

Abstract
The field of pharmacogenomics is focused on the characterization of genetic factors contributing to the response of patients to pharmacological interventions. Drug response and toxicity are complex traits; therefore the effects are likely influenced by multiple genes. The investigation of the genetic basis of drug response has evolved from a focus on single genes to relevant pathways to the entire genome. Preclinical (cell-based models) and clinical genome-wide association studies (GWAS) in oncology provide an unprecedented opportunity for a comprehensive and unbiased assessment of the heritable factors associated with drug response. The primary challenge with attempting to identify pharmacogenomic markers from clinical studies is that they require a homogenous population of patients treated with the same dosage regimen and minimal confounding variables. Therefore, the development of cell-based models for pharmacogenomic marker identification has utility for the field since performing these types of studies in humans is difficult and costly. The scope of this review is to provide a current report on the status of genomic studies in oncology, the methods for discovery and implications for patient care. We present a perspective and summary of the challenges and opportunities in translating heritable genomic discoveries to patients.
Introduction
While undergoing chemotherapy, most patients have a “watch and wait” period before the effectiveness of the treatment is assessed. Some patients endure side effects that can be severe, possibly even life threatening, with an ineffective chemotherapeutic agent. If physicians could better predict which individuals are at greatest risk of experiencing chemotherapy-related toxicities and/or non-response, then use this information to choose chemotherapy regimens, the overall care of cancer patients would be greatly improved. Pharmacogenetics involves the study of the genetic basis for individual differences in response to drug efficacy and adverse effects. Although pharmacogenetics and pharmacogenomics are interchangeable terms, in general pharmacogenetics refers to single genes or pathways and pharmacogenomics refers to more comprehensive genome wide studies to identify signatures of markers involved in drug response. In this era of genomic medicine, clinicians and health practitioners need to understand how to use genetic biomarkers to personalize cancer therapy. The scope of this review is to provide an up to date report on the status of clinical and preclinical genomic studies of heritable markers in oncology, their early discoveries, limitations, and implications for patient care.
Role of germline genomic investigation in oncology
Clinical drug response has multiple facets, including impact on tumor growth and survival, extent of adverse events, and changes in quality of life. These response “phenotypes” result from the interaction of several factors related to the patient (“the host”), the tumor, and the environment. Although non-genetic factors such as diet, age, gender and concomitant medications can influence a patient’s response to chemotherapeutic treatments, understanding the contribution of genetics may be the key to maximizing drug efficacy and minimizing adverse side effects. Germline DNA is more accessible than tumor DNA, and there is accumulating evidence that optimization of treatment strategies may require stratification based on germline genetic factors. Most studies have focused on single nucleotide polymorphisms (SNPs), a DNA sequence variation occurring at a single base in the genome. Elucidation of the unique sets of genetic variables (both common SNPs and rare alleles) that contribute to the patient’s at risk for severe toxicity (or non-response) will provide critical information required for developing personalized therapy for cancer patients.
Family and epidemiology studies suggest that hypertension, coronary artery disease, asthma, and other chronic diseases have a significant heritable component (1, 2). In cancer patients, the contribution of germline variants to patient survival is difficult to assess due to the presence of acquired tumor-related factors with prognostic impact. The tumor DNA is thought to be far more important in the assessment of genetic variables contributing to treatment response. However, recent epidemiologic studies suggest an inherited component for prognosis (3-5) and that even tumor grade and metastatic potential might be, in part, under germline control (6, 7).
The fundamental problem in the field of pharmacogenetics of anticancer agents is that we currently have only a few pharmacogenetic markers to predict the best course of therapy for patients (8). For the most part, these genetic variants are within drug metabolizing genes that have a large effect on the degree or rate at which a drug is converted to its metabolites. Examples include thiopurine methyltransferase (TPMT) variants that account for greater than 90% of cases with low or intermediate TPMT enzyme activity and lead to increased risk for severe myelosuppression after 6-mercaptopurine treatment (9); UDP-glucuronosyltransferase (UGT)1A1*28 associated with a decrease in UGT1A1 expression and increased risk of severe neutropenia when irinotecan is administered (10); and the still controversial lack of response to tamoxifen in CYP2D6 poor metabolizers (11-15). These studies generally used a candidate gene or pathway-centric approach; both make assumptions about which genes are most important in the drug’s activity. The consequence has been that only the strongest signals (i.e., the Mendelian markers with the strongest penetrance) were then replicated across multiple independent studies. Aside from these discoveries, the literature is saturated with under-powered studies demonstrating or refuting the relationship between single genetic variants within “candidate genes or pathways” resulting in some clinical outcome.
Drug response is a complex trait and its heritable component is likely to result from the contribution of multiple genes, each of them conferring a modest effect. Even for the single-gene toxicity traits described above, the predictive power of those genetic tests is not optimal. Over the past 10 years, technology has evolved allowing for changes in the focus of research from single genes to the entire genome, thereby replacing the term pharmacogenetics with pharmacogenomics. This expansion has been made possible by the advent of SNP platforms for interrogating the entire genome of patients through genome-wide association studies (GWAS). This unbiased approach is expected to provide novel candidate genes associated with outcome that might provide unexpected insights into the pathophysiology of the disease (cancer) or the pharmacology of the drugs (chemotherapy). GWAS can also be applied to preclinical studies for discovery of genetic variants associated with pharmacological phenotypes.
GWAS studies in preclinical models
Employing GWAS to identify genetic variants associated with drug response in humans is challenging because of the requirement for large numbers of patients treated homogeneously (single drug/single dosage regimen) to provide enough power to detect true genetic signals in the presence of multiple confounding factors such as concomitant medications, dosage, and diet. For oncologists, this is especially difficult because the standard of care tends to change as new therapies emerge and the vast majority of patients receive multiple drugs. For this reason, there has been an effort to develop cell-based models evaluating pharmacologic phenotypes to identify heritable “predictive genetic markers” of chemotherapy-induced response and toxicity.
One cell-based model employs Epstein Barr Virus (EBV)-transformed lymphoblastoid cell lines (LCLs) derived from individuals from different geographic locations to identify genetic variants that contribute to pharmacologic phenotypes such as cytotoxicity, apoptosis, intracellular drug concentration or drug transport (16-18). These cell lines are part of the International HapMap project with more than 2 million SNPs publicly available (minor allele frequency >5%).
