

Abstract
Amino acid substitution matrices are central to protein-comparison methods. In most commonly used matrices, the substitution scores take a log-odds form, involving the ratio of “target” to “background” frequencies derived from large, carefully curated sets of protein alignments. However, such matrices often are used to compare protein sequences with amino acid compositions that differ markedly from the background frequencies used for the construction of the matrices. Of course, the target frequencies should be adjusted in such cases, but the lack of an appropriate way to do this has been a long-standing problem. This article shows that if one demands consistency between target and background frequencies, then a log-odds substitution matrix implies a unique set of target and background frequencies as well as a unique scale. Standard substitution matrices therefore are truly appropriate only for the comparison of proteins with standard amino acid composition. Accordingly, we present and evaluate a rationale for transforming the target frequencies implicit in a standard matrix to frequencies appropriate for a nonstandard context. This rationale yields asymmetric matrices for the comparison of proteins with divergent compositions. Earlier approaches are unable to deal with this case in a fully consistent manner. Composition-specific substitution matrix adjustment is shown to be of utility for comparing compositionally biased proteins, including those of organisms with nucleotide-biased, and therefore codon-biased, genomes or isochores.
Amino acid substitution matrices are a key component of protein-comparison methods, with the quality of sequence alignments assessed by scores that are the sum of substitution and gap scores. Such scores also provide a starting point for evolutionary distance estimates. It is desirable to produce alignments that reflect as accurately as possible the physicochemical correspondences and evolved mutational differences between amino acid sequences. For this purpose, optimal substitution scores have been developed, which best distinguish such “true alignments” of a given class from chance. However, such substitution scores, developed in a standard context, are widely used to compare the large proportion of proteins that have nonstandard compositions. In this article, we address the long-standing problem of how to restore consistency in such circumstances.
Although a wide variety of rationales have been used to construct amino acid substitution matrices, the great majority implicitly have the same underlying mathematical structure. At least in the context of ungapped local alignments, this structure defines the class of alignments for which any given matrix is optimal. Given a model in which amino acids occur by chance with “background frequencies” pi, any substitution matrix with negative expected score and at least one positive entry may be written in the “log-odds” form
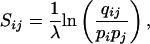 |
where the qij are positive “target frequencies” that sum to 1, and λ is a natural scale factor for the matrix. If the target qij reflect the frequencies with which the various amino acids are aligned within a given class of true alignments, then the scoring system is optimal for discriminating this class (1, 2). Notably, different amino acid substitution matrices are optimal for detecting different classes of alignment. For example, graded series of substitution matrices have been developed, with the target frequencies of each matrix tailored to a particular range of evolutionary divergence (3–10). Any such series implies a model of protein evolution, but current evolutionary theory provides no basis for calculating target frequencies a priori. Accordingly, methods have been developed to derive these target frequencies from large collections of alignments of homologous proteins. There is a degree of circularity in this, because these alignments themselves are generally constructed with the aid of a substitution matrix. The two most widely used series of matrices are based on alternative strategies for mitigating this circularity.
The classic PAM matrices (3, 4) were based on robustly accurate alignments of closely related sequences from which target frequencies for any desired evolutionary distance were estimated by extrapolation using a time-reversible Markov model. More recently, the data underpinning this model have been updated (5, 6), and the theoretical basis for deriving the model has been reworked (7–9). The strategy for the BLOSUM matrices (10) avoided such extrapolation by estimating target frequencies directly for different evolutionary distances by using the ungapped segments of multiple sequence alignments of protein families. Careful curatorial work has gone into the construction of the PAM and BLOSUM matrices, and these or related matrices are used by default in popular database search programs such as fasta (11) and blast (12, 13).
