

Abstract
Carbon monoxide dehydrogenase (CODH) from Oligotropha carboxydovorans catalyzes the oxidation of carbon monoxide to carbon dioxide, providing the organism both a carbon source and energy for growth. In the oxidative half of the catalytic cycle, electrons gained from CO are ultimately passed to the electron transport chain of the Gram-negative organism, but the proximal acceptor of reducing equivalents from the enzyme has not been established. Here we investigate the reaction of reduced enzyme with various quinones, and find them to be catalytically competent. Benzoquinone has a kox of 125.1 s-1 and Kd of 48 μM; ubiquinone-1 has a kox/Kd value of 2.88 × 105 M-1s-1; 1,4-napthoquinone has a kox of 38 s-1 and Kd of 140 μM; and 1,2-napthoquinone-4-sulfonic acid a kox/Kd of 1.31 × 105 M-1s-1. An extensive effort to identify a cytochrome that was reducible by CO/CODH was unsuccessful. Steady-state studies with benzoquinone indicate that the rate-limiting step is in the reductive half of the reaction (that is, the reaction of oxidized enzyme with CO). On the basis of the inhibition of CODH by diphenyliodonium chloride we conclude that quinone substrates interact with CODH at the enzyme’s flavin site. Our results strongly suggest that CODH donates reducing equivalents directly to the quinone pool without using a cytochrome as an intermediary.
Molybdenum-containing enzymes are very broadly distributed in biology, and members of the xanthine oxidoreductase (XOR) family comprise a large and important group of these enzymes. Family members generally catalyze the oxidative hydroxylation of aromatic heterocycles and aldehydes, and the reducing equivalents generated in this process pass from the molybdenum center, where catalysis takes place, through two [2Fe-2S] clusters and (in most cases) on to an FAD where the electrons are passed on to an oxidizing substrate such as NAD+ or O2 (1).
Carbon monoxide dehydrogenase (CODH) from aerobic, chemolithotrophic organisms such as Oligotropha carboxydovorans and Hydrogenophaga pseudoflava is clearly a member of the xanthine oxidase family based on its overall amino acid sequence and three-dimensional structure (2-8). The functional enzyme is a (αβγ)2 hexamer that consists of a small 17.8 kDa subunit (CoxS) containing two [2Fe-2S] clusters, a medium 30.2 kDa subunit (CoxM) containing an FAD cofactor, and a large 88.7 kDa subunit (CoxL) that possesses the molybdenum center. CODH is encoded by the mega plasmid pHCG3 in the CoxMSL cluster (9, 10). The overall protein fold notwithstanding, two aspects make CODH unique in the XOR family: first, the reaction itself is not strictly speaking a hydroxylation and does not involve the cleavage of a C-H bond; and second, the active site consists of a unique binuclear Mo-Cu center rather than a mononuclear molybdenum center such as is seen in all other family members. As shown in Figure 1, the active site is an LMoVIO2-(μS)-CuI-SCys cluster, where L represents the pyranopterin cofactor found in all molybdenum (and tungsten) containing enzymes other than nitrogenase (4, 5, 11). The Mo/Cu-containing CODH from O. carboxydovorans and Hydrogenophaga pseudoflava is structurally and mechanistically distinct from the Fe/Ni-containing CODH of the acetogen Moorella thermoacetica or the methanogen Methanosarcina barkerii (12).
Figure 1. The active site of CODH.
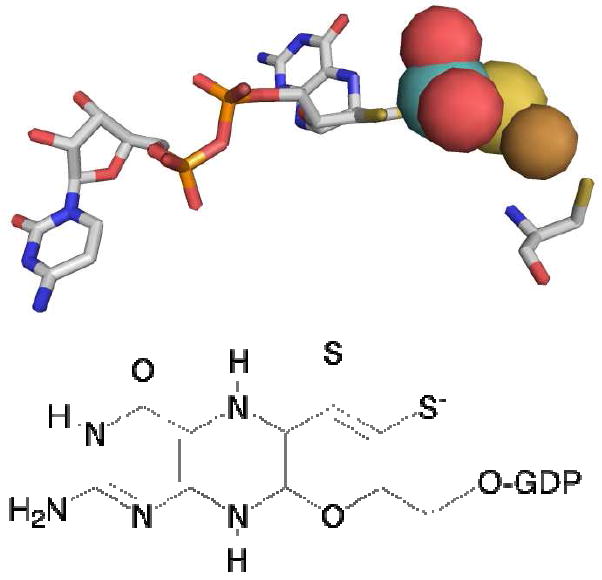
Top, the active site of the enzyme as rendered using PDB file 1N5W. Atom colors are CPK, with the molybdenum atom rendered in teal and the copper in copper color. Cys 388, which coordinates the copper, is shown at far right. Bottom, the structure of the pyranopterin cofactor of the binuclear center, which is present as the dinucleotide of guanine.
In the reaction carried out by CODH, CO is oxidized to CO2, yielding two reducing equivalents according to the following stoichiometry:
The reducing equivalents thus obtained by the enzyme are passed to the electron transport chain to provide energy for cell growth. A portion of the CO2 generated as a product of the reaction is fixed non-photosynthetically by the pentose phosphate cycle (13, 14) with an overall stoichiometry of:
The overall reaction is of profound environmental importance, since aerobic bacteria such as O. carboxydovorans clear an estimated 2× 108 metric tons of CO from the atmosphere annually (15).
It has been suggested that reducing equivalents are removed from CODH by either cyt b561 or the quinine pool, although no definitive evidence has been provided in support of either (13, 16). In the present study we have investigated the kinetics involved with the oxidative half-reaction of the enzyme with various quinones, and find that they serve very effectively as oxidizing substrates for CODH, reacting with reduced enzyme with rate constants large enough to support catalysis. By contrast, cytochrome b561 is found not to readily accept reducing equivalents from reduced CODH, and we are unable to identify any other cytochrome from O. carboxidovorans capable of doing so. We conclude that quinones are the likely physiological oxidant of CODH.