The first phase of the HapMap includes 270 samples from 4 geographic populations: the Yoruba in Ibadan, Nigeria; Japanese in Tokyo, Japan; Han Chinese in Beijing, China; and the CEPH (U.S. Utah residents with ancestry from northern and western Europe). In addition to SNP genotype data that are currently available for 270 HapMap LCLs using multiple genotyping platforms, copy number variation (CNV) data are also publicly available from the Wellcome Trust Sanger Institute (19). More recently, data from the international sequencing effort, the 1000 Genomes Project (http://www.1000genomes.org) with the most detailed catalogue of human genetic variation, are emerging on these 270 HapMap LCLs. Since there is considerable suspicion that rare variants, which are not measured in GWAS, contribute to phenotypic outcomes (20-22), the recent development of next generation sequencing technologies makes it possible to identify rare variants that contribute to pharmacologic outcomes. In addition, basal gene expression data is available for a portion of the 270 LCLs from Affymetrix Focus array (23, 24), Affymetrix Human Exon 1.0 ST array (exon array) (25, 26) and Illumina BeadChips (19, 27). Thus, these LCLs are an extraordinarily rich sample set in terms of genotypic information that simply requires evaluating pharmacologic phenotypes to characterize the genetic variation and/or genes implicated in these phenotypes.
The first question addressed using cell lines derived from individuals within multi-generation, large CEPH pedigrees pertained to whether chemotherapeutic-induced cytotoxicity is a heritable trait (28, 29). Studies showed that a significant genetic component (38-47%) contributed to cisplatin-induced cytotoxicity (28), as well as docetaxel (26-65%) and 5-fluorouracil (21-70%) toxicity (29). Follow-up studies included more pedigrees to provide greater power (30) and additional drugs such as daunorubicin (31), etoposide (32), and carboplatin (33).
With the high-density genotype data available for the HapMap samples, GWAS to search for novel pharmacogenomic markers can easily be performed. However, there is a challenge with prioritizing the genetic variants associated with pharmacologic phenotypes. Thus, one approach has been to evaluate the function of SNPs associated with pharmacologic phenotypes to identify whether these SNPs were disproportionately likely to be within a functional class such as coding (consisting of missense, nonsense, or frameshift polymorphisms), noncoding (such as 3’ untranslated regions or splice sites), or expression quantitative trait loci (eQTLs; indicating that a SNP genotype is associated with the transcript abundance level of a gene) (25). Chemotherapeutic drug susceptibility-associated SNPs are more likely to be eQTLs, and, in fact, more likely to be associated with the transcriptional expression level of multiple genes (n > or = 10) as potential master regulators, than a random set of SNPs in the genome (34). The implication for these findings is that genetic variants playing an important role in drug sensitivity may be those that regulate the expression of many different genes.
The “triangle” approach is a cell based model that takes into consideration genetics, expression and pharmacologic phenotypes (Figure 1). The first arm of the triangle is an evaluation of significant associations between SNPs and drug sensitivity to a specific drug. Then from this list of SNPs, eQTL analysis is performed to find the subset of SNPs associated with expression of transcripts (second arm of the triangle). In the final arm, the expression of the list of target genes is evaluated for significant linear correlation to drug sensitivity. This type of genome-wide analysis has been successfully used to identify novel genetic variants predicting sensitivity to a variety of chemotherapeutics, including etoposide (17), cisplatin (35), carboplatin (18), daunorubicin (36), and cytarabine (16). Since GWAS are susceptible to false positives due to the large number of comparisons, stringent p-values are used and relationships found in the discovery set should be replicated in an independent set of LCLs (16, 36). The ability to replicate these findings in independent sets of cells greatly decreases the likelihood that these associations are spurious but instead could be potentially useful pharmacogenetic markers in a clinical setting. While there are limitations to LCL-based systems including that they are an artificial system representing one cell type, they have the potential to advance the field of pharmacogenomics immensely.
Figure 1.
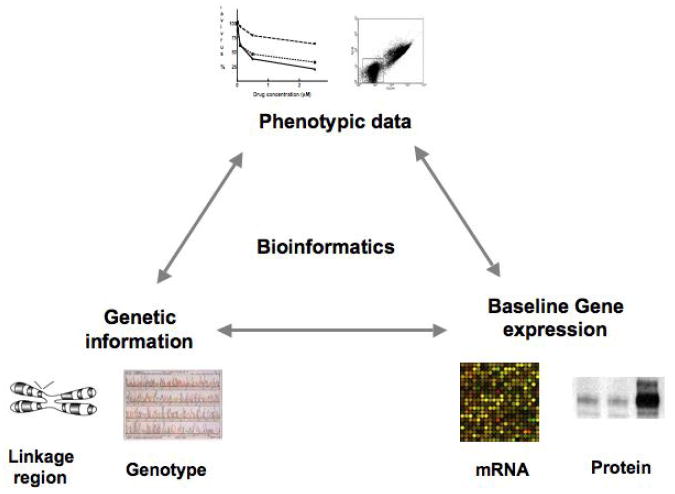
Strategy for discovery and prioritizing SNPs associated with pharmacologic phenotypes referred to as the “triangle approach”.
Another cell line panel that has served as an important pharmacogenomic resource is the NCI60 tumor cell lines (http://dtp.nci.nih.gov/index.html). These cells are derived from tumors (60 cancer cell lines derived from 9 different human organs), thus have the added ability to examine the roles of acquired mutations in drug response. Similar to LCLs, there is an immense amount of publicly available data including mRNA expression (37, 38), CNV (39), and SNP genotype (39) data for these cell lines. Additionally, extensive data on microRNA expression (40, 41) and proteomic data (42) are available (http://discover.nci.nih.gov/cellminer/). The NCI60 cell line panel also has publicly available data on toxicities associated with over 40,000 compounds (43) making them highly valuable for data mining.
Application of this cell-based disease model to pharmacogenetics discovery has resulted in determination of transcription profiles predicting sensitivity to chemotherapeutics (44-49), as well as proteomic (50-52) and microRNA (49, 53) profiles predicting drug response. Studies using these cell lines have successfully identified polymorphisms in candidate genes associated with drug response in vitro (54-57). Methylation of certain promoter CpG islands predicts toxicity to antimetabolites and alkylating agents in the NCI-60 lines (57, 58). In addition, relationships between sensitivity to a panel of EGFR inhibitors and EGFR amplification, EGFR mutations and EGFR mRNA expression have been studied (59). The main drawback is that the limited number of cell lines of different tumor types results in low statistical power for genome wide studies.
