

The combination in advances in MALDI-TOF MS instrumentation(1) and serum sample preparation techniques,(2-4) has lead to the emergence of MALDI serum protein expression profiling as a promising tool for biomarker discovery.(5) However, three factors still pose significant challenges for MALDI profiling of serum: the limited mass window of MALDI, serum protein complexity,(6) and analytical reproducibility.(7, 8) MALDI has a small mass preference due to the ionization efficiency of intact protein molecules and the focusing of flight of ions towards the detector. In linear mode, TOF analyzers have limited sensitivity for masses above 20 kDa. To address this, we previously reported the enhancement of MALDI broad mass range detection of protein signals.(9) Spanning a 100 kDa mass range, we improved the sensitivity of detection by an order of magnitude through the combined optimization of sample preparation, instrument parameters and data processing procedures.
The complexity of the blood proteome is very high, with protein concentrations differing by up to ten orders of magnitude.(10) This large dynamic range exceeds current proteomic analytical capabilities; thus analysis of easily prepared subproteomes of serum or plasma is essential. Here, we shift our focus back to the enhancement of MALDI analysis in the low mass regime. The low molecular weight (LMW) subcomponent of serum promises to be a rich source of undiscovered biomarkers, as biological processes give rise to a plethora of proteolytic protein fragments.(11) Currently, there is no consensus on what constitutes the mass limits of this derivative proteome (also termed peptidome). However, “<15 kDa” is often cited in the literature based on a serum MALDI study using 20 MWCO filters.(12)
To date, small native protein/peptide mass measurements have been mainly conducted in reflectron mode (< 4 kDa),(13) and linear mode up to 10 kDa.(14) However, no systematic comparison has been described for a combination of preparation steps: spotting, filtering and matrix choice, to optimize the performance. A rigorous reproducibility study for different preparation strategies is likewise missing, which prevents extension of the technique to clinical proteomics applications. The purpose of this work was to provide such a systematic comparison, using rigorous performance metrics to optimize sample preparation for LMW serum proteome profiling by MALDI-TOF MS analysis up to 20 kDa. We explored a combination of MALDI sample preparation and spotting methods. Procedures that gave the best results included: 1) MALDI spotting with a thin layer technique using sinapinic acid on a ground steel plate, and 2) centrifugal ultrafiltration with 50000 MWCO filters in the presence of 2% TFA, followed by desalting with C3 magnetic beads. Ultrafiltration has been utilized previously to improve MALDI profiling of serum.(2, 13, 15-17) However, our approach extensively improves MALDI peak intensities of higher MW peptides and small proteins, thus increasing biomarker coverage in the 3-20 kDa range. Reproducibility studies of a protein standard and serum samples gave excellent results, all of which suggests that this procedure can be a useful tool for proteome profiling of the LMW fraction of serum.
Experimental Procedures
All chemicals and solvents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Protein standard 1 (PS1), MALDI matrices and targets, and ClinProt MB-C3 magnetic beads (C3) were obtained from Bruker Daltonics (Leipzig, Germany). Amicon-4 and Amicon-0.5 Ultra centrifugal filter devices were obtained from Millipore (Billerica, MA). A quality control (QC) serum sample was prepared by pooling human serum from a controlled normal group as previously described. (18) Six different MALDI target preparations and 23 different serum fractionations were prepared to compare MALDI profiling in the LMW range (3-20 kDa). Details are in following section.
Mass spectra were acquired in linear and reflectron positive ion mode using an Ultraflex III® MALDI-TOF/TOF mass spectrometer (Bruker Daltonics). The instrument is equipped with a smartbeam™ laser and acquisition laser power was optimized using the PS1 calibration mixture before the collection of sample data. Instrument settings, optimized for mass range m/z 0-20000, were as follows: ion source 1 = 25.0 kV, ion source 2 = 23.7 kV, lens voltage = 6.0 kV, pulsed ion extraction time = 200 ns, matrix suppression mass cut off = 1500, ADC offset = 50, pre-amplifier filter bandwidth = high, digitizer sampling frequency = 500 MHz. All spectra were generated by averaging 1000 shots from 10 non-overlapping positions (100 shots/position).
