

Abstract
Myocardial ischemia damages the electron transport chain and augments cardiomyocyte death during reperfusion. To understand the relationship between ischemic mitochondrial damage and mitochondrial-driven cell death, the isolated perfused heart underwent global stop-flow ischemia with and without mitochondrial protection by reversible blockade of electron transport. Ischemic damage to electron transport depleted bcl-2 content and favored mitochondrial permeability transition (MPT). Reversible blockade of electron transport preserved bcl-2 content and attenuated calcium-stimulated mitochondrial swelling. Thus, the damaged electron transport chain leads to bcl-2 depletion and MPT opening. Chemical inhibition of bcl-2 with HA14-1 also dramatically increased mitochondrial swelling, augmented by exogenous H2O2 stress, indicating that bcl-2 depleted mitochondria are poised to undergo MPT during the enhanced oxidative stress of reperfusion.
1.0 Introduction
Mitochondrial dysfunction contributes to myocardial injury during ischemia-reperfusion [1]. Ischemia results in damage to the electron transport chain (ETC) and decreased rates of oxidative phosphorylation [2,3]. Reperfusion after ischemia does not result in additional damage to electron transport [4,5], although, in contrast to mitochondria, substantial cardiomyocyte injury occurs during reperfusion [6-8]. Mitochondrial-dependent cardiac injury involves the increased production of reactive oxygen species (ROS) [9-12], the depletion of anti-apoptotic proteins from mitochondria [13,14], and increased susceptibility to opening of the mitochondrial permeability transition pore (MPT) [14-17]. Protection of mitochondria against ischemic damage to the ETC by the reversible blockade of electron transport during ischemia [18,19] or other pharmacological treatments [20-22] decreases myocardial injury assessed following reperfusion [4,23,24], thus establishing a link between damage to electron transport during ischemia and cardiomyocyte death during reperfusion.
Although decreased activity of the electron transport chain could contribute to myocardial injury during reperfusion via decreased respiration and energy production, reperfused myocardium can be protected by intervention only during reperfusion. Brief, reversible blockade of electron transport during reperfusion [23,25] or the use of postconditioning consisting of brief periods of intermittent ischemia [26], protect reperfused myocardium despite the persistence of ischemia-induced ETC damage during reperfusion [26, 27]. Thus, mitochondrial-dependent processes other than decreased oxidative phosphorylation must account for the mitochondrial-dependent injury observed during reperfusion. The ETC-dependent processes that generate cardiac injury during reperfusion remain unclear.
The mitochondrial permeability transition pore (MPT) is a non-selective pore spanning the inner and outer mitochondrial membranes. MPT opening is a key contributor to cardiac injury during ischemia-reperfusion [28]. MPT opening is favored at the onset of reperfusion due to increased oxidative stress, rapid normalization of intracellular pH, and mitochondrial calcium loading. [15,16,28,29]. Ischemic damage to the electron transport chain increases ROS generation during re-oxygenation [10,30], whereas prevention of ischemic damage decreases ROS generation during reperfusion [4,31]. Thus, ischemic damage to the ETC may contribute to cardiac injury during reperfusion via ROS generation that facilitates MPT opening. The permeability of the outer mitochondrial membrane is also regulated by the expression of bcl-2 family proteins [13,32]. A decreased content of anti-apoptotic proteins (bcl-2, bcl-xl) and/or the increased content of pro-apoptotic proteins (bax and bak) will lead to permeation of the outer membrane and cytochrome c loss [13,32]. Ischemia-reperfusion decreases myocardial bcl-2 content in the isolated heart [33] and bcl-2 inhibition with the small molecule HA14-1 abrogates cardioprotection [34]. However, the potential electron transport chain dependence of bcl-2 depletion is unknown.