However, the important problem of compositional adjustment remains. The need for adjustment arises when the amino acid frequencies of the sequences being compared are significantly different from the standard background frequencies used to construct the matrices. Such nonstandard amino acid frequencies are not unusual, as with the large sets of “compositionally drifted” proteins encoded by AT- or GC-rich genomes or isochores (14–16) or numerous physicochemically specialized (e.g., hydrophobic or cysteine-rich) proteins. In these cases, naive use of standard substitution matrices may be inappropriate because, as shown below, an inherent inconsistency between target and background frequencies arises. Restoring consistency requires a rationale for the compositional adjustment of target frequencies and therefore of amino acid substitution scores.
The crux of this article is our demonstration, with a proof presented in the Appendix, that any log-odds substitution matrix implies a unique or canonical set of target and background frequencies. We then develop and evaluate a rationale for using the information implicit in any standard substitution matrix to derive variant matrices suitable for altered background frequencies, thus taking advantage of the extensive data analysis embodied in the PAM or BLOSUM series. Neither the PAM nor the BLOSUM approach to matrix construction is directly applicable to the comparison of sequences with differing compositions, whereas our method yields consistent, asymmetric matrices for such comparisons.
Valid Substitution Matrices Imply Canonical Background Frequencies
Although it is possible to specify any arbitrary substitution matrix, let us assume for the moment that we have constructed such a matrix explicitly as a log-odds matrix from a set of alignment data. Specifically, we start with a set of target frequency data qij for the amino acid pairs, consisting of positive numbers that sum to 1; we do not require these qij to be symmetric. For consistency, we define two sets of background frequencies pi and 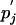 as the marginal sums of the qij:
as the marginal sums of the qij:
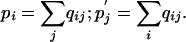 |
[1] |
The substitution matrix scores are then defined as
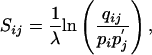 |
[2] |
where λ is an arbitrary positive scale factor. Such a matrix we will call valid in the context of the pi and 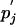 . Note that up to rounding errors, both the PAM and BLOSUM series of substitution matrices are valid by this definition in the context of their implicit background frequencies. Because the PAM and BLOSUM target frequencies qij are symmetric by construction, they imply a single set of background frequencies
. Note that up to rounding errors, both the PAM and BLOSUM series of substitution matrices are valid by this definition in the context of their implicit background frequencies. Because the PAM and BLOSUM target frequencies qij are symmetric by construction, they imply a single set of background frequencies 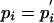 as well as symmetric scores sij = sji, but we will require no such symmetry. In practice, this more general case is readily accommodated by blast (12, 13) and various other database search implementations.
as well as symmetric scores sij = sji, but we will require no such symmetry. In practice, this more general case is readily accommodated by blast (12, 13) and various other database search implementations.
Although a substitution matrix is valid in the context of the background frequencies pi and 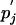 used for its derivation, it is often used to compare sequences characterized by different background frequencies Pi and
used for its derivation, it is often used to compare sequences characterized by different background frequencies Pi and 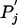 . As long as the expected score
. As long as the expected score 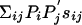 remains negative, the matrix sij still can always be written in the log-odds form
remains negative, the matrix sij still can always be written in the log-odds form 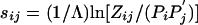 . In other words, in the new background-frequency context, sij is still a log-odds matrix, with a new set of target frequencies Zij and a new scale factor λ. However, it is no longer necessarily the case that Pi = ΣjZij and
. In other words, in the new background-frequency context, sij is still a log-odds matrix, with a new set of target frequencies Zij and a new scale factor λ. However, it is no longer necessarily the case that Pi = ΣjZij and 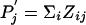 . Thus, although sij remains a log-odds matrix, it may no longer be valid in the new context.
. Thus, although sij remains a log-odds matrix, it may no longer be valid in the new context.
In the Appendix we show that, in fact, sij can be a valid log-odds matrix only in the unique context of the qij used for its construction with their implied background frequencies. Furthermore, given only a matrix that is valid in some context, its implicit scale factor λ, as well as its implicit target frequencies qij, with their implied background frequencies, may all be retrieved effectively and efficiently.
Given a matrix valid in some context, the procedure described in the Appendix allows one to express it in the form of Eqs. 1 and 2. Further, we have developed an efficient numerical approach, to be described elsewhere, for determining whether an arbitrary matrix can be valid. Of course, any matrix that is constructed explicitly as a log-odds matrix with consistent target and background frequencies is valid in the context of these frequencies.