Results and Discussion
MALDI spotting
Clinical profiling of body fluids by MALDI-MS is highly influenced by the choice of MALDI spotting and sample preparation techniques. Our goal was to determine an optimal method for MALDI profiling of LMW serum protein/peptide ions, based on analytical performance measurements of resolution, sensitivity, reproducibility and broad mass range coverage. For sample/matrix crystal preparation, α-cyano-4-hydroxycinnamic acid (CHCA), and sinapinic acid (SA) are most commonly used. Resolution and sensitivity comparisons of these two matrices are well documented in the literature; however, reproducibility studies are limited. Because, reproducibility is one of the most critical elements in profiling studies, our main focus was to optimize reproducibility over the LMW serum mass range (< 20kDa), without undermining other performance factors. We evaluated a variety of matrices, sample/matrix ratios, ionic liquid additives, target plates and spotting methods. We narrowed down the principal comparisons to CHCA vs. SA matrix (with no additives); Bruker AnchorChip (600 μm) vs. ground steel targets; and dried droplet vs. thin layer spotting methods. Six MALDI spotting methods were compared in detail (target – matrix – spotting technique):
Anchorchip – CHCA – dried droplet
Anchorchip – SA – dried droplet
Steel – CHCA – dried droplet
Steel – SA – dried droplet
Steel – CHCA – thin layer
Steel – SA – thin layer
For the dried droplet method (DD), matrices were dissolved in 50% ACN, 0.1% TFA, (5 mg/ml CHCA, or saturated SA). Samples were mixed 1:5 with matrix, then 1 μl was spotted onto an AnchorChip or ground steel plate. For our modified thin layer (TL) method, 0.3 μl of matrix solution (CHCA saturated in MeOH, or SA saturated in EtOH) was deposited onto the ground steel target as a seed layer. Protein sample is mixed with matrix solution as above, followed by 1 μl deposited onto the seed layer.
Protein Standard 1 (PS1) contains a good spread of ion signals in the range of 3-20 kDa and was used to compare the different MALDI spotting methods. The PS1 mixture contains insulin (5734 Da), ubiquitin (8565 Da), cytochrome C (12361 Da), and myoglobin (16952 Da). PS1 was spotted 10 times for each preparation. A quick visual inspection showed the TL method to be far superior to the more common DD method in the 3-20 kD range. An advantage of the TL method is the very homogenous size of microcrystals.(19) On deposition of the seed layer, the solution quickly spreads and evaporates almost instantaneously (< 2 sec) leaving an ultra-thin uniform coating. The method generally yields higher resolution spectra and the detection limit is increased compared to the DD method.
Of the six preparations, three were then statistically compared: CHCA – DD on AnchorChip, CHCA – TL on steel plate, and SA – TL on steel plate. Signal processing was performed on raw PS1 spectra to enhance signal-to-noise. Processing algorithms, described by Malyarenko, et al(20) included analytical-model-baseline subtraction, integrative down-sampling optimal linear filtering, pedestal removal, peak detection and alignment. Noise level spectra were estimated by finding the standard deviation of the noise in the down-sampled spectra and adjusting for the expected effect of filtering. Poisson dependence on baseline amplitude was included to account for the observed dark current amplification for early TOF. Integrated down-sampled signal intensities were compared to exclude the influence of the originally different peak-widths. Implementation details and Matlab toolbox for signal processing are available from [matlabcentral/fileexchange/24469]. Metrics for 9 m/z peaks common to each of the three preparations included: measured ion intensity, estimated noise, signal-to-noise (SNR), normalized intensity, normalized SNR and %CV.
Figure 1 shows the mean raw spectra for each of the three preparations. The top spectrum (SA spotted with the TL method on ground steel) proved to be of highest quality based on SNR and reproducibility. Compared to CHCA on Anchorchip, SA on steel produced 3.5X greater average signal intensity and 10X greater average SNR (based on the average values for all peaks and replicates for each sample preparation); while thin layer methods on steel produced similar ion signals for both CHCA and SA. Noise was greatly reduced with SA (1/3.5) giving an overall 3X increase in SNR for SA over CHCA on steel. Comparing individual ion signals in Figure 1 shows that 5 of the 9 ions have higher signals for CHCA on steel than for SA on steel. However, only two of these ions (ubiquitin 3+ and 2+), have higher SNR (with only 1.5X increase).
