation 3736–1G/A was seen in the neighboring countries Greece and Turkey, whereas 2542 del‐G mutation occurs only in the Japanese. p.R1339C appeared in Northern Europeans and Scandinavians, whereas p.Q378X was present worldwide. The splice site mutation 2787 + 1G/T was found in French Canadians and Northern Europeans.
The findings presented above, together with previous observations on ABCC6 mutation (Refs. 17, 23, and24), suggest future screening strategies that incorporate the patient’s ethnic background. Such diagnostic strategy should be especially efficient for the European and Mediterranean populations in which a number of PXE mutations occur frequently (p.R1141X and del exons 23–29). Our systematic approach begins with detection of two frequent mutations using PCR and enzymatic digestion of exon 24. Employing a simplified flow chart ( Figure 1 ), a series of questions is then presented to direct the investigator. Analysis of recurring mutations that are frequently present in certain ethnic groups enabled us to provide guidelines for screening specific exons. It should be noted, however, that several of the mutations were found in different ethnic population, possibly reflecting historic migrations of such populations. Nevertheless, evidence for a founder effect has been previously reported in the Afrikaner population in South Africa, in which 53% of the PXE alleles harbored pathogenetic p.R1339C mutation. 18
Figure 1.
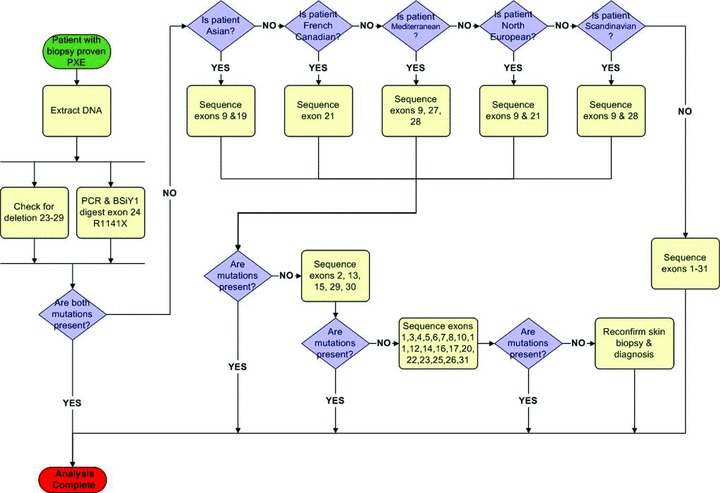
Streamlined ethnicity‐based strategy for detection of mutations in the ABCC6 gene in patients with PXE.
In summary, we have identified a set of recurrent mutations in the ABCC6 gene among five different ethnic groups. Knowledge of a patients’ ethnic origin can facilitate an efficient screening strategy. However, one of the limitations of this study is that not all ethnic origins were analyzed. Because our international database did not include all ethnicities, further investigations of patients with PXE are needed to determine if different or additional recurrent mutations exist. If clinical findings and skin biopsy suggest PXE and the patient’s ethnicity is known, this information may be helpful for clinicians and provide further insight for genetic counseling. This will ultimately facilitate the identification of individuals at risk, improving their care to prevent significant ophthalmological and vascular complications of this, currently intractable, disease.
Acknowledgments
The authors thank Carol Kelly and Lauren Fuchsel for assistance. This study was supported by the NIH/NIAMS grants R01 AR‐28450 and R01 AR‐55225, and by the Dermatology Foundation.
References
- 1. Neldner KH. Pseudoxanthoma elasticum. Clin Dermatol. 1988; 6: 1–92. [DOI] [PubMed] [Google Scholar]
- 2. Li Q, Jiang Q, Pfendner E, Váradi A, Uitto J. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Derm. 2009; 18: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Uitto J, Li Q, Jiang Q. Pseudoxanthoma elasticum—molecular genetics and putative pathomechanisms. J Invest Dermatol. 2010; 130: 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dreyer R, Green WR. The pathology of angioid streaks: a study of twenty‐one cases. Trans Pa Acad Ophthalmol Otolaryngol. 1978; 31: 158–167. [PubMed] [Google Scholar]
- 5. Lebwohl M, Halperin J, Phelps RG. Brief report: occult pseudoxanthoma elasticum in patients with premature cardiovascular disease. N Engl J Med. 1993; 329: 1237–1239. [DOI] [PubMed] [Google Scholar]
- 6. Nolte KB. Sudden cardiac death owing to pseudoxanthoma elasticum: a case report. Hum Pathol. 2000; 31: 1002–1004. [DOI] [PubMed] [Google Scholar]
- 7. Köblös G, Andrikovics H, Prohászka Z, Tordai A, Váradi A, Arányi T. The R1141X loss‐of‐function mutation of the ABCC6 gene is a strong genetic risk factor for coronary artery disease. Genet Test Mol Biomarkers. 2010; 14: 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J. Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP‐binding cassette (ABC) transporter. Proc Natl Acad Sci U S A. 2000; 97: 6001–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bergen AA, Plomp AS, Schuurman EJ, Terry S, Breuning M, Dauwerse H, Swart J, Kool M, van Soest S, Baas F, ten Brink JB, de Jong P. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000; 25: 228–231. [DOI] [PubMed] [Google Scholar]