A Strategy to Adapt Substitution Matrices to Noncanonical Background Frequencies
As shown above, there is an underlying inconsistency to using standard amino acid substitution matrices such as the PAM or BLOSUM series to compare proteins with substantially divergent background frequencies. Moreover, it is not feasible to develop new substitution matrices de novo for every new compositional context by reworking the original PAM or BLOSUM strategies based on many carefully curated alignments. Therefore, we have developed the following rationale for adapting any existing log-odds matrix to nonstandard contexts.
One way to formulate this problem is to suppose one is given a substitution matrix of the form of Eq. 2 and satisfying the consistency conditions of Eq. 1. A nonstandard context can be understood as the specification of new background amino acid frequencies Pi and 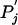 . We then seek a new set of target frequencies Qij that is as “close” to the original target frequencies qij as possible but that satisfies the consistency conditions
. We then seek a new set of target frequencies Qij that is as “close” to the original target frequencies qij as possible but that satisfies the consistency conditions
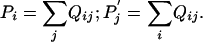 |
[3] |
To measure the idea of close, it is natural to use the relative entropy, or Kullback–Liebler distance, of the frequency distribution Qij from qij:
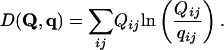 |
[4] |
The requirement that the Qij sum to 1 makes the space of possible target frequencies 399-dimensional. The consistency conditions (Eq. 3) impose 38 additional, independent conditions on the Qij, reducing the space to 361 dimensions. In the context of nucleic acid comparison, the space of consistent Qij is nine-dimensional. Using Lagrange multipliers, we have developed an efficient Newtonian procedure for finding the Qij that minimize D(Q, q) of Eq. 4. This procedure will be described in detail elsewhere.
If one chooses, one may place additional constraints on the Qij. For example, a major factor influencing the effectiveness of a substitution matrix is its relative entropy (2, 10). Therefore, it may be useful to control the implicit relative entropy H of the substitution matrix sought, thereby imposing the additional constraint
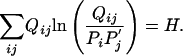 |
[5] |
One may wish H to equal the relative entropy of the original matrix in the context of the original background frequencies pi and 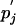 or, as below, in the context of the new background frequencies Pi and
or, as below, in the context of the new background frequencies Pi and 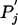 . By adding one more Lagrange multiplier to the optimization procedure, it is a simple matter to impose this extra constraint. Further study may suggest other ways to constrain the Qij or more biologically appropriate measures to optimize than that of Eq. 4.
. By adding one more Lagrange multiplier to the optimization procedure, it is a simple matter to impose this extra constraint. Further study may suggest other ways to constrain the Qij or more biologically appropriate measures to optimize than that of Eq. 4.
Comparison of Standard and Composition-Adjusted Substitution Matrices
To study the effects of adjusting substitution matrices for amino acid composition, we consider proteins from organisms with very biased AT- or GC-rich genomes. Many such organisms, including several important pathogens and parasites, show widespread biases in codon and amino acid usage, reflecting genome-wide or isochore-specific directional mutation pressures (14–16). The proteins of AT-rich organisms tend to have a greater background content of phenylalanine, leucine, isoleucine, asparagine, lysine, tyrosine, and methionine (FLINKYM), encoded by AU-rich codon sets, and a lesser content of proline, arginine, alanine, tryptophan, and glycine (PRAWG), encoded by GC-rich codon sets. The proteins of GC-rich organisms show the reverse bias.
For this study, we constructed three test sets of sequence pairs for which “orthology” provided extrinsic evidence for alignment quality and a fourth test set supported by three-dimensional structural evidence (Tables 2–5, which are published as supporting information on the PNAS web site, www.pnas.org). The COG (clusters of orthologous groups) relation of three-lineage reciprocal best match (17) was used to define the “ortholog-pair” sets, which were from: (i) Clostridium tetani (AT-rich) and Mycobacterium tuberculosis (GC-rich), with contrasting strong biases; (ii) Bacillus subtilis and Lactococcus lactis, both with relatively unbiased genomes and average amino acid frequencies close to those underpinning BLOSUM-62; and (iii) M. tuberculosis and Streptomyces coelicolor with strong biases in the same, GC-rich direction. We included only sequence pairs that had a BLOSUM-62 alignment score <100 bits and only one pair among mutually homologous orthologs. As a negative control, for each pair of test organisms, we compared all test sequences from one organism with those from the other, excluding the orthologous pairs.