Figure 1.
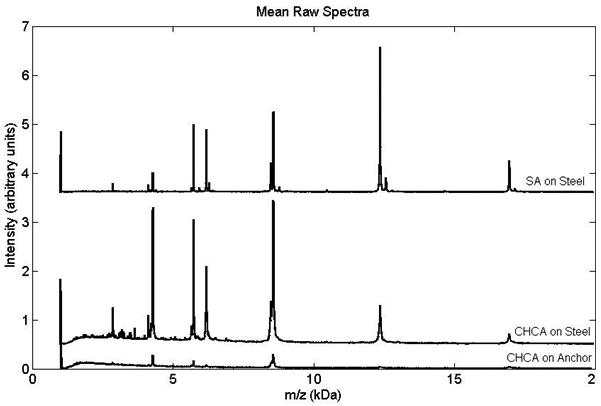
Averaged raw MALDI-TOF spectra (offset for clarity) of PS1 standard prepared by different spotting methods: (top) SA thin layer on steel (10 replicates), (middle) CHCA thin layer on steel (10 replicates), (bottom) CHCA dried droplet on AnchorChip (9 replicates).
In the literature, utilization of thin layer preps have focused generally on increasing signal and reproducibility of peptides (< m/z 4000) using CHCA(21, 22) or higher mass proteins (> m/z 10000) using SA.(23, 24) However, our interest is in the middle range – m/z 3000-20000 – where LMW serum protein peaks are commonly found. As Figure 1 demonstrates, we determined SA is a better matrix over CHCA in our modified thin layer method, due to the reduced noise, higher SNR and resolution that SA offers in the 3-20 kD range.
Coefficients of variation (%CV) were calculated for experimental replicates for each m/z peak based on normalized intensity and normalized SNR. Table 1 presents the %CV for each peak and for each surface preparation. The average %CVs for a given preparation are shown at the bottom of the table. Reproducibility was comparable whether using normalized intensity or SNR. All preps gave good within day reproducibility, however the thin layer prep with SA on steel was slightly better (%CV = 9.5). Our previous studies for reproducibility of calibration spectra (29) indicated that under carefully preserved instrumental settings the %CV does not exceed 10% in the course of several months. This confirmed that the major contribution to the variability of MS signals comes from the sample preparation step rather than the instrument or time of the study.
Table 1.
%CVs for PS1 ions (10 replicate average) for the studied MALDI preparations
| Peak | Matrix/Surface | ||||||
|---|---|---|---|---|---|---|---|
| m/z ( Da) |
Species | CHCA on Anchor dried droplet |
CHCA on Steel thin layer |
SA on Steel thin layer |
|||
| Intensity | SNR | Intensity | SNR | Intensity | SNR | ||
| 2855 | Ub+3 | 17.0 | 16.4 | 7.8 | 6.5 | 17.3 | 18.5 |
| 2867 | In+2 | 16.3 | 16.9 | 9.9 | 11.7 | 13.4 | 11.4 |
| 4283 | Ub+2 | 6.0 | 5.9 | 5.2 | 5.2 | 9.1 | 9.7 |
| 5734 | In+1 | 25.3 | 25.6 | 7.7 | 8.2 | 7.8 | 7.0 |
| 6181 | Cyto+2 | 17.7 | 17.5 | 8.1 | 8.3 | 5.7 | 5.9 |
| 8476 | Myo+2 | 7.5 | 7.2 | 17.1 | 16.8 | 9.6 | 9.5 |
| 8565 | Ub+1 | 5.6 | 5.7 | 5.3 | 5.7 | 6.6 | 7.1 |
| 12360 | Cyto+1 | 9.0 | 8.3 | 7.5 | 7.5 | 3.7 | 3.8 |
| 16952 | Myo+1 | 8.0 | 7.1 | 19.1 | 18.0 | 12.3 | 12.0 |
|
| |||||||
| Ave %CV | 12.5 | 12.3 | 9.8 | 9.8 | 9.5 | 9.4 | |
Ultrafiltration
Human QC serum samples(25) were used with Millipore Amicon ultrafiltration devices to optimize MALDI profiling of the LMW serum fraction (30). Devices were used according to the manufacturer’s recommendations. Twenty-three comparisons were made depending on: 1) MWCO – 3, 10, 30, 50, 100 K, 2) collecting filtrate or retentate, 3) filter size – 0.5 ml or 4 ml, 4) double filter combinations, 5) denaturation and 5) MB-C3 purification. A list of these combination preparations can be found in Supplementary Material 1. All samples were MALDI spotted using the thin layer method with SA on ground steel.