MATERIALS AND METHODS
Materials
Carbon monoxide gas was obtained from Air, Inc. at a purity of 99.5%. 1,4-benzoquinone, 1,2-naphthoquinone-4-sufonic acid, 1,4-naphthoquinone, and ubiquinone-1 were purchased from Sigma-Aldrich. Isotopically enriched D2O was obtained from Cambridge Isotope Lab, Inc. All other chemicals and reagents were obtained at the highest quality and purity commercially available and used without additional purification.
Bacterial cultivation and enzyme purification
O. carboxydovorans (ATCC 49405) cells were grown at 30° C, pH 7 in a 20 L fermentor (BioFlo 415, New Brunswick) containing Minimal Medium and CO as the carbon source (introduced as a mixture of 50% CO and 50% air). Cells were harvested in late log phase (OD436 >5), washed in 50 mM HEPES (pH 7.2) and stored at -80° C until needed (17). CODH was purified according to the procedure described by Zhang et al. (18), using a combination of Q-Sepharose and Sephacryl S-300 FPLC chromatography.
Cytochrome b561 was purified from O. carboxydovorans grown as described above. 100 g of thawed cells were resuspended in 50 mM HEPES (pH 7.2) containing 1 mM EDTA, 5 mg DNase, 0.2 mM PMSF and broken open by French press (FA-078A, Thermo Electron Co.). Cell debris were separated by ultra centrifugation at 100k × g for 2 h. Cell membranes were solubilized in 50 mM HEPES (pH 7.2) containing 1 mM EDTA, 0.2 mM PMSF and 10% v/v Triton X-100. The non-solubilized membranes were separated by ultra centrifugation at 100k × g for 2 h. The soluble fraction was loaded onto a CM anion exchange column (11cm × 1.5cm) using an ÄKTA FPLC apparatus (GE Healthcare) at 4°C; the column was pre-equilibrated with 50 mM HEPES (pH 7.2) containing 0.1 mM EDTA and 0.2% Triton X-100. Elution was carried out over 10 column volumes in a linear gradient from 0 mM to 500 mM NaCl. Fractions containing cytochrome b561 were pooled and concentrated using an Amicon concentrator equipped with a 10 kDa cutoff filter. The identity of cytochrome b561 was verified by UV-vis spectroscopy with an absorbance peak at 415 nm in the oxidized cytochrome and 425 nm, 530 nm, and 561 nm in the reduced cytochrome (Supplementary Material, Figure 1). Approximately 20-50 μg of cytochrome b561 was obtained from 100 g of cells. Fractions containing other cytochromes were also saved and examined for activity with CODH.
Protein determination and activity assay
Enzyme quantity was determined by the absorbance at 450 nm (ε450=70 mM-1cm-1) and 550 nm and purity assessed by the ratios of absorbance at 280 nm, 420, nn, 450 nm and 550 nm: the purified enzyme had absorbance ratios of A280/A450 ~5.5, A450/A550 ~2.9 and A450/A420 > 1 (18). Routine activity was determined by the CO-dependent reduction of methylene blue (ε615=37.11 mM-1cm-1) at 30° C (3,18). A second assay utilized 1,4-benzoquinone as oxidizing substrate, following reduction of, e.g., 50 μM 1,4-benzoquinone at 246 nm using an HP 8452A UV-visible spectrophotometer. A serum-stoppered cuvette (Starna Cells, inc) containing 50 μM 1,4-benzoquinone in 2mL of 50 μM HEPES, pH 7.2, was bubbled with 100% CO for 10-15 min in the dark, after which 10-20 μL of 4 μM anaerobic stock CODH solution was added by Hamilton syringe. The specific activity in units per mg was determined using an extinction change for 1,4-benzoquinone reduction, obtained from the extinction coefficient ε247=20.6 mM-1cm-1 for the oxidized 1,4-benzoquinone (20), a reduced coefficient of ε247=0.42 mM-1cm-1 determined by the equation: εred=(Absred)(εox)/(Absox) to give Δε247=20.2 mM-1cm-1 for 1,4-benzoquinone, with 1 unit of activity being defined as 1 μmol CO oxidized per min at 30° C. The enzyme used in the present work exhibited a specific activity of approximately 8.
All enzyme preparations were reconstituted with sulfur and copper using a modification of the procedure of Resch et al. (19). Ca. 100 μM CODH in 1.0 mL of 50 mM Tris-HCl, pH 8.2 was made anaerobic by alternately evacuating and flushing with O2-scrubbed Ar gas over the course of an hour. Appropriate volumes of stock solutions of 10 mM methyl viologen and 100 mM Na2S were added to give final concentrations of 0.1 mM and 2.0 mM, respectively, followed by the addition of a sufficient volume of a ~0.1 M dithionite stock solution to sustain the blue color of the reduced viologen. This was incubated at 20° C for 12-18 hr under an atmosphere of argon gas. The enzyme was then passed through a G-25 chromatography column equilibrated with anaerobic 50 mM Tris-HCl, pH 8.2 to remove excess Na2S, dithionite and methyl viologen. A stock solution of 10 mM Cu(I)-thiourea was prepared by dissolving Cu(I)Cl, thiourea, and sodium ascorbate in a 1:3:1 (w/w) ratio in anaerobic water, and the enzyme solution then made 0.2 mM in Cu(I) using this solution and incubated 5-10 hr at 20° C. A final G-25 column, again equilibrated with anaerobic 50 mM Tris-HCl, pH 8.2, was used to remove excess Cu(I). The enzyme was assayed for activity as described above, and the degree of functionality independently determined by comparing the extent of enzyme bleaching, as observed at 450 nm, by CO (which reduces only the fully functional enzyme) with that seen using dithionite (which reduces both functional and non-functional enzyme) enzyme (5). Our enzyme was approximately 40% active, comparable to the levels seen previously (19). Unless otherwise stated, enzyme concentrations are given as functional enzyme, corrected for the fraction of nonfunctional enzyme present.