Comparing sequences from biased organisms presents a choice: One may adjust a substitution matrix for amino acid frequencies calculated from the entire proteome of each organism, or one may rely on the frequencies manifest in the actual sequence pair being aligned. This latter approach is attractive, because it requires no data extrinsic to the two sequences themselves, and because it accommodates any isochore or protein family-specific biases implicit in these sequences. One can mitigate potential inaccuracies caused by small sample size by adding “pseudocounts” to the amino acid counts from the actual proteins, as in the examples below.
We compared the ortholog pairs and their negative controls by using a scaled version of the standard BLOSUM-62 matrix (a) and composition-adjusted BLOSUM-62 matrices based on background frequencies from the whole organisms (b) and the actual pair of sequences compared (c). For both b and c, the adjusted matrix was constrained to have relative entropy, in the context of the new background frequencies, equal to that of BLOSUM-62 in this context (Eq. 5). This controls for the possibility that improved performance may be ascribable merely to more appropriate relative entropy.
The results showed enhanced performance of the composition-adjusted matrices vis a vis BLOSUM-62, manifest both as increases in bit score and statistical significance (Table 1) and as improved alignment length and quality (Fig. 1 and Table 5). Adjusting background frequencies for organism proteome frequencies (the rows denoted “Organism” in Table 1, column 5) yielded improvements in most cases, and adjusting to conform to the actual sequence pairs gave even better results (rows denoted “Sequence” highlighted in bold, Table 1). For all three ortholog-pair test sets, the median increase in bit score was >2 bits, corresponding to a >4-fold increase in statistical significance, with 85–100% of the cases showing improvement. Of the 74 alignments from organisms with skewed compositions, the statistical significance improved by a factor of >10 for 26 while worsening by a similar factor for only a single alignment (Table 1, right-hand column). Moreover, for the organisms with near-standard compositions, substantial improvements were seen often enough that our method may prove to be of use for general-purpose database searches.
Table 1. Performance of composition-adjusted substitution matrices.
| Organisms compared
|
No. of sequence pairs
|
Mean BLOSUM-62 bit score*
|
Background frequencies specified
|
Median change in bit score* with respect to BLOSUM-62
|
Cases improved (%)
|
Cases (%) with statistical significance improved/worsened by a factor >10†
|
||
|---|---|---|---|---|---|---|---|---|
| Sequence pairs | Absolute | Relative (%) | ||||||
| Related | C. tetani and M. tuberculosis | 40 | 68.3 | Organism | +1.6 | +2.7 | 58 | 20/8 |
| Sequence‡ | +2.3 | +3.3 | 85 | 38/3 | ||||
| B. subtilis and L. lactis | 37 | 59.8 | Organism | +1.1 | +1.8 | 84 | 16/3 | |
| Sequence‡ | +2.1 | +3.6 | 95 | 11/3 | ||||
| M. tuberculosis and S. coelicolor | 34 | 58.6 | Organism | +1.4 | +2.6 | 76 | 24/3 | |
| Sequence‡ | +2.7 | +4.1 | 100 | 32/0 | ||||
| Unrelated (negative control) | C. tetani and M. tuberculosis | 1,560 | 16.7 | Organism | -0.02 | -0.1 | 49 | 0.4/0.1 |
| Sequence‡ | -0.05 | -0.3 | 47 | 0.6/0.4 | ||||
| B. subtilis and L. lactis | 1,332 | 15.7 | Organism | +0.00 | +0.0 | 50 | 0.0/0.0 | |
| Sequence‡ | +0.04 | +0.3 | 52 | 0.2/0.4 | ||||
| M. tuberculosis and S. coelicolor | 1,122 | 16.4 | Organism | +0.05 | +0.3 | 53 | 0.0/0.1 | |
| Sequence‡ | +0.06 | +0.4 | 53 | 0.6/0.2 | ||||
| Structural | Various | 32 | 50.4 | Sequence‡ | +1.3 | +3.2 | 72 | 22/0 |
Bit scores for all comparisons were calculated by using composition-based statistics (19), and experimentally determined gapped statistical parameters (18, 19), as is now standard in blast (12, 13). All matrices were scaled to have ungapped λ = 0.00635 and used in conjunction with gap costs of -550 -50k for a gap of length k.