For the MWCO, we found that serum filtrate from the 50K filter gave the best profile in the 3-20 kD range. This was enhanced by pre-filtration protein denaturation with 2% TFA, and post-filtration C3 magnetic bead desalting. Other filtrations which gave complimentary profiles (but less overall coverage in the 3-20 kD range) included the 10K retentate and combination 50K filtrate followed by 3K retentate (data not shown). The filter size did not matter unless double filtrations were performed (0.5 ml filter for the second filtration).
Figure 2 shows MALDI spectra of processed serum. Figure 2a is a typical profile of C3-magnetic bead processed QC serum. Figure 2b is 50K filtered QC serum followed by C3 bead cleanup. In comparison, noise is greatly reduced for the filtered sample which enhanced peaks in the m/z 2000-6000 range. Adding upfront 2% TFA denaturation (Figure 2c), noise is further reduced with enhanced peaks in the m/z 7000-10000 range.
Figure 2.
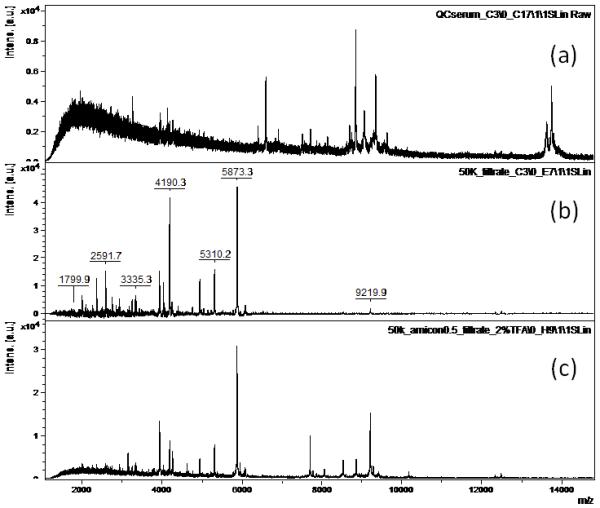
Raw MALDI-TOF spectra of processed QC serum: (a) C3 magnetic bead fractionation, (b) 50K ultrafiltration + C3 purification, (c) 2% TFA denaturation + 50K ultrafiltration + C3 purification.
The reproducibility of the two-step serum preparation was determined based on variation in peak intensities and normalized SNR. The systematic reproducibility study was performed only for the best preparation method (Figure 2c), since other methods did not provide the desired signal gain over the full LMW range. Eight separate aliquots of QC serum (8 biological replicates) underwent 50 kDa centrifugal ultrafiltration and C3 magnetic bead purification. Prepared aliquots were spotted 10 times (10 experimental replicates) on a MALDI target. Raw QC spectra were processed using the same procedures (with slightly different parameters) as the PS1 spectra. Sixteen m/z peaks were selected for comparison. From the data, we found that intensity CVs were not correlated to the m/z value or mean intensity value of a peak. In other words, the variation of a peak’s intensity is independent of the absolute value of m/z and intensity. This has been observed previously in MALDI profiling studies.(16, 26)
Table 2 presents %CV values for each peak in each aliquot based on normalized SNR. The ranges for %CV for each peak across all aliquots are given in the second column from the right. The column to the far right contains between sample %CV values calculated using data from all replicates, both experimental and biological (74 replicates). The average within and between sample %CVs are summarized at the bottom of the table. No single aliquot yielded the highest or lowest %CV for all peaks. There was no relationship found between %CV and average intensity, noise or SNR.
Table 2.