Combining cell based models and clinical findings
Traditional approaches to validating clinical genetic associations require identification of the same genotype to phenotype relationship in multiple independent populations of individuals or in cell based models and clinical studies. For example, clinical studies have shown that low intracellular concentrations of the chemotherapeutic cytarabine in leukemia cells predict poorer outcome to therapy (60, 61). Similarly, low mRNA levels of deoxycytidine kinase (DCK), an enzyme involved in the rate-limiting step of cytarabine activation, in blast cells predict shorter disease free survival as well as overall survival in an AML population treated with cytarabine (62). Cell based models were able to identify SNPs associated with these observations. Examination of LCLs from HapMap populations determined that SNPs within DCK resulted in decreased enzyme activity and were associated with increased intracellular concentrations of metabolite (ara-CTP), higher basal levels of DCK, and increased sensitivity to cytarabine (16, 63). Additional SNPs in the 3’UTR of DCK (positions +36113 and +35708) were also associated with DCK basal expression and cytarabine sensitivity in the HapMap cell lines and ara-CTP levels in leukemic cell samples from AML patients, respectively (16, 63). Thus, for cytarabine pharmacogenomics, clinical studies were successful in identifying a biomarker (ara-CTP levels) and candidate gene (DCK), while cell-based models identified candidate SNPs associated with these phenotypes. The ultimate goal is to develop a genetic signature that can be applied clinically to identify patients at risk for either increased or decreased susceptibility to cytarabine.
GWAS in clinical oncology studies
GWAS for cancer risk
To survey the common variations in the genome, SNP chips have been designed to interrogate up to millions of SNPs. In oncology, GWAS has been applied extensively to the elucidation of heritable determinants of cancer risk in case-control studies (recently reviewed in (64, 65)). To briefly summarize their main results in non familial disease, the application of GWAS has led to novel genetic variants conferring small to moderate risk of cancer, usually a 2-fold risk or less, with a few exceptions. For many cancer types, the risk conferred by these SNPs is smaller than other well known and already used risk factors. A signal in 8q24 has been reproducibly observed in several tumor types, including breast, colorectal, and prostate cancer (65). Challenges remain significant in establishing validation in heterogeneous populations, molecular assessment of the observed effects, and translation into clinical utility. However, the focus of this review is on GWAS for treatment outcome as opposed to disease susceptibility, and we will focus on this in the remainder of this review.
GWAS for treatment outcome in patients treated with chemotherapy
The GWAS catalog (66) is a repository of GWAS published since November 2008. The catalogued studies have used platforms with at least 100,000 SNPs and the results had a significant p < 10-5. These studies have been performed to identify the genetic basis of chronic diseases, drug response, risk of cancer, and other complex traits. Out of the 618 GWAS conducted so far, only 30 studies have investigated drug response as an outcome measure. Out of those, only two oncology studies are currently listed. These studies have used SNP platforms with at least 500,000 SNPs. A summary of their features and results is reported in Table 1, where we also report published preliminary findings from additional GWAS in treatment outcomes of oncology studies.
Table 1. GWAS of treatment outcome in cancer patients.
Effect sizes are reported as median values.
| Patients | Endpoint | Treatment | Sample size | Genes/rs# (effect size, p value) | Replication, sample size | Reference |
|---|---|---|---|---|---|---|
| Childhood ALL | MRD | Induction therapy | 318 | IL15 (OR 2.67, p 8.85×10-7a) | Yes, 169 | (67) |
| SCLC | Response rate | Carboplatin or cisplatin plus etoposide | 245 | BTBD3, STXBP5, BCR (p 0.043b) | Yes, 183 | (72) |
| Advanced pancreatic cancer | Overall survival | Gemcitabine with either placebo or bevacizumab | 294 | IL17F (p 2.7×10-8c) | No | (75) |
| NSCLC | Overall survival | Carboplatin, paclitaxel | 117 | EIF4E2 (HR 3.58, p 4.2×10-7) ETS2 (HR 4.95, p 9.2×10-8) rs10074374 (HR 23.2, p 2.2×10-6) rs2063681 (HR 5.2, p 2.2×10-8) | No | (78) |
| Breast cancer | Grade 2-4 Neuropathy | Cyclophosphamide, doxorubicin, paclitaxel | 859 | EPHA5 (OR 1.81, p 2.2×10-6) FGD4 (OR 1.69, p 1.9×10-5) NDRG1 (OR 0.58, p 1.9×10-5) | No | (79) |
in combined analysis
effect sizes are not reported. The p value has been computed according to a joint significance analyses of all SNPs in the validation data (see (72) for details)
effect size is not reported
The first GWAS was conducted in childhood acute lymphoblastic leukemia (ALL) patients (mainly of European ancestry) treated with chemotherapy (67). The main outcome measure in this study was minimal residual disease (MRD) at the end of induction therapy, a strong predictive marker of survival (68). Two groups of patients who differed in post-remission therapy were enrolled (318 and 169 patients), each serving as the discovery and training set in a bidirectional validation approach. Out of 72 unique SNPs (after removing SNPs due to linkage disequilibrium), the strongest SNP associated with MRD was in the ST8SIA6 gene. This SNP was not in linkage disequilibrium with other surrounding SNPs, had a relatively low allele frequency (q 0.04), and insights on the putative biological mechanism have not been reported by the authors. The second most significant SNP was in IL15, a gene on which several ex vivo observations from subsets of the same patients (gene expression in pre-treatment blasts) and in other series (69, 70) build mechanistic hypotheses about its role as a determinant of MRD in childhood ALL. Molecular effects of this IL15 SNP increasing gene expression (both at protein and mRNA level) have been reported (71). Hence, this study postulated that increased IL15 expression driven by germline variation leads to increased risk of MRD. In light of the numerous drugs taken by the ALL patients in the induction regimen, it cannot be established whether IL15 has a drug-specific effect or rather a prognostic effect. As the induction regimens used in the two patient cohorts are quite different, this gene is more likely to have a prognostic effect with respect to MRD.
A GWAS in Chinese patients with small-cell lung cancer (SCLC) proposed three novel candidate genes associated with objective response after first-line treatment with an etoposide-platinum doublet (72). In the discovery set of 245 patients, 20 SNPs have been selected for genotyping in an additional replication set of 183 patients. The identified genes (BTBD3, STXBP5, and BCR) have not previously been tested in candidate gene studies of lung cancer patients treated with platinum (73). This is the first GWAS ever conducted in solid tumor patients treated with chemotherapy. Due to the relatively small sample size, this study should be regarded as exploratory and to be confirmed in subsequent studies evaluating the impact of the gene variations on overall survival in SCLC patients.
A series of GWAS studies have been recently reported in preliminary form, awaiting independent replication in external validation sets. In CALGB 80303 (74), advanced pancreatic cancer patients were randomized to gemcitabine plus either bevacizumab or placebo. In 294 genetically-determined European patients, a nonsynonymous SNP in the IL17F gene was associated with a significantly shorter overall survival compared to patients without this allele (75). There was no evidence of an interaction with bevacizumab, suggesting that this SNP is prognostic rather than predictive of a drug effect. This IL17F allele might mediate its effect on overall survival by altering the metastatic potential of tumors through an effect mediated by angiogenesis (76, 77).