Equivalent to a change of >3.322 bits.
Twenty pseudocounts proportional to the amino acid frequencies implicit in BLOSUM-62 were added to the actual amino counts from the proteins compared.
Fig. 1.

Example of an alignment extension yielded by compositional adjustment of the scoring system. The sequences compared are P. falciparum putative asparagine synthase (NCBI gi 16805184) (top lines) and M. tuberculosis PurF protein (NCBI gi 15607948) (bottom lines). In the central lines, aligned identical residues are echoed, and aligned residues with positive substitution score are indicated by + symbols. (a) The alignment yielded by a scaled version of the standard BLOSUM-62 substitution matrix (see * footnote in Table 1). The alignment has a normalized score of 29.7 bits. (b) The alignment yielded by a composition-adjusted matrix derived from BLOSUM-62 (see * and ‡ footnotes in Table 1). The normalized score of the alignment is 31.8 bits. The alignment in b corresponds very closely to the three-dimensional structural superposition of the entire domain fold (NCBI CDD 9909, COG 0034) that is shared between the PurF and asparagine synthase families. Secondary structure elements were assigned by using the known crystal structures of E. coli asparagine synthetase B (PDB ID 1CT9 chain A) and B. subtilis PurF protein (PDB ID 1GPH chain 3). β-strands (straight bars) and α-helices (zig-zags) are indicated above and below their respective homologous sequences.
To assess alignment length and quality, we used the test set of protein pairs for which three-dimensional structural data provided an objective standard. At least one of each such “structural pair” of sequences was chosen from a strongly biased organism, and many of these pairs represent the “twilight zone” of borderline alignment statistical significance. As for the ortholog pairs, the composition-adjusted matrices gave improvements in bit score and statistical significance (Table 1, bottom row). Moreover, 13 of the 32 cases (41%) showed substantial alignment extensions compared with the standard BLOSUM-62 alignments and in 6 cases (19%) by >50 amino acids (Table 5). These extensions were judged by inspection to be generally compatible with the three-dimensional structural superpositions inferred for the protein pairs. Fig. 1 shows an example of such an extended alignment and its consistency with the structural evidence for the AT-biased Plasmodium falciparum asparagine synthase sequence aligned with the GC-biased M. tuberculosis PurF protein. The normalized scores (18, 19) of the alignments yielded by unadjusted and adjusted BLOSUM-62 matrices were 29.7 and 31.8 bits, respectively. This 2.1-bit change is equivalent to an increase in statistical significance of a factor of >4 for this twilight-zone example.
In reference to the example in Fig. 1, Tables 6–9, which are published as supporting information on the PNAS web site, provide the amino acid frequencies of the sequences compared, the scaled original BLOSUM-62 matrix and composition-adjusted matrix used, and the differences between these two matrices. Notable changes include decreased scores for most aligned pairs of residues involving amino acids that are biasedly rare in one of the proteins and increased scores for pairs that include biasedly abundant amino acids. One case is alanine, which comprises ≈5% of the P. falciparum protein and ≈14% of the M. tuberculosis protein, compared with a background frequency of ≈7% for BLOSUM-62. This is one factor in the increased length and score of the optimal alignment of Fig. 1b, which contains 6 additional substituted alanines from the P. falciparum protein but 24 from the M. tuberculosis protein, compared with the alignment of Fig. 1a.