Steady-state kinetics
Steady-state studies monitoring the reduction of ubiquinone-1 and 1,4-benzoquinone were performed by bubbling anaerobic solutions of each in 50 mM HEPES (pH 7.2) with CO for 15 min to give 1 mM CO (previous studies having shown that concentrations of CO above 100 μM were saturating in steady-state assays; 18). The concentration ranges used were 11 μM to 97 μM for ubiquinone-1 and 8.4 μM to 191 μM for 1,4-benzoquinone. The reaction was initiated by the addition of 10 μL of 2 μM CODH, and the reaction monitored by the spectral change at 275 nm or 246, respectively, over 300 s at 25°C. Activities were obtained from the initial slopes of each assay, calculated using ε278=14.7 mM-1cm-1 for ubiquinone-1 (22) and Δε247=20.2 mM-1cm-1 for 1,4-benzoquinone.
A kinetic isotope study was performed under the same conditions as described above using 50 mM HEPES in D2O (pD 7.6) with the addition of varying concentrations of ubiquinone-1 in D2O. CODH in D2O prepared by anaerobic buffer exchange G-25 column into 50 mM HEPES in D2O (pD 7.6).
Rapid reaction kinetics
The oxidative half-reaction of CODH was monitored by stopped-flow spectroscopy (using an Applied Photophysics, Inc. SX-18MV. Standard reaction conditions were 50 mM HEPES, pH 7.2, 25°C. Enzyme at a concentration of ~10 μM before mixing was placed in a glass tonometer equipped with a sidearm cuvette and made anaerobic by repeated evacuation and flushing with O2-scrubbed Ar over the course of an hour. The anaerobic enzyme was then titrated with an anaerobic solution of ~0.1 M sodium dithionite in 50 mM HEPES, pH 7.2, monitoring enzyme reduction spectrophotometrically. The reduced enzyme was then mounted on the stopped-flow apparatus and mixed with varying concentrations of anaerobic, oxidized quinone substrate in 50 mM HEPES, pH 7.2, the reaction being monitored by the absorbance increase observed at 450 nm or 550 nm. Kinetic transients thus obtained were fit using the ProData Viewer package to obtain the rate constants, which were averaged and plotted as a function substrate concentration. When saturating kinetic behavior for kobs as a function of [quinone] was observed, values for kox, the limiting rate constant at high [S], and dissociation constant, Kd, were obtained from hyperbolic fits to these plots using SigmaPlot (Systat Software, Inc.). When linear behavior was observed, the ratio kox/Kd was determined directly from the slope.
Titrations of CODH with quinones
Titrations were performed using 4 μM or 8 μM CODH in 50 mM HEPES, pH 7.2 at 20°C. Titrations of oxidized enzyme with oxidized quinone were carried out aerobically, following the spectral change observed in the vicinity of 450 nm. Titration of oxidized enzyme with reduced quinone (prepared by directly bubbling Ar in a cuvette for 15 min to make anaerobic and titrated to reduction with 0.1 M sodium dithionite using a Hamilton syringe, following quinone reduction at 247 nm) was carried out anaerobically, by alternately evacuating and flushing the enzyme solution in an anaerobic cuvette with O2-scrubbed Ar gas for 1 hour. Aliquots of reduced 1,4-benzoquinone were then added with a Hamilton syringe and the spectral change in the visible monitored spectrophotometrically. For 1,4-benzoquinone, plots of absorbance change vs. [quinone] were used to obtain Kd, which was determined by fitting the data to the hyperbolic equation:
Inhibition of CODH by diphenyliodonium chloride
Inactivation of the FAD cofactor of CODH was accomplished by covalent modification of the flavin with diphenyliodonium chloride using a modification of the procedure of Chakraborty and Massey (21). 10 μM CODH in 50 mM HEPES, pH 7.2 was flushed with Ar for 1 hr and reduced with ~2 fold excess sodium dithionite. Diphenyliodonium chloride was added to a final concentration of 1 mM and incubated at 20°C for 2 hr. Excess diphenyliodonium chloride was removed by G-25 column and the enzyme assayed for activity. Spectral changes and specific activity of the enzyme with ubiquinone-1 and methylene blue were used to assess the degree of inhibition, as described above.
Electron paramagnetic resonance spectroscopy
EPR spectra were recorded using a Brüker Instruments ER 300 spectrometer equipped with an ER 035M gaussmeter and HP 5352B microwave frequency counter. Temperature was controlled at 150 K using a Brüker ER 4111VT liquid N2 cryostat. Samples were prepared by reducing 50 μM anaerobic CODH in 50 mM HEPES, pH 7.2 with a stock solution of 0.1 M dithionite, followed by addition of 0.5 equivalents of 1,4-benzoquinone or ubiquinone-1. Samples were immediately frozen in liquid N2. Controls of 50 μM CODH in 50 mM HEPES, pH 7.2, either fully oxidized or fully reduced by titration with dithionite, were also prepared. A final sample of 50 μM CODH was prepared by first reducing the enzyme with 0.1 M dithionite and re-oxidizing with 12 equivalents of quinone prior to freezing.
RESULTS
CODH reactivity toward cytochrome b561
Cytochrome b561 was isolated from O. carboxydovorans as described in Materials and Methods and examined for reactivity toward CODH in both steady-state and rapid-reaction experiments. In the assays performed, cytochrome b561 in 50 mM HEPES, pH 7.2, containing 0.2% Triton X-100 or N,N-dimethyldodecylamine-N-oxide was made anaerobic by repeated evacuation and flushing with O2-scrubbed Ar followed by CO bubbling. The activity assay was carried out over 10 min observing total spectral change with an emphasis near 415 nm and 561 nm, but no reduction of cytochrome b561 was seen in these experiments. Other cytochrome-containing fractions isolated from soluble and membrane cell fractions included multiple cytochromes c, and cytochrome a, and these were also examined for activity toward CODH; in no case was cytochrome reduction observed (supplemental figure 2). Only when 1,4-benzoquinone (at a concentration of 5 μM) was added to the assay with cytochrome c was reduction observed, as reflected in the absorbance increase at 550 nm and a shift of the Soret band from 410 nm to 415 nm. These results suggest that cytochrome reduction was mediated by the quinone, which served as the proximal oxidant for the enzyme.