Blockade of the proximal electron transport chain protects mitochondria during ischemia [19], providing an experimental model to identify and study the mechanisms of ETC-dependent cardiac injury. Mitochondria were studied at the end of ischemia, in order to exclude potential contributions of in situ reperfusion to mitochondrial damage. The current study found that bcl-2 depletion from mitochondria during ischemia is indeed ETC dependent. Decreased bcl-2 content, perhaps in concert with increased ROS generation from the damaged ETC, increases the probability of mitochondrial permeability transition. Thus, an increased predisposition to permeability transition and activation of programmed cell death are complimentary, reinforcing mechanisms that translate ETC damage from ischemia into cardiomyocyte death during reperfusion.
2.0 Methods
2.1 Isolated rabbit heart model of ischemia and reperfusion
The Animal Care and Use Committees of the Louis Stokes VA Medical Center and Case Western Reserve University approved the protocol. The isolated rabbit heart perfusion protocol was performed as described previously [3,5] (Supplemental Methods). Untreated ischemic hearts were first perfused with Krebs-Henseleit buffer for 15 min. followed by 30 min. stop-flow ischemia. In amobarbital treated ischemic hearts, amobarbital (2.5 mM) [18] in oxygenated Krebs-Henseleit buffer was infused for 1 min. immediately before ischemia. Time control hearts were perfused for 45 min. without ischemia [2].
There were no differences in hemodynamic parameters between time control, untreated ischemia, and amobarbital treated ischemia groups at the end of the 15 min. equilibration period before the infusion of amobarbital (Supplemental Table 1). Developed pressure was maintained during 45 min perfusion in time control hearts (88±6 at 15 min equilibration and 85±1 mmHg at end of 45 min perfusion). Ischemia led to myocardial contracture and markedly increased diastolic pressure compared to the pre-ischemic value. Amobarbital treatment significantly attenuated the increase in diastolic pressure compared to the untreated heart as previously described (Supplemental Table 1).
2.2 Isolation and analysis of two populations of cardiac mitochondria
At the end of ischemia, hearts were removed from the perfusion column and placed into Chappel-Perry buffer [(in mM) 100 KCl, 50 MOPS, 1 EGTA, 5 MgSO4•7 H2O, and 1 ATP; pH 7.4] at 4°C. Cardiac mitochondria were isolated according to Palmer et al. [35], with minor modifications as previously described [3] (Supplemental Methods). Oxygen consumption in mitochondria was measured using a Clark-type oxygen electrode at 30°C (Stratkelvin, Scotland, UK) in a solution containing 80 mM KCl, 50 mM MOPS, 1 mM EGTA, 5 mM KH2PO4, and 1 mg/ml BSA, at pH 7.4. Glutamate (complex I substrate), and TMPD-ascorbate plus rotenone (complex IV substrate) were used as electron donors [3]. Cytochrome contents were measured in mitochondria solubilized in 2% deoxycholate in 10 mM sodium phosphate buffer using the difference of sodium dithionite reduced and air oxidized spectra [3,36]. H2O2 production from intact mitochondria was measured using the oxidation of the fluorogenic indicator amplex red in the presence of horseradish peroxidase (HRP) [37]. Glutamate (10 mM) and succinate (10 mM) were used as substrates.
2.3 Western blot analysis
SSM were solubilized and boiled for 5 min in buffer including 4% (w/v) SDS, 1 mM 2-mercaptoethanol, 10 mM Tris/HCl (pH 6.8) and 10% (w/v) glycerol. Equal amounts of protein (100 μg) were loaded onto 14% SDS-PAGE, run and transferred to PVDF membrane. The membranes were first blotted by 5% non-fat milk for one hour. Then membranes were blotted with primary antibodies for one hour or overnight. Antibodies to bcl-2 (catalog number: 2876) bax (2722), and subunit 4 of cytochrome oxidase (4844) were purchased from Cell Signaling Technology (Danvers, MA). The blots were incubated with peroxidase conjugated anti-rabbit antibody (1:5000) for 1 hour prior to ECL detection. The intensities of blotting were quantified by densitometer (GS800 densitometer, Golden Valley, MN).