Discussion and Conclusion
We have shown that log-odds substitution matrices are valid, in the sense of having consistent target and background frequencies, only in the unique context of the background frequencies implicit in the data used for their construction. Consequently, standard amino acid substitution matrices are not appropriate for the comparison of proteins or protein domains with nonstandard amino acid composition. We have developed one rationale for transforming the target frequency data implicit in standard substitution matrices for application to nonstandard compositional contexts. This transformation can be accomplished efficiently (in a small fraction of a second on standard workstations) by using a multidimensional Newtonian optimization procedure.
We evaluated the performance of the resulting compositionally adjusted matrices by using test sets of sequence pairs with low-scoring alignments, including many cases with borderline statistical significance. For all the test sets, context-specific adjusted matrices showed improved performance in detecting biologically appropriate alignments of biased sequences, consistent with COG orthology relationships or structural evidence. We also found (data not shown) that adjusted matrices gave generally enhanced bit scores for the less demanding cases of more closely related ortholog pairs, with BLOSUM-62 alignment scores of 100–2,000 bits. In contrast, for the negative controls of unrelated sequence pairs (Table 1), the unadjusted and adjusted BLOSUM-62 matrices showed only small unsystematic differences in alignment bit scores, as expected from the theory of normalized scoring systems for random sequence alignment (1, 20). Taken together, these results demonstrate the substantially enhanced power of compositionally adjusted substitution scores to discriminate biological alignments from chance.
Other efforts have been made to improve the sensitivity of sequence alignment by constructing specialized substitution matrices specific for particular protein classes: notable examples are the PHAT (21) and SLIM (22) matrices, which were derived from curated collections of transmembrane proteins. Our strategy differs from these in two important respects. First, it generates asymmetric matrices that maintain consistency between the background and target frequencies. Second, it requires as input only the pair of sequences being compared and a valid general-purpose substitution matrix. Our method avoids extensive curatorial work with collections of compositionally biased proteins and is readily implementable in a sequence-comparison procedure. We note that matrix-construction strategies starting from curated alignments, as for the PAM and BLOSUM series, cannot in principle yield valid asymmetric target frequencies and substitution scores. This is because the initial aligned sequences are treated symmetrically, with no justifiable distinction between “query” and “subject.” Moreover, a time-reversible Markov model or its variants cannot generate asymmetric target frequencies. Indeed, before our treatment in this article, we have not found a systematic way to construct asymmetric log-odds matrices that maintain consistency between background and target frequencies.
Amino acid content bias can reflect both directional mutation pressures at the genomic level and constraints specific to classes of proteins. Our data show that compositional adjustment of substitution matrices is beneficial in both cases. Whereas the average proteome amino acid composition of such organisms as B. subtilis, L. lactis, and Homo sapiens is very close to that implied by BLOSUM-62, the different protein families found in these organisms show a wide distribution of distances from this BLOSUM-62 standard. Indeed, these compositional distances, measured by relative entropy or other metrics, can form part of a heuristic to determine whether compositional adjustment of matrices would likely be advantageous for a given sequence pair. Other simple heuristics can be readily applied to enable domain-specific adjustment of scoring matrices for cases of multidomain sequences with internal compositional heterogeneity. Such additional procedures, and the systematic application of compositional adjustment to the comparison of proteins from organisms with nucleotide-biased genomes, and to general-purpose database searching, will be described more extensively elsewhere.
Supplementary Material
Acknowledgments
We are particularly indebted to J. L. Spouge for valuable suggestions for the mathematical development and numerous stimulating discussions. Y.-K.Y. acknowledges support from National Science Foundation Grant DMR-0110903 during the early stage of this work. He also thanks T. P. Doerr for technical support.
Appendix: The Scale and Background and Target Frequencies of a Valid Substitution Matrix
Here we will first explain how to extract the scale λ and target and background frequencies implied by a valid substitution matrix and then prove that these associated numbers are in fact unique.