A preliminary study in non-small cell lung cancer (NSCLC) patients with stage IIIB-IV treated with first-line carboplatin and paclitaxel has been recently reported (78). In 117 patients, the GWAS scan selected four SNPs associated with shortened overall survival, two of them being located in the EIF4E2 and ETS2 genes.
CALGB study 90401 addressed a different endpoint, i.e. grade ≥2 paclitaxel-induced neuropathy in 1,040 breast cancer patients treated with adjuvant cyclophosphamide, doxorubicin, and paclitaxel (79). This GWAS has identified three genes (EPHA5, FGD4, NRDG1) implicated in congenital or experimental paclitaxel neuropathy. Pathway analysis has also shown an enrichment for SNPs annotated in pathways of neuronal development.
Lessons from clinical GWAS of response in cancer chemotherapy
Relative to the experience in GWAS of cancer risk, the experience of GWAS of outcome of chemotherapy is still limited, but it provides interesting insights. Although immediate translation of these findings into clinical utility may not occur in the short term, they have led to the discovery of novel genes outside of those previously thought to be involved in the pathophysiology of the investigated traits. This is clearly a major contribution to the field, opening new venues to biological discoveries. At the present stage, the most benefit from GWAS derives from postulating new biological mechanisms and directing research on novel pathways and novel targets for therapeutic intervention. It is quite interesting to observe that the interleukin pathway emerges from a few GWAS studies, not only in cancer GWAS (67, 75), but also in GWAS of response to interferon and ribavirin in chronic hepatitis C (80).
These studies found several highly significant SNPs that are in inter-genic regions, in genes with poor knowledge of the biology of coded proteins, and in genes that are not annotated yet. Because of the way the GWAS platforms have been designed, it is unlikely that the SNPs selected through GWAS will be the causative SNPs. So far, only one study (67) has provided mechanistic evidence at an in vitro level to support the statistical association with phenotype. In oncology, mechanistic studies of heritable SNPs are also challenging because of the intrinsic genomic instability of cancer cell lines and the potential confounder of acquired alterations in oncogenes and tumor suppressor genes in tumors.
It has been hypothesized that the genes involved in the occurrence of non familial cancers are potentially also determinants of survival (81). Germline features that drive carcinogenesis might also drive a biological response to therapy and aggressiveness of disease, affecting prognosis. These initial GWAS cannot confirm or refute this hypothesis. However, the SNPs identified so far do not seem to be involved in predisposing to cancer. It is without doubt that GWAS is a “treasure trove” of information, and large randomized clinical trials of patients treated with chemotherapy could be used to identify cancer predisposition genes by using publicly available non cancer controls that have been already “GWASed”. Conversely, case-control studies on cancer predisposition could be used to identify prognostic markers of survival. A few case-control cancer risk GWAS have served as the platform to test the effect of heritable prognosticators of survival in glioblastoma multiforme (82) and prostate cancer (83-85). A GWAS performed in more than 600 pancreatic cancer patients in a cancer risk cohort study has enabled investigation of the effect of the ligand sonic (SHH gene, an activator of the hedgehog pathway) on patient survival. In this preliminary study (86) one SNP in SHH resulted in increased overall survival and reduced SHH expression in primary tumors. The hedgehog signaling pathway is a promising stromal target, and attempts to target the stroma in pancreatic cancer are ongoing (87).
When GWAS is used to examine toxicity of chemotherapy, significant advantages exist compared to GWAS of patient survival. First, the effect is expected to be host-derived and therefore germline, and the confounder of acquired tumor instability does not exist. Second, the effect size of the associated SNPs should be larger than those observed in complex diseases. Particularly in the context of controlled trials, where the information on treatment and drug dosing is carefully annotated, and patients are subjected to the strong environmental influence of drug therapy, which can be taken into account and controlled for. The experience with candidate gene studies of treatment toxicity reports large effects sizes of associated SNPs, in the range of 5-50 genotype relative risks (88). Those large risks are also the ones that can be more easily reproduced and have the most impact in the clinic. Several recent GWAS of drug response in areas outside of oncology indicate that genome-wide significance (p<10-7) can be achieved with 50-85 cases in a case-control design (89, 90). Considering that traditional chemotherapy is administered at doses that are close to those maximally tolerated by patients, GWAS of toxicity of chemotherapy is expected to have a major impact in personalizing patient care.
Current opportunities and challenges in the translation of genome-wide information into the clinic
The opportunities for discoveries provided by GWAS are unprecedented. Repositories for public deposit of GWAS information allow comparisons across studies and meta-analyses, as many of these studies might have overlapping characteristics, i.e. tumor types, drugs, drugs classes, toxicities, and toxicities that are specific to a particular drug class. Such comparisons might clarify prognostic vs. predictive effects, and even identify prognostic factors that are tumor-type independent, similar to the locus on 8q24 for cancer risk.
For analysis of heritable genetic factors, it is expected that full genome sequencing will soon replace genomic interrogation through genotyping. In reality, this is currently happening, as sequencing platforms have already generated refined and more accurate maps of population variation in non phenotyped subjects (91). Large international projects launched to entirely sequence the tumor of thousands of subjects will provide the matching heritable information from each study participant. Integrated analyses of acquired alterations (from the tumor) and matching heritable information (from the host) will define genetic signatures of survival for each sequenced tumor type. They will also clarify the role of rare variants, improving the understanding of “missing heritability” of traits.
In terms of challenges, the pace at which genomic discoveries are obtained is much faster than the ability to test those discoveries for clinical utility and select those of significant impact in patient care. Performing GWAS is a challenge because of the cost and infrastructure it requires. Replication of the initial GWAS findings is even more difficult. Although some criteria have been recently proposed (NCI-NHGRI Working Group on Replication in Association Studies), there are no accepted “gold standard” guidelines about what constitutes a finding deserving replication, an adequate replication study, and a replication or refutation. The replication of GWAS findings from clinical trials has intrinsic difficulties, as there will never be an existing trial (to be used for replication) with the same eligibility criteria and drug treatment of the initial trial used for discovery. Although phase III randomized trials will have adequate sample size and can discern predictive from prognostic effects, a new phase III trial for prospective validation is not likely to be feasible.