2.4 Mitochondrial swelling analysis
Opening of the mitochondrial pore was determined by Ca2+ induced swelling of isolated mitochondria measured as a reduction in A520. Isolated SSM or IFM (0.25 mg/ml) were resuspended in swelling buffer (120 mM KCl, 5 mM KH2PO4, 50 mM MOPS, pH 7.4) with succinate (10 mM) as substrate. Absorbance was determined at 520 nm and MPT induced by 200 uM CaCl2. Cyclosporine A (1 μM) inhibited swelling by approximately 90% (Figure 4), confirming the decrease in absorbance was due to MPT. Results were expressed as Δ change of absorbance /mg protein [38]. To test the effect of HA14-1 on MPT opening, isolated control rabbit SSM were incubated with HA14-1 (20 uM) [39] in the absence and presence of H2O2 (100 uM) [40] for 5 min. at 30 °C and mitochondrial swelling was determined.
Figure 4. Inhibition of bcl-2 with the small molecule inhibitor HA14-1 increases mitochondrial swelling in SSM isolated from non-ischemic control rabbit hearts.
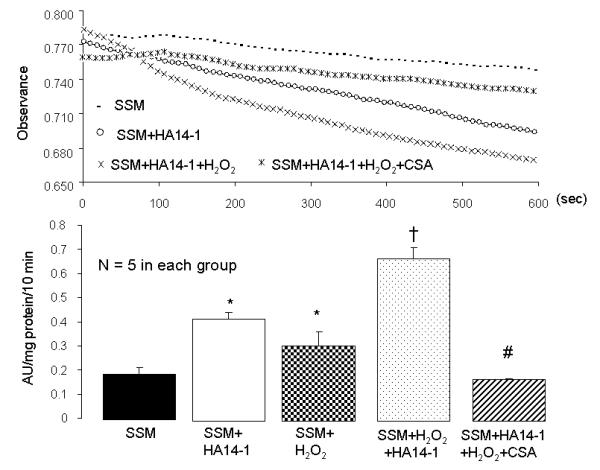
In addition, there is an additive effect when H2O2 is applied in the presence of HA14-1. The upper panel shows representative tracings (to preserve clarity, only select tracings are shown). The lower panel shows the summed results of mitochondrial swelling induced by the bcl-2 inhibitor, H2O2 treatment, bcl-2 inhibitor + H2O2 treatment, and bcl-2 inhibitor + H2O2 + cyclosporine A (CSA). (Mean±SEM; *p<0.05 vs. control SSM; † p<0.05 vs. other groups; # p<0.05 vs HA14-1 + H2O2). HA14-1 and H2O2 stimulation results in CSA-sensitive mitochondrial swelling, indicting that these treatments increase MPT opening.
2.5 Statistical Analysis
Data are expressed as the mean ± standard error of the mean. Not every sample was available for each assay and led to different animal numbers in different assays. Differences among groups were compared by one-way analysis of variance with post hoc comparisons performed using the Student-Newman-Keuls test of multiple comparisons. A difference of p<0.05 was considered significant (SigmaStat for Windows Version 1.0, Jandel Corporation).
3.0 Results
3.1 Blockade of electron transport protects oxidative phosphorylation in SSM during ischemia
Thirty minutes of ischemia decreased the maximal rate of ADP-stimulated respiration in SSM and IFM with glutamate as substrate, as previously described in this model [2,3]. In contrast, glutamate oxidation in SSM isolated from hearts with blockade of electron transport during ischemia due to amobarbital-treatment was similar to rates observed in SSM from time control hearts (Table 1). Ischemia also decreased dinitrophenol-uncoupled respiration in SSM and IFM compared to time control, and this defect was prevented by amobarbital treatment (Table 1). Oxidative phosphorylation through cytochrome oxidase also decreased in SSM following ischemia. Amobarbital treatment preserved respiration through cytochrome oxidase, indicating that blockade of the proximal electron transport chain protects the distal electron transport chain against ischemic damage (Table 1). Ischemia did not damage the distal electron transport chain in IFM (Table 1), consistent with previous findings [2,3].
Table 1.