Extracting the Scale and Background and Target Frequencies. From the definition of a valid substitution matrix (Eq. 2), we have 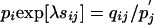 , and with (Eq. 1) we obtain
, and with (Eq. 1) we obtain
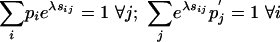 |
[6] |
together with the constraints Σi pi = 1, 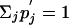 and pi>0 and
and pi>0 and 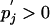 for all i and j.
for all i and j.
To extract an unknown λ from a valid substitution matrix with scores sij, we proceed as follows. Define a matrix M(τ) depending on the parameter τ, with matrix element Mij(τ) given by Mij(τ) = exp[τsij]. The conditions (Eq. 6) then lead to
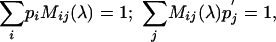 |
[7] |
where the first equation in Eqs. 7 can be viewed as multiplying the matrix M(λ) by a row vector {pi} from the left and the second equation in Eqs. 7 as multiplying the matrix M(λ) by a column vector 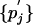 from the right. Let matrix Y(λ) be the inverse of M(λ). We then have
from the right. Let matrix Y(λ) be the inverse of M(λ). We then have
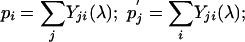 |
[8] |
and the condition 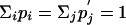 implies simply that
implies simply that
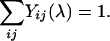 |
[9] |
One can easily use numerical tools to invert the matrix M(τ) and vary the parameter τ until the condition (Eq. 9) is fulfilled. Once λ is found, one obtains pi and 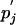 by Eq. 8, and qij is then
by Eq. 8, and qij is then 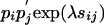 . As shown below, any sensible solution, i.e., one with positive pi and
. As shown below, any sensible solution, i.e., one with positive pi and 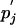 , must be unique.
, must be unique.
Uniqueness of the Background and Target Frequencies. Can there exist more than one set of target frequencies corresponding to a valid substitution matrix? The answer is no: Every valid matrix implies a unique set of target frequencies.
We will use Hölder's inequality to prove this uniqueness. Hölder's inequality, in the form we need, states (23): Let r > 1, s > 1, and 1/r + 1/s = 1, and assume {an} and {bn} are nonnegative numbers. We then have
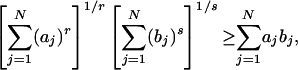 |
[10] |
and equality holds only when
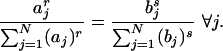 |
[11] |
Given a set of scores sij, assume that qij and Qij are two distinct corresponding sets of target frequencies. Without loss of generality, we assume the qij correspond to scale parameter λ = 1, and the Qij correspond to a scale parameter 1/x < 1. That is to say,
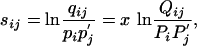 |
[12] |
where pi = Σj qij, 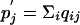 , Pi =Σ j Qij, and
, Pi =Σ j Qij, and 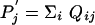 Consequently, we have
Consequently, we have
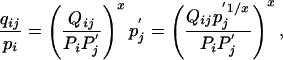 |
[13] |
which implies
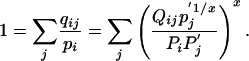 |
[14] |
Multiplying Eq. 14 by the identity
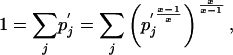 |
we obtain
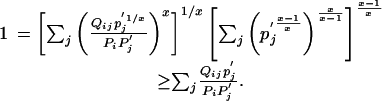 |
[15] |
Equality holds only when
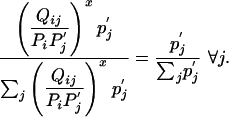 |
[16] |
By Eq. 14 and the definition of probability, both denominators in the above equation are equal to 1, so the condition for equality to hold, excluding 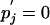 , in Eq. 15 becomes
, in Eq. 15 becomes
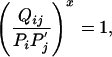 |
which, after excluding 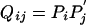 , can only be true if x = 0. Because x > 1 by assumption, we know that equality can never be reached. Eq. 15 therefore leads to
, can only be true if x = 0. Because x > 1 by assumption, we know that equality can never be reached. Eq. 15 therefore leads to
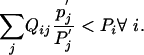 |
[17] |
We now show that Eq. 17 cannot be true. Using 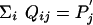 ,
, 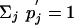 , and Σi Pi = 1, we find the contradictory result, 1 < 1, after summing over i on both the left-hand side and right-hand side of Eq. 17. We therefore have proved that a scoring system can never have more than one valid set of target and background frequencies.