It is conventional to use a p value of 10-7 or 10-8 as the threshold after Bonferroni correction for multiple testing in the discovery GWAS. Statistical significance of the initial discovery should not be the only parameter for further testing in validation, as demonstrated by the Wellcome Trust Case Control Consortium (WTCCC) study, the largest GWAS ever conducted (92). In the WTCCC study, SNPs associated at p 10-5 or 10-6 were subsequently replicated. The selection of SNPs for replication could be dictated also by availability of DNA coupled with phenotypic clinical information, and funding for genotyping and data analysis.
Even if markers are independently validated through a very lengthy and expensive process, is this information sufficient to apply them in the clinic? So far, the uptake of genomic information from the cancer risk and outcome studies in the clinic has been limited (93). Oncologists will be inclined to add genetic information to their decision-making process if the performance of the genetic test is robust enough to add meaningful information to the traditional methods based upon staging, family history, and other characteristics. Existing cancer risk algorithms have uncertain clinical validity. Increased oversight by the US Food and Drug Administration and other regulatory agencies on genetic and genomic tests will increase the confidence of clinicians in genomic medicine.
As a clear indication of how the field of genomic medicine is changing rapidly, the American Society of Clinical Oncology has already revised twice its policy statements of genetic and genomic testing for cancer susceptibility since 1996 (94). The proliferation of for-profit companies for personal genomics profiling has increased the patient demand for competence on direct-to-consumer tests and confidence in their interpretation and their implications. Training in genomic medicine is pivotal in increasing such competence of health practitioners.
Conclusion and look at the future of genomic technologies in oncology
Clinically, genomic medicine is expected to provide tools that will enhance the traditional approaches for estimating the risk/benefit of an intervention in cancer patients. The development of comprehensive, cost-efficient and high-throughput technologies will result in large amounts of data for incorporation into electronic medical records. One might anticipate a future where computerized systems will be available to physicians for selecting the drug regimen that has the highest likelihood of therapeutic success along with the lowest risk of toxicities based on genomic and non-genomic variables. In oncology, these assessments will require an understanding of both germline background of the patient and somatic changes of the tumor, scoring each drug or regimen with the optimal toxicity/efficacy ratio. For pharmacogenomics to be successful in the field of oncology, there needs to be alternative therapies with different mechanisms available for patients whose genetics show that they are at risk for severe toxicity or non response with standard chemotherapy. The increase in pharmacogenomic information will require a concomitant increase in education of physicians to best implement improvements in the care of patients.
Acknowledgments
The work performed by the authors of this manuscript is supported by Pharmacogenetic of Anticancer Agents Research (PAAR) Group (http://pharmacogenetics.org) NIH/NIGMS UO1GM61393 (FI, NJC, MED), the University of Chicago Breast Cancer SPORE grant P50 CA125183 (NJC, MED), NIH/NCI grant CA136765 (MED), and NIH/NCI K07CA140390-01 (FI).
Footnotes
Financial disclosure or conflict of interest statements The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barrett JH, Sheehan NA, Cox A, Worthington J, Cannings C, Teare MD. Family based studies and genetic epidemiology: theory and practice. Hum Hered. 2007;64(2):146–8. doi: 10.1159/000101993. [DOI] [PubMed] [Google Scholar]
- 2.Hopper JL, Bishop DT, Easton DF. Population-based family studies in genetic epidemiology. Lancet. 2005;366(9494):1397–406. doi: 10.1016/S0140-6736(05)67570-8. [DOI] [PubMed] [Google Scholar]
- 3.Lindstrom LS, Hall P, Hartman M, Wiklund F, Gronberg H, Czene K. Familial concordance in cancer survival: a Swedish population-based study. Lancet Oncol. 2007;8(11):1001–6. doi: 10.1016/S1470-2045(07)70282-6. [DOI] [PubMed] [Google Scholar]
- 4.Lindstrom LS, Hall P, Hartman M, Wiklund F, Czene K. Is genetic background important in lung cancer survival? PLoS One. 2009;4(5):e5588. doi: 10.1371/journal.pone.0005588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartman M, Lindstrom L, Dickman PW, Adami HO, Hall P, Czene K. Is breast cancer prognosis inherited? Breast Cancer Res. 2007;9(3):R39. doi: 10.1186/bcr1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia-Closas M, Hall P, Nevanlinna H, Pooley K, Morrison J, Richesson DA, et al. Heterogeneity of breast cancer associations with five susceptibility loci by clinical and pathological characteristics. PLoS Genet. 2008;4(4):e1000054. doi: 10.1371/journal.pgen.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunter KW. Allelic diversity in the host genetic background may be an important determinant in tumor metastatic dissemination. Cancer Lett. 2003;200(2):97–105. doi: 10.1016/s0304-3835(03)00420-8. [DOI] [PubMed] [Google Scholar]
- 8.Coate L, Cuffe S, Horgan A, Hung RJ, Christiani D, Liu G. Germline genetic variation, cancer outcome, and pharmacogenetics. J Clin Oncol. 2010;28(26):4029–37. doi: 10.1200/JCO.2009.27.2336. [DOI] [PubMed] [Google Scholar]
- 9.Lennard L, Lilleyman JS, Van Loon J, Weinshilboum RM. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336(8709):225–9. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- 10.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22(8):1382–8. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 11.Borges S, Desta Z, Jin Y, Faouzi A, Robarge JD, Philip S, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol. 2010;50(4):450–8. doi: 10.1177/0091270009359182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goetz MP, Toft D, Reid J, Ames M, Stensgard B, Safgren S, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23(6):1078–87. doi: 10.1200/JCO.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 13.Higgins MJ, Stearns V. CYP2D6 polymorphisms and tamoxifen metabolism: clinical relevance. Curr Oncol Rep. 2010;12(1):7–15. doi: 10.1007/s11912-009-0076-5. [DOI] [PubMed] [Google Scholar]