Steady state kinetic studies
Based on the above results suggesting that 1,4-benzoquinone is an oxidizing substrate of CODH, a steady-state study was performed using 1,4-benzoquinone as oxidizing substrate. The assay was performed under the standard conditions of 25°C in 50 mM HEPES pH 7.2, with solutions of 8.4 μM to 191 μM 1,4-benzoquinone placed in a serum-stoppered cuvette and bubbled first with argon and then CO to give a concentration of 400 μM. The reaction was followed by the spectral change at 246 nm associated with reduction of the quinone. Significant catalytic rates were observed. A plot of observed catalytic velocity versus [1,4-benzoquinone] was hyperbolic and a fit to the data yielded a kcat of 104 s-1, Km of 16.4 μM and kcat/Km of 6.37 × 106 M-1s-1 (Supplementary Material, Figure 3). This is essentially identical to the previously observed kcat of 93.3 s-1 using methylene blue as oxidizing substrate, consistent with the previous conclusion that the reductive half-reaction was principally rate-limiting (18). Ubiquinone-1 was also examined as substrate, but in this case it proved impossible to obtain sufficiently high concentrations of substrate to yield saturating kinetics given the limited water-solubility of ubiquinone-1. A plot of the observed catalytic velocity versus [ubiquinone-1] thus yielded a straight line, and from the slope a value for the ratio kcat/Km of 5.81 × 104 M-1s-1 was obtained. When the steady-state kinetics with ubiquinone-1 were repeated in D2O, a solvent isotope effect of 1.4 was obtained from the ratio of H(kcat/Km)/D(kcat/Km) (Supplementary Material, Figure 4).
With other enzymes of the XOR family, reducing equivalents enter at the molybdenum center in the reductive half of the catalytic sequence and leave via the FAD (after intramolecular electron transfer involving the iron-sulur clusters) (1). In order to establish that quinone substrates interacted with CODH at its FAD site, the enzyme was reacted with diphenyliodonium chloride to covalently modify the FAD and render the cofactor redox-inert (21). Treatment of enzyme in this way lowered the steady state rates of ubiquinone-1 reduction by 83% and methylene blue by 80%; modification of the flavin was confirmed by comparing the absorption spectrum of the reoxidized, modified enzyme with that of the original (Figure 2). The unmodified minus modified difference spectra revealed features at 370 nm and 450 nm constant with flavin modification seen in xanthine oxidase (Figure 2, inset) (21). With the modified enzyme, reoxidation of the iron-sulfur clusters is observed after exposure to air, while the additional absorbance increase expected for reoxidation of the FAD is not observed. The reaction was carried out to longer time periods in order to achieve a greater degree of inhibition, but no further inhibition was observed. The incomplete inactivation was most likely due to incomplete covalent modification of the flavin due to the lower solvent accessibility of the FAD of CODH relative to xanthine oxidase. We observed, for example, that CODH inhibited with diphenyliodonium chloride regained slowly regained activity over time, suggesting that the inhibited flavin-diphenyliodonium chloride enzyme complex was slowly returned back to functional enzyme. Consistent with this, the absorbance of oxidized FAD is slowly recovered over the next 18 hr, reflecting the slow breakdown of the flavin-diphenyliodonium chloride complex to yield the oxidized cofactor (21).
Figure 2. Diphenyliodonium chloride inhibition of CODH.
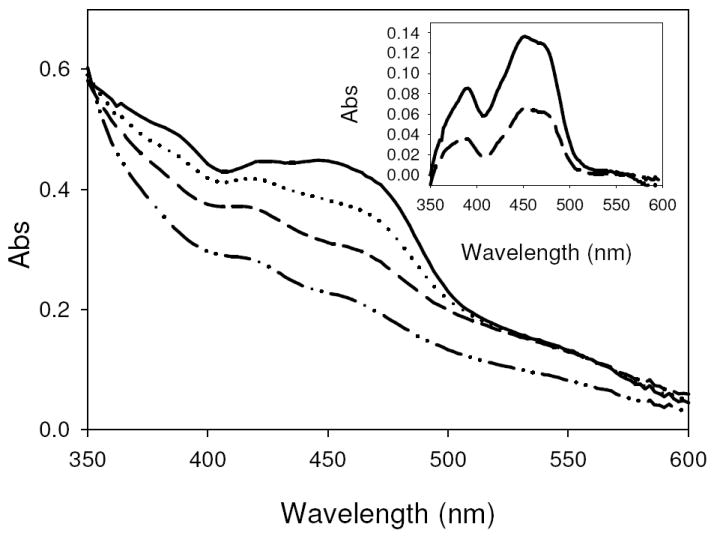
A, The spectral change over the course of inhibition of CODH in 50 mM HEPES, pH 7.2, with diphenyliodonium chloride. Spectra are for oxidized CODH (—), dithionite-reduced CODH (- · · –), air oxidized CODH following diphenyliodonium chloride inhibition (- -), and diphenyliodonium chloride-inhibited CODH after 18hrs (⋯·). Inset, The oxidized-minus-inhibited difference spectrum following diphenyliodonium chloride inhibition (—) and after an 18 hr incubation (- -).