Within and between samples %CVs based on replicate spotting of 8 aliquots of QC serum processed by ultrafiltration. (%CV values calculated for 16 peaks normalized by SNR)
| Peak m/z (Da) |
QC Aliquot | Range %CV (within sample) |
between sample %CV* |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A 8 reps |
D 9 reps |
G 10 reps |
H 10 reps |
J 8 reps |
K 10 reps |
M 10 reps |
N 9 reps |
|||
| 2371 | 24.1 | 13.9 | 15.3 | 23.5 | 10.3 | 16.1 | 16.6 | 13.7 | 10.3 - 24.1 | 23.2 |
| 2596 | 9.2 | 13.7 | 7.3 | 11 | 12.5 | 13.1 | 13.1 | 12.7 | 7.3 - 13.7 | 21.8 |
| 2924 | 19.8 | 10 | 8.7 | 12.3 | 16.8 | 21.2 | 32 | 38.2 | 8.7 - 38.2 | 47.3 |
| 3182 | 17.7 | 11.6 | 9.1 | 7.8 | 19 | 14.7 | 25.5 | 18.5 | 7.8 - 25.5 | 42.6 |
| 3252 | 28 | 8.1 | 6.5 | 6.8 | 19.5 | 13.4 | 19.2 | 14.9 | 6.5 - 28.0 | 45.6 |
| 3338 | 17.3 | 12.6 | 8.8 | 10.3 | 13.6 | 12.6 | 11.6 | 10.5 | 8.8 - 17.3 | 19.3 |
| 3938 | 17.2 | 9.7 | 9.1 | 4.8 | 7.2 | 5.4 | 5.8 | 4.4 | 4.4 - 17.2 | 20.3 |
| 4075 | 35.5 | 8.2 | 5.7 | 6.1 | 5.6 | 4.6 | 5.2 | 8.3 | 4.6 - 35.5 | 21.7 |
| 4194 | 14.2 | 8.3 | 4.2 | 4.8 | 2.9 | 3.9 | 6.6 | 6.9 | 2.9 - 14.2 | 18.4 |
| 4766 | 17.6 | 11.1 | 11.1 | 5.1 | 10.2 | 13.9 | 12.5 | 11.7 | 5.1 - 17.6 | 17.7 |
| 4942 | 25.2 | 7.3 | 9.6 | 8.5 | 13.9 | 10.8 | 18.1 | 10.5 | 7.3 - 25.2 | 15.9 |
| 5311 | 29 | 5.4 | 3.8 | 5.4 | 4.5 | 5.4 | 5.4 | 5 | 3.8 - 29.0 | 13.4 |
| 5874 | 30.6 | 9.1 | 7.4 | 4.8 | 3.5 | 2.8 | 4.4 | 7.4 | 2.8 - 30.6 | 14.5 |
| 6036 | 21.7 | 8.7 | 8.3 | 5.4 | 4.9 | 5.7 | 5.1 | 7.4 | 4.9 - 21.7 | 12.9 |
| 6628 | 22.5 | 12.8 | 10.1 | 14.8 | 12.1 | 17.7 | 14.7 | 23.6 | 10.1 - 23.6 | 26.4 |
| 9218 | 35.1 | 25.4 | 11.2 | 5.9 | 13.8 | 5.8 | 18.5 | 8.8 | 5.8 - 35.1 | 42.4 |
|
| ||||||||||
|
within sample Ave %CV |
22.8 | 11 | 8.5 | 8.6 | 10.6 | 10.4 | 13.4 | 12.7 | 8.5 – 22.8 | 25.2 |
based on 74 replicates
mean CV 12.3% within sample
mean CV 25.2% between samples
The average CV per experimental replicate ranged from 8 – 23%, with a mean CV of 12% within samples. The mean CV between samples ranged between 13 – 47%, with an average of 25%. While this sample-to-sample reproducibility of 25% is not very noteworthy, it is within the range of CVs regularly observed in serum MS profiling assays.(16, 18, 26, 27) Of greater note is that the additional sample processing steps – ultrafiltration, C3 – did not significantly impact within sample reproducibility (12% for serum with ultrafiltration/C3 compares well with 9% for PS1 without). Plus, the MALDI profiling is greatly enhanced in the LMW serum protein range.