- 14.Kiyotani K, Mushiroda T, Hosono N, Tsunoda T, Kubo M, Aki F, et al. Lessons for pharmacogenomics studies: association study between CYP2D6 genotype and tamoxifen response. Pharmacogenet Genomics. 2010;20(9):565–8. doi: 10.1097/FPC.0b013e32833af231. [DOI] [PubMed] [Google Scholar]
- 15.Schroth W, Hamann U, Fasching PA, Dauser S, Winter S, Eichelbaum M, et al. CYP2D6 polymorphisms as predictors of outcome in breast cancer patients treated with tamoxifen: expanded polymorphism coverage improves risk stratification. Clin Cancer Res. 2010;16(17):4468–77. doi: 10.1158/1078-0432.CCR-10-0478. [DOI] [PubMed] [Google Scholar]
- 16.Hartford CM, Duan S, Delaney SM, Mi S, Kistner EO, Lamba JK, et al. Population-specific genetic variants important in susceptibility to cytarabine arabinoside cytotoxicity. Blood. 2009;113(10):2145–53. doi: 10.1182/blood-2008-05-154302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang RS, Duan S, Bleibel WK, Kistner EO, Zhang W, Clark TA, et al. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc Natl Acad Sci U S A. 2007;104(23):9758–63. doi: 10.1073/pnas.0703736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang RS, Duan S, Kistner EO, Hartford CM, Dolan ME. Genetic variants associated with carboplatin-induced cytotoxicity in cell lines derived from Africans. Mol Cancer Ther. 2008;7(9):3038–46. doi: 10.1158/1535-7163.MCT-08-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315(5813):848–53. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305(5685):869–72. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 21.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8(1):e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, Dickson SP, Stolle CA, Krantz ID, Goldstein DB, Hakonarson H. Interpretation of association signals and identification of causal variants from genome-wide association studies. Am J Hum Genet. 2010;86(5):730–42. doi: 10.1016/j.ajhg.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spielman RS, Bastone LA, Burdick JT, Morley M, Ewens WJ, Cheung VG. Common genetic variants account for differences in gene expression among ethnic groups. Nat Genet. 2007;39(2):226–31. doi: 10.1038/ng1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Storey JD, Madeoy J, Strout JL, Wurfel M, Ronald J, Akey JM. Gene-expression variation within and among human populations. Am J Hum Genet. 2007;80(3):502–9. doi: 10.1086/512017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan S, Huang RS, Zhang W, Bleibel WK, Roe CA, Clark TA, et al. Genetic architecture of transcript-level variation in humans. Am J Hum Genet. 2008;82(5):1101–13. doi: 10.1016/j.ajhg.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, Duan S, Bleibel WK, Wisel SA, Huang RS, Wu X, et al. Identification of common genetic variants that account for transcript isoform variation between human populations. Hum Genet. 2009;125(1):81–93. doi: 10.1007/s00439-008-0601-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, et al. Population genomics of human gene expression. Nat Genet. 2007;39(10):1217–24. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dolan ME, Newbold KG, Nagasubramanian R, Wu X, Ratain MJ, Cook EH, Jr, et al. Heritability and linkage analysis of sensitivity to cisplatin-induced cytotoxicity. Cancer Res. 2004;64(12):4353–6. doi: 10.1158/0008-5472.CAN-04-0340. [DOI] [PubMed] [Google Scholar]
- 29.Watters JW, Kraja A, Meucci MA, Province MA, McLeod HL. Genome-wide discovery of loci influencing chemotherapy cytotoxicity. Proc Natl Acad Sci U S A. 2004;101(32):11809–14. doi: 10.1073/pnas.0404580101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shukla SJ, Duan S, Badner JA, Wu X, Dolan ME. Susceptibility loci involved in cisplatin-induced cytotoxicity and apoptosis. Pharmacogenet Genomics. 2008;18(3):253–62. doi: 10.1097/FPC.0b013e3282f5e605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duan S, Bleibel WK, Huang RS, Shukla SJ, Wu X, Badner JA, et al. Mapping genes that contribute to daunorubicin-induced cytotoxicity. Cancer Res. 2007;67(11):5425–33. doi: 10.1158/0008-5472.CAN-06-4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bleibel WK, Duan S, Huang RS, Kistner EO, Shukla SJ, Wu X, et al. Identification of genomic regions contributing to etoposide-induced cytotoxicity. Hum Genet. 2009;125(2):173–80. doi: 10.1007/s00439-008-0607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shukla SJ, Duan S, Wu X, Badner JA, Kasza K, Dolan ME. Whole-genome approach implicates CD44 in cellular resistance to carboplatin. Hum Genomics. 2009;3(2):128–42. doi: 10.1186/1479-7364-3-2-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gamazon ER, Huang RS, Cox NJ, Dolan ME. Chemotherapeutic drug susceptibility associated SNPs are enriched in expression quantitative trait loci. Proc Natl Acad Sci U S A. 2010;107(20):9287–92. doi: 10.1073/pnas.1001827107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang RS, Duan S, Shukla SJ, Kistner EO, Clark TA, Chen TX, et al. Identification of genetic variants contributing to cisplatin-induced cytotoxicity by use of a genomewide approach. Am J Hum Genet. 2007;81(3):427–37. doi: 10.1086/519850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang RS, Duan S, Kistner EO, Bleibel WK, Delaney SM, Fackenthal DL, et al. Genetic variants contributing to daunorubicin-induced cytotoxicity. Cancer Res. 2008;68(9):3161–8. doi: 10.1158/0008-5472.CAN-07-6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet. 2000;24(3):236–44. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- 38.Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24(3):227–35. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 39.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436(7047):117–22. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 40.Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, et al. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007;67(6):2456–68. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- 41.Blower PE, Verducci JS, Lin S, Zhou J, Chung JH, Dai Z, et al. MicroRNA expression profiles for the NCI-60 cancer cell panel. Mol Cancer Ther. 2007;6(5):1483–91. doi: 10.1158/1535-7163.MCT-07-0009. [DOI] [PubMed] [Google Scholar]