Oxidative-Half Reaction of CODH with Quinone Substrates
The rapid reaction kinetics of the reoxidation of reduced CODH by several quinones was next examined by stopped-flow spectrophotometry at 25°C, following the reoxidation of enzyme at 450 nm. The substrates used were 1,4-benzoquinone, 1,4-naphthoquinone, 1,2-naphthoquinone-4-sulfonic acid and ubiquinone-1. Figure 3 shows a typical time course for the reaction with 25 μM 1,4-benzoquinone with 5 μM CODH after mixing in the stopped flow apparatus. At low substrate concentrations the reaction appeared biphasic with apparent rate constants of 37 s-1 for the first phase (with an associated absorbance change of 0.01 OD, 24% of the total absorbance change) and 3.33 s-1 for the second (with amplitude of 0.03 OD, 73% of the total absorbance change). (A third phase was sometimes observed at low substrate concentrations, with a rate constant 0.5 s-1 and absorbance change of ~0.001 OD, 2% of the total absorbance change, that was attributed to oxygen contamination. This phase was neglected on the basis of the very small absorbance change associated with it.) The overall kinetic complexity of the reaction is a reflection of the fact that three equivalents of quinone must react with the fully reduced enzyme in turn for full reoxidation. We attribute the first phase to the reaction of fully reduced (i.e., six-electron-reduced) CODH with a first equivalent of 1,4-benzoquinone to yield four-electron reduced enzyme and 1,4-benzoquinone-H2, and the second rate constant the subsequent reaction of four-electron reduced enzyme with two additional equivalents of 1,4-benzoquinone in turn (which are kinetically unresolved). At higher substrate concentrations, the amplitude of the faster process increases at the expense of the slower, the latter eventually disappearing by 500 μM. Plots of the observed rate constant versus [1,4-benzoquinone] were hyperbolic for both phases of the reaction; the first phase yielded a kox of 125.1 s-1, Kd of 48 μM and kox/Kd (i.e., the slope of the plot of kobs versus [1,4 BQ]) of 2.60 × 106 M-1s-1, while the second phase showed a kox of 32.7 s-1 and Kd of 154 μM. Again, the second phase was only clearly resolved below 500 μM 1,4-benzoquinone when 5 μM enzyme is used.
Figure 3. Oxidation of reduced CODH by 25μM 1,4-benzoquinone.

Oxidation of dithionite-reduced 5 μM CODH by 1,4-benzoquinone in 50 μM HEPES, pH 7.2 25° C, over 0.6 s. The traces shown are those recored at 0.004 s, 0.007 s, 0.015 s, 0.027 s, 0.042 s, 0.076 s, 0.098 s, 0.0193 s, 0.0382 s and 0.0571 s increasing in absorbance from 0.004 s to 0.0571 s at 450 nm. Inset, Time course of absorbance change at 450 nm over 0.6 s (absorbance measurements shown are for 0.004 s, 0.007 s, 0.015 s, 0.027 s, 0.042 s, 0.076 s, 0.098 s, 0.0193 s, 0.0382 s and 0.0571 s) during the course of CODH oxidation. Fits to the data yield observed rate constants of 37 s-1 for the first phase and 3.3 s-1 for the second phase (with an R2 value for the fit of 0.993).
The reaction of reduced CODH with 1,2-naphthoquinone-4-sulfonic acid was also biphasic, with amplitudes of 0.02 OD (28% of the total observed spectral change) seen for the first phase and 0.04 OD (57% of the total spectral change) for the second when 5 μM CODH was used. A third very slow phase (0.01 OD, 14% of the total spectral change) was observed that was attributed to photosensitivity of the quinone, as confirmed by monitoring the spectral change of 1,2-naphthoquinone-4-sulfonic acid alone in the observation cell of the stopped-flow apparatus. This phase became negligible at substrate concentrations above 125 μM owing to the short time scale of the reaction. Due to the high extinction of 1,2-naphthoquinone-4-sulfonic acid, concentrations under 1 mM had to be used and it was not possible to reach saturating concentrations; only the ratio kox/Kd could be obtained from the linear plot of kobs versus [1,2-naphthoquinone-4-sulfonic acid], with values of 1.31 × 105 M-1s-1 and 3.90 × 104 M-1s-1 for the faster and slower phases, respectively.
1,4-naphthoquinone also exhibited biphasic behavior, with amplitudes of 0.04 OD (40% of the total observed spectral change) and 0.06 OD (60% of the observed spectral change) for the first and second phases, respectively, with 5 μM enzyme. 1,4-naphthoquinone (which is structurally similar to menaquinone) yielded somewhat slower kinetics than seen with 1,4-benzoquinone, with kox = 38.1 s-1, Kd = 140 μM, and kox/Kd = 2.72 × 105 M-1s-1 for the faster phase of the reaction; the slower phase gave kox = 6.0 s-1, Kd 213 μM and kox/Kd = 2.82 × 104 M-1s-1.
Ubiquinone-1 also showed biphasic behavior with amplitudes of 0.02 OD (14% of the total observed spectral change) and 0.12 (86% of the total spectral change) OD for the first and second phases, respectively, with 5 μM CODH. Due to the limited solubility of ubiquinone-1 it was again not possible to approach saturating conditions, and only a ratio kox/Kd could be determined from the slope of the linear plot of kobs versus [ubiquinone-1], with values of 2.88 × 105 M-1s-1 and 1.99 × 104 M-1s-1 obtained for the faster and slower phases, respectively. Plots of observed rate constant versus [quinone] for each substrate is shown in Figure 4, with the kinetic parameters thus determined summarized in Table 1. For the purposes of comparison, attention was focused on the faster phases of the overall reaction which reflected the reaction of fully reduced enzyme with the first equivalent of quinone in the course of reoxidation and thus represented the intrinsic reactivity of the fully reduced flavin site with quinone. It was evident that of the three quinones examined, 1,4-benzoquinone (which is structurally similar to ubiquinone) was the most effective substrate having a kox/Kd an order of magnitude higher than the other two.
Figure 4. Substrate concentration dependence of reoxidation of dithionite-reduced CODH by quinones.