- 10. Le Saux O, Urban Z, Tschuch C, Csiszar K, Bacchelli B, Quaglino D, Pasquali‐Ronchetti I, Pope FM, Richards A, Terry S, Bercovitch L, de Paepe A, Boyd CD. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000; 25: 223–227. [DOI] [PubMed] [Google Scholar]
- 11. Struk B, Cai L, Zäch S, Ji W, Chung J, Lunsden A, Stumm M, Huber M, Schaen L, Kim CA, Goldsmith LA, Viljoen D, Figuera LE, Fuchs W, Munier F, Ramesar R, Hohl D, Richards R, Neldner KH, Lindpaintner K. Mutations of the gene encoding the transmembrane transporter protein ABC‐C6 cause pseudoxanthoma elasticum. J Mol Med. 2000; 78: 282–286. [DOI] [PubMed] [Google Scholar]
- 12. Borst P, Evers R, Kool M, Wijnholds J. The multidrug resistance protein family. Biochim Biophys Acta. 1999; 1461: 347–357. [DOI] [PubMed] [Google Scholar]
- 13. Belinsky MG, Kruh GD. MOAT‐E (ARA) is a full‐length MRP/cMOAT subfamily transporter expressed in kidney and liver. Br J Cancer. 1999; 80: 1342–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kool M, Van Der Linden M, de Haas M, Baas F, Borst P. Expression of human MRP6, a homologue of the multidrug resistance protein gene MRP1, in tissues and cancer cells. Cancer Res. 1999; 59: 175–182. [PubMed] [Google Scholar]
- 15. Madon J, Hagenbuch B, Landmann L, Meier PJ, Stieger B. Transport function and hepatocellular localization of mrp6 in rat liver. Mol Pharmacol. 2000; 57: 634–641. [DOI] [PubMed] [Google Scholar]
- 16. Hu X, Plomp A, Wijnholds J, ten Brink J, van Soest S, Van Den Vorn LI, Leys A, Peek R, de Jong P, Bergen AA. ABCC6/MRP6 mutations: further insights into the molecular pathology of pseudoxanthoma elasticum. Eur J Hum Genet. 2003; 11: 215–224. [DOI] [PubMed] [Google Scholar]
- 17. Pfendner E, Vanakker O, Terry SF, Vourthis S, McAndrew P, McClain MR, Fratta S, Marais AS, Hariri S, Coucke PJ, Ramsay M, Viljoen D, Terry PF, DePaepe A, Uitto J, Bercovitch LG. Mutation detection in the ABCC6 gene and genotype/phenotype analysis in a large international case series affected by pseudoxanthoma elasticum. J Med Genet. 2007; 44: 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Le Saux O, Beck K, Sachsinger C, Treiber C, Göring HH, Curry K, Johnson EW, Bercovitch L, Marais AS, Terry SF, Viljoen DL, Boyd CD. Evidence for a founder effect for pseudoxanthoma elasticum in the Afrikaner population of South Africa. Hum Genet. 2002; 111: 331–338. [DOI] [PubMed] [Google Scholar]
- 19. Chassaing N, Martin L, Mazereeuw J, Barrié L, Nizard S, Bonafé JL, Calvas P, Hovnanian A. Novel ABCC6 mutations in pseudoxanthoma elasticum. J Invest Dermatol. 2004; 122: 608–613. [DOI] [PubMed] [Google Scholar]
- 20. Gheduzzi D, Guidetti R, Anzivino C, Tarugi P, Di Leo E, Quaglino D, Pasquali Ronchetti I. ABCC6 mutations in Italian families affected by pseudoxanthoma elasticum. Hum Mutat. 2004; 24: 438–439. [DOI] [PubMed] [Google Scholar]
- 21. Hendig D, Schulz V, Eichgrün J, Szliska C, Gotting C, Kleesiek K. New ABCC6 gene mutations in German pseudoxanthoma elasticum patients. J Mol Med. 2005; 83: 140–147. [DOI] [PubMed] [Google Scholar]
- 22. Noji Y, Inazu A, Higashikata T, Nohara A, Kawashiri M, Yu W, Todo Y, Nozue T, Uno Y, Hifumi S, Mabuchi H. Identification of two novel missense mutations (p.R1221C and p.R1357W) in the ABCC6 (MRP6) gene in a Japanese patient with pseudoxanthoma elasticum. Intern Med. 2004; 43: 1171–1176. [DOI] [PubMed] [Google Scholar]
- 23. Le Saux O, Beck K, Sachsinger C, Silvestri C, Treiber C, Göring HH, Johnson EW, de Paepe A, Pope FM, Pasquali‐Ronchetti I, Bercovitch L, Marais AS, Viljoen DL, Terry SF, Boyd CD. A spectrum of ABCC6 mutations is responsible for pseudoxanthoma elasticum. Am J Hum Genet. 2001; 69: 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pfendner E, Uitto J, Gerard GF, Terry SF. Pseudoxanthoma elasticum: genetic diagnostic markers. Expert Opinion Med Diagn. 2008; 2: 1–17. [DOI] [PubMed] [Google Scholar]