- 42.Nishizuka S, Charboneau L, Young L, Major S, Reinhold WC, Waltham M, et al. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci U S A. 2003;100(24):14229–34. doi: 10.1073/pnas.2331323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6(10):813–23. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 44.Staunton JE, Slonim DK, Coller HA, Tamayo P, Angelo MJ, Park J, et al. Chemosensitivity prediction by transcriptional profiling. Proc Natl Acad Sci U S A. 2001;98(19):10787–92. doi: 10.1073/pnas.191368598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinstein JN, Myers TG, O’Connor PM, Friend SH, Fornace AJ, Jr, Kohn KW, et al. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275(5298):343–9. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 46.Blower PE, Yang C, Fligner MA, Verducci JS, Yu L, Richman S, et al. Pharmacogenomic analysis: correlating molecular substructure classes with microarray gene expression data. Pharmacogenomics J. 2002;2(4):259–71. doi: 10.1038/sj.tpj.6500116. [DOI] [PubMed] [Google Scholar]
- 47.Huang R, Wallqvist A, Thanki N, Covell DG. Linking pathway gene expressions to the growth inhibition response from the National Cancer Institute’s anticancer screen and drug mechanism of action. Pharmacogenomics J. 2005;5(6):381–99. doi: 10.1038/sj.tpj.6500331. [DOI] [PubMed] [Google Scholar]
- 48.Dai Z, Barbacioru C, Huang Y, Sadee W. Prediction of anticancer drug potency from expression of genes involved in growth factor signaling. Pharm Res. 2006;23(2):336–49. doi: 10.1007/s11095-005-9260-y. [DOI] [PubMed] [Google Scholar]
- 49.Salter KH, Acharya CR, Walters KS, Redman R, Anguiano A, Garman KS, et al. An integrated approach to the prediction of chemotherapeutic response in patients with breast cancer. PLoS ONE. 2008;3(4):e1908. doi: 10.1371/journal.pone.0001908. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Ma Y, Ding Z, Qian Y, Shi X, Castranova V, Harner EJ, et al. Predicting cancer drug response by proteomic profiling. Clin Cancer Res. 2006;12(15):4583–9. doi: 10.1158/1078-0432.CCR-06-0290. [DOI] [PubMed] [Google Scholar]
- 51.Shankavaram UT, Reinhold WC, Nishizuka S, Major S, Morita D, Chary KK, et al. Transcript and protein expression profiles of the NCI-60 cancer cell panel: an integromic microarray study. Mol Cancer Ther. 2007;6(3):820–32. doi: 10.1158/1535-7163.MCT-06-0650. [DOI] [PubMed] [Google Scholar]
- 52.Stevens EV, Nishizuka S, Antony S, Reimers M, Varma S, Young L, et al. Predicting cisplatin and trabectedin drug sensitivity in ovarian and colon cancers. Mol Cancer Ther. 2008;7(1):10–8. doi: 10.1158/1535-7163.MCT-07-0192. [DOI] [PubMed] [Google Scholar]
- 53.Blower PE, Chung JH, Verducci JS, Lin S, Park JK, Dai Z, et al. MicroRNAs modulate the chemosensitivity of tumor cells. Mol Cancer Ther. 2008;7(1):1–9. doi: 10.1158/1535-7163.MCT-07-0573. [DOI] [PubMed] [Google Scholar]
- 54.Jarjanazi H, Kiefer J, Savas S, Briollais L, Tuzmen S, Pabalan N, et al. Discovery of genetic profiles impacting response to chemotherapy: application to gemcitabine. Hum Mutat. 2008;29(4):461–7. doi: 10.1002/humu.20732. [DOI] [PubMed] [Google Scholar]
- 55.Le Morvan V, Bellott R, Moisan F, Mathoulin-Pelissier S, Bonnet J, Robert J. Relationships between genetic polymorphisms and anticancer drug cytotoxicity vis-a-vis the NCI-60 panel. Pharmacogenomics. 2006;7(6):843–52. doi: 10.2217/14622416.7.6.843. [DOI] [PubMed] [Google Scholar]
- 56.Puyo S, Le Morvan V, Robert J. Impact of EGFR gene polymorphisms on anticancer drug cytotoxicity in vitro. Mol Diagn Ther. 2008;12(4):225–34. doi: 10.1007/BF03256288. [DOI] [PubMed] [Google Scholar]
- 57.Sasaki S, Kobunai T, Kitayama J, Nagawa H. DNA methylation and sensitivity to antimetabolites in cancer cell lines. Oncol Rep. 2008;19(2):407–12. [PubMed] [Google Scholar]
- 58.Shen L, Kondo Y, Ahmed S, Boumber Y, Konishi K, Guo Y, et al. Drug sensitivity prediction by CpG island methylation profile in the NCI-60 cancer cell line panel. Cancer Res. 2007;67(23):11335–43. doi: 10.1158/0008-5472.CAN-07-1502. [DOI] [PubMed] [Google Scholar]
- 59.Liu W, Wu X, Zhang W, Montenegro RC, Fackenthal DL, Spitz JA, et al. Relationship of EGFR mutations, expression, amplification, and polymorphisms to epidermal growth factor receptor inhibitors in the NCI60 cell lines. Clin Cancer Res. 2007;13(22 Pt 1):6788–95. doi: 10.1158/1078-0432.CCR-07-0547. [DOI] [PubMed] [Google Scholar]
- 60.Raza A, Gezer S, Anderson J, Lykins J, Bennett J, Browman G, et al. Relationship of [3H]Ara-C incorporation and response to therapy with high-dose Ara-C in AML patients: a Leukemia Intergroup study. Exp Hematol. 1992;20(10):1194–200. [PubMed] [Google Scholar]
- 61.Estey E, Plunkett W, Dixon D, Keating M, McCredie K, Freireich EJ. Variables predicting response to high dose cytosine arabinoside therapy in patients with refractory acute leukemia. Leukemia. 1987;1(8):580–3. [PubMed] [Google Scholar]
- 62.Galmarini CM, Thomas X, Graham K, El Jafaari A, Cros E, Jordheim L, et al. Deoxycytidine kinase and cN-II nucleotidase expression in blast cells predict survival in acute myeloid leukaemia patients treated with cytarabine. Br J Haematol. 2003;122(1):53–60. doi: 10.1046/j.1365-2141.2003.04386.x. [DOI] [PubMed] [Google Scholar]
- 63.Lamba JK, Crews K, Pounds S, Schuetz EG, Gresham J, Gandhi V, et al. Pharmacogenetics of deoxycytidine kinase: identification and characterization of novel genetic variants. J Pharmacol Exp Ther. 2007;323(3):935–45. doi: 10.1124/jpet.107.128595. [DOI] [PubMed] [Google Scholar]
- 64.Galvan A, Ioannidis JP, Dragani TA. Beyond genome-wide association studies: genetic heterogeneity and individual predisposition to cancer. Trends Genet. 2010;26(3):132–41. doi: 10.1016/j.tig.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stadler ZK, Thom P, Robson ME, Weitzel JN, Kauff ND, Hurley KE, et al. Genome-wide association studies of cancer. J Clin Oncol. 2010;28(27):4255–67. doi: 10.1200/JCO.2009.25.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hindorff LA JH, Hall PN, Mehta JP, Manolio TA. A Catalog of Published Genome-Wide Association Studies. 2010 abstract. [Google Scholar]