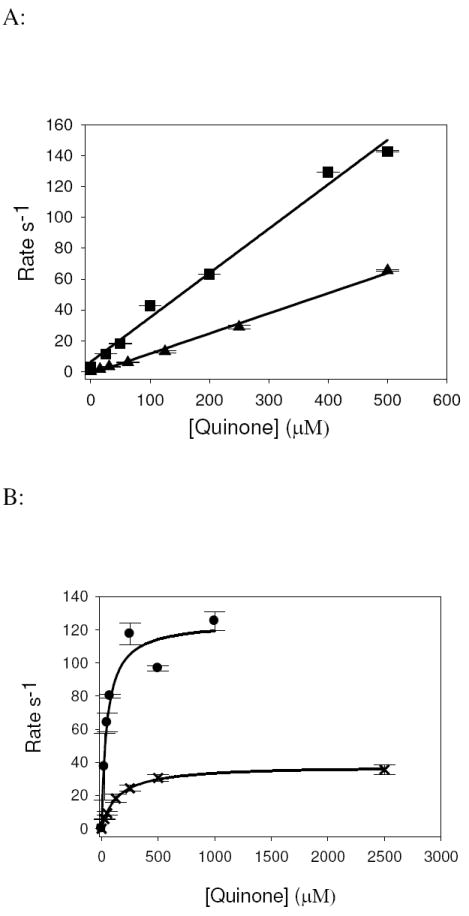
A, plots of kfast versus [quinone] for the reaction of 5 μM enzyme with ubiquinone-1 (■) and 1,2-naphthoquinone-4-sulfonic acid (▲) in 50 mM HEPES, pH 7.2, 25°C. Solid Line, fit with a linear equation using SigmaPlot (Systat Software, Inc.). The ratio kox/Kd (corresponding to the second-order reaction of reduced enzyme with substrate in the low-[Q] regime) was determined by the slope of the fitted line, which for ubiquinone-1 and 1,2-naphthoquinone-4-sulfonic acid were 2.88 × 105 M-1 s-1 (R2=0.990) and 1.31 × 105 M-1 s-1 (R2=0.995), respectively. B, plots of kfast versus [quinone] for the reaction of 5 μM enzyme with 1,4-benzoquinone (●) and 1,4-naphthoquinone (x) in 50 mM HEPES, pH 7.2, 25°C. Solid line, fit using the hyperbolic equation kobs = kox[Q]/(Kd + [Q]) using SigmaPlot, where kox is the limiting rate constant for reoxidation at high [Q] and Kd is the dissociation constant for substrate binding. Kinetic parameters thus determined for 1,4-benzoquinone are kox, 125.1 s-1, Kd, 46.7 μM, kox/Kd, 2.60 × 106 M-1 s-1 (line fit R2= 0.955) and for 1,4-napthoquinone are kox, 38.1 s-1, Kd, 140 μM, and kox/Kd of 2.72 × 105 M-1 s-1 (line fit R2 = 0.999).
Table 1.
Kinetic parameters of CODH oxidation by various quinones.
| Kd (μM) | kox (s-1) | kox/Kd (M-1s-1) | R2 value for oxidative half fit data | Km (μM) | kcat (s-1) | kcat/Km (M-1s-1) | R2 value for steady state fit data | KIE | |
|---|---|---|---|---|---|---|---|---|---|
| 1,4-benzoquinone | 47.6 | 125.1 | 2.60 × 106 | 0.955 | 16.4 | 104.5 | 6.37 × 106 | 0.995 | |
| 1,4-naphthoquinone | 140 | 38.1 | 2.72 × 105 | 0.999 | |||||
| 1,2-naphthoquinone-4-sufonic acid | 1.31 × 105 | 0.995 | |||||||
| Ubiquinone-1 H20 | 2.88 × 105 | 0.990 | 5.81 × 104 | 0.980 | 1.41 | ||||
| Ubiquinone-1 D20 | 4.13 × 104 | 0.996 |
Titration of CODH with oxidized and reduced quinone
In order to establish the affinity of CODH for 1,4-benzoquinone, titrations of CODH with oxidized and reduced 1,4-benzoquinone were carried out. Figure 5 shows the titration of oxidized 1,4-benzoquinone with 8 μM CODH, with the greatest spectral change seen at 424 nm. The inset to Figure 5 inset shows a plot of the change in absorbance at 424 nm vs. [1,4-benzoquinone] from which a Kd of 100 μM can be determined. A comparison with the kinetically determined Kd for binding of oxidized 1,4-benzoquinone with reduced CODH of 48 μM indicates that oxidation of the enzyme flavin reduces affinity for the quinone by a factor of two. Titration of oxidized CODH with pre-reduced 1,4-benzoquinone under anaerobic conditions yielded an oxidized minus-reduced difference spectrum indicating that the FAD and Fe/S centers became fully reduced, as reflected by bleaching at 450 nm and 550 nm (Supplementary Material, Figure 5).
Figure 5. Titration of oxidized CODH with oxidized 1,4-benzoquinone.
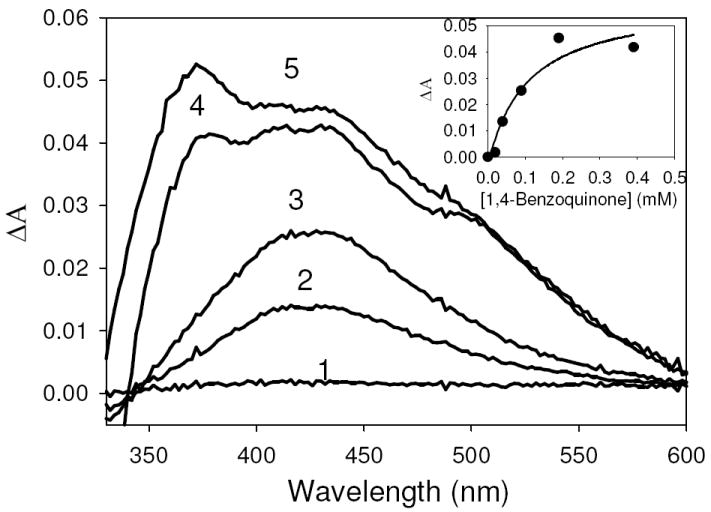
Difference spectra of CODH with bound 1,4-benzoquinone in 50 mM HEPES, pH 7.2, minus oxidized CODH when CODH is titrated with oxidized 1,4-benzoquinone. Additions of 1,4 benzoquinone yielded final concentrations of 20 μM (1), 40 μM (2), 90 μM (3), 190 μM (4), 390 μM(5). Inset, concentration dependence of the spectral change at 424 nm produced by 1,4-benzoquinone titration with CODH. Kd was found to be 104 μM, determined by a fit using the hyperbolic equation Aobs=(ΔAmax•x)/(Kd+x) (R2= 0.9359).