In conclusion, currently there is debate as to the diagnostic utility of low molecular weight protein/peptide signatures since all the potential markers to date correspond to proteolytic fragments of the most abundant plasma proteins.(8, 28) However, our ultrafiltration/MALDI method extensively improves peptide/protein peak intensities and reproducibility in the 3-20 kDa range. Our preliminary results (data not shown) comparing prostate cancer to normal serum samples indicate significant peak differences in the LMW range. For future work, we plan to conduct a larger LMW serum clinical profiling study of prostate cancer-related samples using our denaturing ultrafiltration technique.
Supplementary Material
Acknowledgements
This investigation was supported by NIH grant CA126118 and its ARRA supplement from the Advanced Proteomics Platforms and Computational Sciences Program of the National Cancer Institute. We thank Drs. Eugene Tracy, William Cooke and Dennis Manos of the College of William and Mary for comments, and Dr. Lisa Cazares of Eastern Virginia Medical School for experimental support.
References
- 1.Vestal ML. Modern MALDI time-of-flight mass spectrometry. J Mass Spectrom. 2009;44:303. doi: 10.1002/jms.1537. [DOI] [PubMed] [Google Scholar]
- 2.Aresta A, et al. Impact of sample preparation in peptide/protein profiling in human serum by MALDI-TOF mass spectrometry. J Pharm Biomed Anal. 2008;46:157. doi: 10.1016/j.jpba.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 3.Callesen AK, et al. Serum protein profiling by solid phase extraction and mass spectrometry: a future diagnostics tool? Proteomics. 2009;9:1428. doi: 10.1002/pmic.200800382. [DOI] [PubMed] [Google Scholar]
- 4.Whiteaker JR, et al. Head-to-head comparison of serum fractionation techniques. J Proteome Res. 2007;6:828. doi: 10.1021/pr0604920. [DOI] [PubMed] [Google Scholar]
- 5.Hortin GL. The MALDI-TOF mass spectrometric view of the plasma proteome and peptidome. Clin Chem. 2006;52:1223. doi: 10.1373/clinchem.2006.069252. [DOI] [PubMed] [Google Scholar]
- 6.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 7.Albrethsen J. Reproducibility in protein profiling by MALDI-TOF mass spectrometry. Clin Chem. 2007;53:852. doi: 10.1373/clinchem.2006.082644. [DOI] [PubMed] [Google Scholar]
- 8.Karbassi ID, et al. Proteomic expression profiling and identification of serum proteins using immobilized trypsin beads with MALDI-TOF/TOF. J Proteome Res. 2009;8:4182. doi: 10.1021/pr800836c. [DOI] [PubMed] [Google Scholar]
- 9.Gatlin-Bunai CL, Cazares LH, Cooke WE, Semmes OJ, Malyarenko DI. Optimization of MALDI-TOF MS detection for enhanced sensitivity of affinity-captured proteins spanning a 100 kDa mass range. J Proteome Res. 2007;6:4517. doi: 10.1021/pr0703526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson L. Candidate-based proteomics in the search for biomarkers of cardiovascular disease. J Physiol. 2005;563:23. doi: 10.1113/jphysiol.2004.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drake RR, Cazares LH, Semmes OJ. Mining the low molecular weight proteome of blood. Proteomics Clin Appl. 2007;1:758. doi: 10.1002/prca.200700175. [DOI] [PubMed] [Google Scholar]
- 12.Richter R, et al. Composition of the peptide fraction in human blood plasma: database of circulating human peptides. J Chromatogr B Biomed Sci Appl. 1999;726:25. doi: 10.1016/s0378-4347(99)00012-2. [DOI] [PubMed] [Google Scholar]
- 13.Chernokalskaya E, Gutierrez S, Pitt AM, Leonard JT. Ultrafiltration for proteomic sample preparation. Electrophoresis. 2004;25:2461. doi: 10.1002/elps.200405998. [DOI] [PubMed] [Google Scholar]
- 14.Baumann S, et al. Standardized approach to proteome profiling of human serum based on magnetic bead separation and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin Chem. 2005;51:973. doi: 10.1373/clinchem.2004.047308. [DOI] [PubMed] [Google Scholar]