- 67.Yang JJ, Cheng C, Yang W, Pei D, Cao X, Fan Y, et al. Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA. 2009;301(4):393–403. doi: 10.1001/jama.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Dongen JJ, Seriu T, Panzer-Grumayer ER, Biondi A, Pongers-Willemse MJ, Corral L, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352(9142):1731–8. doi: 10.1016/S0140-6736(98)04058-6. [DOI] [PubMed] [Google Scholar]
- 69.Cario G, Izraeli S, Teichert A, Rhein P, Skokowa J, Moricke A, et al. High interleukin-15 expression characterizes childhood acute lymphoblastic leukemia with involvement of the CNS. J Clin Oncol. 2007;25(30):4813–20. doi: 10.1200/JCO.2007.11.8166. [DOI] [PubMed] [Google Scholar]
- 70.Tinhofer I, Marschitz I, Henn T, Egle A, Greil R. Expression of functional interleukin-15 receptor and autocrine production of interleukin-15 as mechanisms of tumor propagation in multiple myeloma. Blood. 2000;95(2):610–8. [PubMed] [Google Scholar]
- 71.Zhang XJ, Yan KL, Wang ZM, Yang S, Zhang GL, Fan X, et al. Polymorphisms in interleukin-15 gene on chromosome 4q31.2 are associated with psoriasis vulgaris in Chinese population. J Invest Dermatol. 2007;127(11):2544–51. doi: 10.1038/sj.jid.5700896. [DOI] [PubMed] [Google Scholar]
- 72.Wu C, Xu B, Yuan P, Ott J, Guan Y, Liu Y, et al. Genome-wide examination of genetic variants associated with response to platinum-based chemotherapy in patients with small-cell lung cancer. Pharmacogenet Genomics. 2010;20(6):389–95. doi: 10.1097/FPC.0b013e32833a6890. [DOI] [PubMed] [Google Scholar]
- 73.Gautschi O, Mack PC, Davies AM, Jablons DM, Rosell R, Gandara DR. Pharmacogenomic approaches to individualizing chemotherapy for non-small-cell lung cancer: current status and new directions. Clin Lung Cancer. 2008;9(Suppl 3):S129–38. doi: 10.3816/CLC.2008.s.019. [DOI] [PubMed] [Google Scholar]
- 74.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303) J Clin Oncol. 2010;28(22):3617–22. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Innocenti F, Owzar W, Cox N, Evans P, Kubo M, Hollis D, et al. Heritable interleukin-17F (IL17F) gene variation and overall survival (OS) in pancreatic cancer patients (pts): Results from a genome-wide association study (GWAS) in CALGB 80303. J Clin Oncol. 2009;27:15s. abstract. [Google Scholar]
- 76.Kawaguchi M, Takahashi D, Hizawa N, Suzuki S, Matsukura S, Kokubu F, et al. IL-17F sequence variant (His161Arg) is associated with protection against asthma and antagonizes wild-type IL-17F activity. J Allergy Clin Immunol. 2006;117(4):795–801. doi: 10.1016/j.jaci.2005.12.1346. [DOI] [PubMed] [Google Scholar]
- 77.Starnes T, Robertson MJ, Sledge G, Kelich S, Nakshatri H, Broxmeyer HE, et al. Cutting edge: IL-17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J Immunol. 2001;167(8):4137–40. doi: 10.4049/jimmunol.167.8.4137. [DOI] [PubMed] [Google Scholar]
- 78.Sato Y YN, Kunitoh H, Ohe Y, Katori N, Sawada J, et al. Genome-wide association scan detected candidate polymorphisms associated with overall survival (OS) in advanced non-small cell lung cancer (NSCLC) treated with carboplatin (CBDCA) and paclitaxel (PTX) J Clin Oncol. 2009;27:15s. abstract. [Google Scholar]
- 79.Kroetz D BR, Owzar K, Jiang C, Zembutsu H, Kubo M, et al. Inherited genetic variation in EPHA5, FGD4, and NRDG1 and paclitaxel (P)-induced peripheral neuropathy (PN): Results from a genome-wide association study (GWAS) in CALGB 40101. J Clin Oncol. 2010;28:15s. abstract. [Google Scholar]
- 80.Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41(10):1105–9. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 81.Spitz MR, Wu X, Mills G. Integrative epidemiology: from risk assessment to outcome prediction. J Clin Oncol. 2005;23(2):267–75. doi: 10.1200/JCO.2005.05.122. [DOI] [PubMed] [Google Scholar]
- 82.Liu Y, Shete S, Etzel CJ, Scheurer M, Alexiou G, Armstrong G, et al. Polymorphisms of LIG4, BTBD2, HMGA2, and RTEL1 genes involved in the double-strand break repair pathway predict glioblastoma survival. J Clin Oncol. 2010;28(14):2467–74. doi: 10.1200/JCO.2009.26.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salinas CA, Koopmeiners JS, Kwon EM, FitzGerald L, Lin DW, Ostrander EA, et al. Clinical utility of five genetic variants for predicting prostate cancer risk and mortality. Prostate. 2009;69(4):363–72. doi: 10.1002/pros.20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zheng SL, Sun J, Wiklund F, Smith S, Stattin P, Li G, et al. Cumulative association of five genetic variants with prostate cancer. N Engl J Med. 2008;358(9):910–9. doi: 10.1056/NEJMoa075819. [DOI] [PubMed] [Google Scholar]
- 85.Klein RJ, Hallden C, Cronin AM, Ploner A, Wiklund F, Bjartell AS, et al. Blood biomarker levels to aid discovery of cancer-related single-nucleotide polymorphisms: kallikreins and prostate cancer. Cancer Prev Res (Phila) 2010;3(5):611–9. doi: 10.1158/1940-6207.CAPR-09-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McWilliams R BW, Fernandez-Zapico M, Sicotte H, Kim G, Burch P, et al. Association of sonic hedgehog variant with survival in pancreatic cancer. Gastrointestinal Cancers Symposium. 2010 abstract. [Google Scholar]
- 87.Garber K. Stromal depletion goes on trial in pancreatic cancer. J Natl Cancer Inst. 2010;102(7):448–50. doi: 10.1093/jnci/djq113. [DOI] [PubMed] [Google Scholar]
- 88.Nelson MR, Bacanu SA, Mosteller M, Li L, Bowman CE, Roses AD, et al. Genome-wide approaches to identify pharmacogenetic contributions to adverse drug reactions. Pharmacogenomics J. 2009;9(1):23–33. doi: 10.1038/tpj.2008.4. [DOI] [PubMed] [Google Scholar]
- 89.Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med. 2008;359(8):789–99. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 90.Daly AK, Day CP. Genetic association studies in drug-induced liver injury. Semin Liver Dis. 2009;29(4):400–11. doi: 10.1055/s-0029-1240009. [DOI] [PubMed] [Google Scholar]
- 91.Siva N. 1000 Genomes project. Nat Biotechnol. 2008;26(3):256. doi: 10.1038/nbt0308-256b. [DOI] [PubMed] [Google Scholar]
- 92.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Innocenti F, Schilsky RL. Translating the cancer genome into clinically useful tools and strategies. Dis Model Mech. 2009;2(9-10):426–9. doi: 10.1242/dmm.004119. [DOI] [PubMed] [Google Scholar]
- 94.Robson ME, Storm CD, Weitzel J, Wollins DS, Offit K. American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2010;28(5):893–901. doi: 10.1200/JCO.2009.27.0660. [DOI] [PubMed] [Google Scholar]