DISCUSSION
Here we have examined the reaction of reduced CODH with several cytochromes and quinones in an effort to determine the oxidizing substrate for the enzyme. Having failed in an exhaustive effort to identify a cytochrome capable of being effectively reduced by CODH, we find that several quinones are very effective substrates, rapidly oxidizing reduced CODH under anaerobic conditions. 1,4-benzoquinone is found to be the most effective oxidizing substrate, with a kox of 125.1 s-1 at pH 7.2, 25°C for the fastest phase of enzyme reoxidation. The multiple phases observed in the oxidative half-reaction kinetics seen here are attributed to the necessarily sequential nature of the reaction of fully (six-electron) reduced enzyme with three successive equivalents of quinone.
Although 1,4-benzoquinone was the most effective (and also most soluble) of the quinones tested, ubiquinone-1 was also a fairly effective substrate, but due to its low solubility in water only kox/Kd, 2.88 × 105 M-1s-1 and kcat/Km, 5.81 × 104 M-1s-1 could be determined experimentally – these were approximately an order of magnitude slower than seen with 1,4-benzoquinone, kox/Kd, 2.60 × 106 M-1s-1. A similar situation has been seen in chromate reductase, a soluble quinone-reducing protein that best utilizes 1,4-benzoquinone over ubiquinone-1 (23). Both 1,4-naphthoquinone and 1,2 naphthoquinone-4-sulfonate were poorer oxidizing substrates for CODH, suggesting that the structurally related menaquinone is a less likely physiological substrate for the enzyme than ubiquinone. No semiquinone EPR signal was observed in EPR experiments monitoring catalytic enzyme turnover, and it appear that the principal oxidizing event is an effective two-electron process (supplemental figure 6). Finally, given the overall effectiveness of quinones as substrate, we conclude that one or another component of the quinone pool of O. carboxidovorans constitutes the proximal oxidizing substrate for CODH, and that cytochromes become reduced only subsequent to the introduction of reducing equivalents into the quinone pool.
Supplementary Material
Supplemental Figure 1, consisting of absorption spectra for oxidized and dithionite reduced cyt b561 in 50mM Hepes (pH 7.2) 0.2% Triton X-100 (supplemental figure 1)
Supplemental Figure 2, consisting of absorption spectra recorded at 1 s, 250 s and 500 s in the course of the reaction of anaerobic reduced CODH with anaerobic cyt b561 in 50mM Hepes (pH 7.2) 0.2% Triton X-100.
Supplemental Figure 3, consisting of a plot of steady-state catalytic velocity as a function of 1,4-benzoquinone concentration for CODH in 50mM Hepes (pH 7.2, 25°C, 1mM CO).
Supplemenatal Figure 4, consisting oftal figure 3), steady-state catalytic velocity as a function of ubiquinone-1 concentration (in H2O and D2O) for CODH in 50 mM Hepes (pH 7.2 or pD 7.6, 25°C, 1mM CO).
Supplemental Figure 5, consisting of an anaerobic titration of 3.5 μM CODH with reduced 1,4-benzoquinone in 50 mM Hepes (pH 7.2, 25° C).
Supplemental Figure 6, consisting of electron paramagnetic resonance spectra of oxidized CODH in 50 mM Hepes (pH 7.2) and enzyme that has been fully reduced with dithionite then re-oxidized with either 0.5 equivalent or 12 equivalents of ubiquinone-1, spectra were obtained at 150 K, 9.5 GHz microwave frequency, 10 milliwatt microwave power and 5 Gauss modulation amplitude.
ABBREVIATIONS USED
- CODH
CO dehydrogenase
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- XOR
xanthine oxidoreductase
Footnotes
This work was supported by the National Institute of Health Grant GM 075036 (to R.H.).
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hille R. The Mononuclear Molybdenum Enzymes. Chem Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 2.Meyer O, Schlegel HG. Oxidation of Carbon Monoxide in Cell Extracts of Pseudomonas carboxydovorans. Arch Microbiol. 1978;118:35–43. doi: 10.1007/BF00406071. [DOI] [PubMed] [Google Scholar]
- 3.Meyer O, Schlegel HG. Carbon Monoxide:Methylene Blue Oxidoreductase from Pseudomonas carboxydovorans. Bacteriol. 1980;141:78–80. doi: 10.1128/jb.141.1.74-80.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dobbek H, Gremer L, Meyer O, Huber R. Crystal structure and mechanism of CO dehydrogenase, a molybdo iron-sulfur flavoprotein containing S-selanylcysteine. Proc Natl Acad Sci USA. 1999;96:8884–888. doi: 10.1073/pnas.96.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobbek H, Gremer L, Kiefersauer R, Huber R, Meyer O. Catalysis at a dinuclear [CuSMo(AO)OH] cluster in a CO dehydrogenase resolved at 1.1-Å resolution. Proc Natl Acad Sci USA. 2002;99:15971–15976. doi: 10.1073/pnas.212640899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hänzelmann P, Dobbek H, Gremer L, Huber R, Meyer O. The Effect of Intracellular Molybdenum in Hydrogenophaga pseudoflava on the Crystallographic Structure of the Seleno-Molybdo-Iron-Sulfur Flavoenzyme Carbon Monoxide Dehydrogenase. J Mol Biol. 2000;301:1221–1235. doi: 10.1006/jmbi.2000.4023. [DOI] [PubMed] [Google Scholar]
- 7.Gremer L, Kellner S, Dobbek H, Huber R, Meyer O. Binding of Flavin Adenine Dinucleotide to Molybdenum-containing Carbon Monoxide Dehydrogenase from Oligotropha carboxidovorans. J Biol Chem. 2000;275:1864–1872. doi: 10.1074/jbc.275.3.1864. [DOI] [PubMed] [Google Scholar]