- 15.Tirumalai RS, et al. Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics. 2003;2:1096. doi: 10.1074/mcp.M300031-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Orvisky E, et al. Enrichment of low molecular weight fraction of serum for MS analysis of peptides associated with hepatocellular carcinoma. Proteomics. 2006;6:2895. doi: 10.1002/pmic.200500443. [DOI] [PubMed] [Google Scholar]
- 17.Aristoteli LP, Molloy MP, Baker MS. Evaluation of endogenous plasma peptide extraction methods for mass spectrometric biomarker discovery. J Proteome Res. 2007;6:571. doi: 10.1021/pr0602996. [DOI] [PubMed] [Google Scholar]
- 18.Semmes OJ, et al. Evaluation of serum protein profiling by surface-enhanced laser desorption/ionization time-of-flight mass spectrometry for the detection of prostate cancer: I. Assessment of platform reproducibility. Clin Chem. 2005;51:102. doi: 10.1373/clinchem.2004.038950. [DOI] [PubMed] [Google Scholar]
- 19.Vorm O, Roepstorff P, Mann M. Improved resolution and very high sensitivity in MALDI TOF of matrix surfaces made by fast evaporation. Anal Chem. 1994;66:3281. [Google Scholar]
- 20.Malyarenko DI, et al. Resampling and deconvolution of linear time-of-flight records for enhanced protein profiling. Rapid Commun Mass Spectrom. 2006;20:1670. doi: 10.1002/rcm.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Padliya ND, Wood TD. Improved peptide mass fingerprinting matches via optimized sample preparation in MALDI mass spectrometry. Anal Chim Acta. 2008;627:162. doi: 10.1016/j.aca.2008.05.059. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Schey KL. Fragmentation of multiply-charged intact protein ions using MALDI TOF-TOF mass spectrometry. J Am Soc Mass Spectrom. 2008;19:231. doi: 10.1016/j.jasms.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleinert P, et al. Mass spectrometry: a tool for enhanced detection of hemoglobin variants. Clin Chem. 2008;54:69. doi: 10.1373/clinchem.2007.089961. [DOI] [PubMed] [Google Scholar]
- 24.Keller BO, Li L. Three-layer matrix/sample preparation method for MALDI MS analysis of low nanomolar protein samples. J Am Soc Mass Spectrom. 2006;17:780. doi: 10.1016/j.jasms.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Adam BL, et al. Serum protein fingerprinting coupled with a pattern-matching algorithm distinguishes prostate cancer from benign prostate hyperplasia and healthy men. Cancer Res. 2002;62:3609. [PubMed] [Google Scholar]
- 26.Zerefos P, Prados J, Kossida S, Kalousis A, Vlahou A. Sample preparation and bioinformatics in MALDI profiling of urinary proteins. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;853:20. doi: 10.1016/j.jchromb.2007.02.063. [DOI] [PubMed] [Google Scholar]
- 27.Rai AJ, et al. Analysis of Human Proteome Organization Plasma Proteome Project (HUPO PPP) reference specimens using surface enhanced laser desorption/ionization-time of flight (SELDI-TOF) mass spectrometry: multi-institution correlation of spectra and identification of biomarkers. Proteomics. 2005;5:3467. doi: 10.1002/pmic.200401320. [DOI] [PubMed] [Google Scholar]
- 28.Diamandis EP. Mass spectrometry as a diagnostic and a cancer biomarker discovery tool: opportunities and potential limitations. Mol Cell Proteomics. 2004;3:367. doi: 10.1074/mcp.R400007-MCP200. [DOI] [PubMed] [Google Scholar]
- 29.Malyarenko DI, Cooke WE, Bunai CL, Manos DM. Automated assignment of ionization states in broad-mass matrix-assisted laser desorption/ionization spectra of protein mixtures. Rapid Commun. Mass. Spectrom. 2010;24:138. doi: 10.1002/rcm.4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bunai CL, et al. Enhancement in MALDI Profiling of the Low Molecular Weight Human Serum Proteome using Ultrafiltration; 58th ASMS Conference on Mass Spectrometry and Allied Topics; Salt Lake City, Utah. May 26, 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.