, and Σi Pi = 1, we find the contradictory result, 1 < 1, after summing over i on both the left-hand side and right-hand side of Eq. 17. We therefore have proved that a scoring system can never have more than one valid set of target and background frequencies.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Karlin, S. & Altschul, S. F. (1990) Proc. Natl. Acad. Sci. USA 87, 2264–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F. (1991) J. Mol. Biol. 219, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dayhoff, M. O., Schwartz, R. M. & Orcutt, B. C. (1978) in Atlas of Protein Sequence and Structure, ed. Dayhoff, M. O. (National Biomedical Research Foundation, Washington, DC), Vol. 5, Suppl. 3, pp. 345–352. [Google Scholar]
- 4.Schwartz, R. M. & Dayhoff, M. O. (1978) in Atlas of Protein Sequence and Structure, ed. Dayhoff, M. O. (National Biomedical Research Foundation, Washington, DC), Vol. 5, Suppl. 3, pp. 353–358. [Google Scholar]
- 5.Gonnet, G. H., Cohen, M. A. & Benner, S. A. (1992) Science 256, 1443–1445. [DOI] [PubMed] [Google Scholar]
- 6.Jones, D. T., Taylor, W. R. & Thornton, J. M. (1992) Comput. Appl. Biosci. 8, 275–282. [DOI] [PubMed] [Google Scholar]
- 7.Benner, S. A., Cohen, M. A. & Gonnet, G. H. (1994) Protein Eng. 7, 1323–1332. [DOI] [PubMed] [Google Scholar]
- 8.Muller, T. & Vingron, M. (2000) J. Comput. Biol. 7, 761–776. [DOI] [PubMed] [Google Scholar]
- 9.Muller, T., Spang, R. & Vingron, M. (2002) Mol. Biol. Evol. 19, 8–13. [DOI] [PubMed] [Google Scholar]
- 10.Henikoff, S. & Henikoff, J. G. (1992) Proc. Natl. Acad. Sci. USA 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pearson, W. R. & Lipman, D. J. (1988) Proc. Natl. Acad. Sci. USA 85, 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 13.Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sueoka, N. (1988) Proc. Natl. Acad. Sci. USA 85, 2653–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan, H. & Wootton, J. C. (2000) Comput. Chem. 24, 71–94. [DOI] [PubMed] [Google Scholar]
- 16.Knight, R. D., Freeland, S. J. & Landweber, L. F. (2001) Genome Biol. 2, research0010.1–0010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tatusov, R. L., Koonin, E. V. & Lipman, D. J. (1997) Science 278, 631–637. [DOI] [PubMed] [Google Scholar]
- 18.Altschul, S. F., Bundschuh, R., Olsen, R. & Hwa, T. (2001) Nucleic Acids Res. 29, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schäffer, A. A., Aravind, L., Madden, T. L., Shavirin, S., Spouge, J. L., Wolf, Y. I., Koonin, E. V. & Altschul, S. F. (2001) Nucleic Acids Res. 29, 2994–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altschul, S. F. (1993) J. Mol. Evol. 36, 290–300. [DOI] [PubMed] [Google Scholar]
- 21.Ng, P. C., Henikoff, J. G. & Henikoff, S. (2000) Bioinformatics 16, 760–766. [DOI] [PubMed] [Google Scholar]
- 22.Muller, T., Rahmann, S. & Rehmsmeier, M. (2001) Bioinformatics 17, Suppl. 1, S182–S189. [DOI] [PubMed] [Google Scholar]
- 23.Gradshteyn, I. S. & Ryzhik, I. M. (1994) Tables of Integrals, Series, and Products (Academic, San Diego), 5th Ed., pp. 1125.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.