- 8.Kang BS, Kim YM. Cloning and Molecular Characterization of the Genes for Carbon Monoxide Dehydrogenase and Localization of Molybdopterin, Flavin Adenine Dinucleotide, and Iron-Sulfur Centers in the Enzyme of Hydrogenophaga pseudoflava. J Bacteriol. 1999;181:5581–5590. doi: 10.1128/jb.181.18.5581-5590.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schübel U, Kraut M, Morsdorf G, Meyer O. Molecular characterization of the gene cluster coxMSL encoding the molybdenum-containing carbon monoxide dehydrogenase of Oligotropha carboxidovorans. J Bacteriol. 1995;177:2197–2203. doi: 10.1128/jb.177.8.2197-2203.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santiago B, Schübel U, Egelseer C, Meyer O. Sequence analysis, characterization and CO-specific transcription of the cox gene cluster on the megaplasmid pHCG3 of Oligotropha carboxidovorans. Gene. 1999;236:115–124. doi: 10.1016/s0378-1119(99)00245-0. [DOI] [PubMed] [Google Scholar]
- 11.Gnida M, Ferner R, Gremer L, Meyer O, Meyer-Klaucke W. A Novel Binuclear [CuSMo] Cluster at the Active Site of Carbon Monoxide Dehydrogenase: Characterization by X-ray Absorption Spectroscopy. Biochemistry. 2003;42:222–230. doi: 10.1021/bi026514n. [DOI] [PubMed] [Google Scholar]
- 12.Ragsdale SW, Pierce E. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim Biophys Acta. 2008;1784:1873–1898. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cypionka H, Meyer O. Carbon Monoxide-Insensitive Respiratory Chain of Pseudomonas carboxydovorans. J Bacteriol. 1983;141:74–80. doi: 10.1128/jb.156.3.1178-1187.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer O, Gremer L, Ferner R, Ferner M, Dobbek H, Gnida M, Meyer-Klaucke W, Huber R. The role of Se, Mo and Fe in the structure and function of carbon monoxide dehydrogenase. Biol Chem. 2000;381:865–876. doi: 10.1515/BC.2000.108. [DOI] [PubMed] [Google Scholar]
- 15.Moersdorf G, Frunzke K, Gadkari D, Meyer O. Microbial growth on carbon monoxide. Biodegredation. 1992;3:61–82. [Google Scholar]
- 16.Kim YM, Hegeman GD. Electron Transport System of an Aerobic Carbon Monoxide-Oxidizing Bacterium. J Bacteriol. 1981;148:991–994. doi: 10.1128/jb.148.3.991-994.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer O, Schlegel HG. Reisolation of the carbon monoxide utilizing hydrogen bacterium Pseudomonas carboxydovorans (Kistner) comb. nov. Arch, Microbiol. 1978;118:35–43. doi: 10.1007/BF00406071. [DOI] [PubMed] [Google Scholar]
- 18.Zhang B, Hermann C, Hille R. Kinetic and Spectroscopic Studies of the Molybdenum-Copper CO Dehydrogenase from Oligotropha carboxidovorans. J Biol Chem. 2010;285:12571–12578. doi: 10.1074/jbc.M109.076851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Resch Dobbek H, Meyer O. Structural and functional reconstruction in situ of the [CuSMoO2] active site of carbon monoxide dehydrogenase from the carbon monoxide oxidizing eubacterium Oligotropha carboxidovorans. J Biol Inorg Chem. 2005;10:518–528. doi: 10.1007/s00775-005-0006-4. [DOI] [PubMed] [Google Scholar]
- 20.Albarran G, Schuler RH. Determination of the spectroscopic properties and chromatographic sensitivities of substituted quinones by hexachlorate(IV) oxidation of hydroquinone. Talanta. 2008;74:844–850. doi: 10.1016/j.talanta.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Chakraborty S, Massey V. Reaction of Reduced Flavins and Flavoproteins with Diphenyliodonium Chloride. J Biol Chem. 2002;277:41507–41516. doi: 10.1074/jbc.M205432200. [DOI] [PubMed] [Google Scholar]
- 22.Kita K, Vibat CRT, Meinhardt S, Guest JR, Gennis RB. One-step Purification from Escherichia coli of Complex II (Succinate:Ubiquinone Oxidoreductase) Associated with Succinate-reducible Cytochrome b556. J Biol Chem. 1989;264:2672–2677. [PubMed] [Google Scholar]
- 24.Gonzalez Claudio F, Ackerley David F, Lynch Susan V, Matin A. ChrR, a Soluble Quinone Reductase of Pseudomonas putida That Defends against H2O2. J Biol Chem. 2005;280:22590–22595. doi: 10.1074/jbc.M501654200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1, consisting of absorption spectra for oxidized and dithionite reduced cyt b561 in 50mM Hepes (pH 7.2) 0.2% Triton X-100 (supplemental figure 1)
Supplemental Figure 2, consisting of absorption spectra recorded at 1 s, 250 s and 500 s in the course of the reaction of anaerobic reduced CODH with anaerobic cyt b561 in 50mM Hepes (pH 7.2) 0.2% Triton X-100.
Supplemental Figure 3, consisting of a plot of steady-state catalytic velocity as a function of 1,4-benzoquinone concentration for CODH in 50mM Hepes (pH 7.2, 25°C, 1mM CO).
Supplemenatal Figure 4, consisting oftal figure 3), steady-state catalytic velocity as a function of ubiquinone-1 concentration (in H2O and D2O) for CODH in 50 mM Hepes (pH 7.2 or pD 7.6, 25°C, 1mM CO).
Supplemental Figure 5, consisting of an anaerobic titration of 3.5 μM CODH with reduced 1,4-benzoquinone in 50 mM Hepes (pH 7.2, 25° C).
Supplemental Figure 6, consisting of electron paramagnetic resonance spectra of oxidized CODH in 50 mM Hepes (pH 7.2) and enzyme that has been fully reduced with dithionite then re-oxidized with either 0.5 equivalent or 12 equivalents of ubiquinone-1, spectra were obtained at 150 K, 9.5 GHz microwave frequency, 10 milliwatt microwave power and 5 Gauss modulation amplitude.