Blockade of electron transport with amobarbital preserves respiration following ischemia.
| Time control (n=7) | Ischemia (n=9) | AMO (n=10) | |
|---|---|---|---|
| Subsarcolemmal Mitochondria (SSM) | |||
| Glutamate-ADP | 175±10 104±8* | 167±8† | |
| Glutamate-DNP | 155±10 | 102±10* | 157±11† |
| TMPD-asc-ADP | 504±17 | 315±21* | 540±31† |
| Interfibillilar Mitochondria (IFM) | |||
| Glutamate-ADP | 252±10 186±18* | 260±12† | |
| Glutamate-DNP | 252±20 | 189±27* | 273±24† |
| TMPD-asc-ADP | 858±53 | 714±22 | 810±49 |
| SSM | IFM | |||
|---|---|---|---|---|
| Glutamate | TMPD-ascorbate | Glutamate | TMPD-ascorbate | |
| Time control (n=7) | 175±10 | 504±17 | 252±10 | 858±53 |
| Ischemia (n=9) | 104±8* | 315±21* | 186±18* | 714±22 |
| AMO (n=10) | 167±8† | 540±31† | 260±12† | 810±49 |
Data are expressed as Mean ± SEM.
P<0.05 vs. time control
P<0.05 vs. ischemia. AMO, amobarbital + ischemia ADP, 2 mM; DNP (dinitrophenol, 0.3 mM).
3.2 Blockade of electron transport during ischemia prevents bcl-2 depletion
Ischemia decreased the content of bcl-2 in SSM compared to time control, whereas blockade of electron transport during ischemia preserved bcl-2 content (Figure 1). In contrast, ischemia did not substantially alter the bax content in SSM compared to time control or amobarbital treated SSM (Figure 1). Equal protein loading was confirmed by the detection of subunit IV of cytochrome oxidase (Figure 1). These results indicate that the ETC lies upstream of and contributes to bcl-2 depletion during ischemia. Bcl-2 could not be detected in IFM, likely secondary to the protease treatment used during isolation.
Figure 1. Blockade of electron transport during ischemia preserves bcl-2 content in SSM.
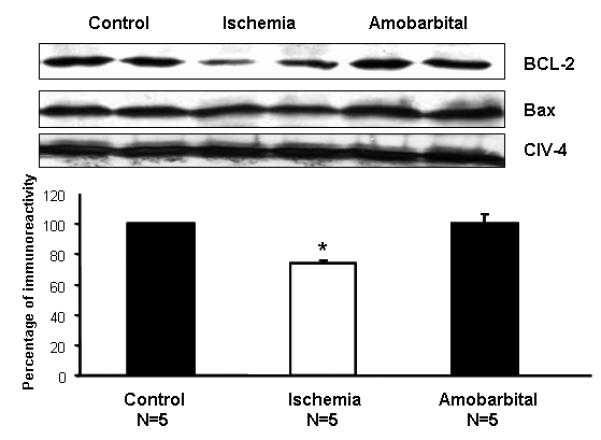
Ischemia markedly decreases the bcl-2 content in SSM isolated from rabbit heart compared to time control, whereas amobarbital prevents bcl-2 depletion during ischemia. Ischemia did not alter bax content compared to time control or amobarbital treated hearts. The upper panel consists of representative immunoblots, and the lower panel is the quantitation using densitometery. (Mean±SEM; *p<0.05 vs. time control; † p<0.05 vs. untreated-ischemia).
3.3 Blockade of electron transport during ischemia decreases net H2O2 production from SSM
Ischemic damage to the ETC increased the net production of H2O2 in SSM oxidizing glutamate as a complex I substrate. The increased H2O2 generation from SSM is also observed following ischemia with succinate as a complex II substrate in the presence of rotenone. Amobarbital treatment during ischemia decreased ROS generation from the ETC (Figure 2). Thus, ischemic damage to electron transport increased the net ROS production. Ischemia also tended to increase H2O2 generation from IFM isolated after ischemia when glutamate is the substrate, but the results did not reach the statistical difference (Figure 2).
Figure 2. The net release of H2O2 from isolated mitochondria in the presence and absence of ischemia.
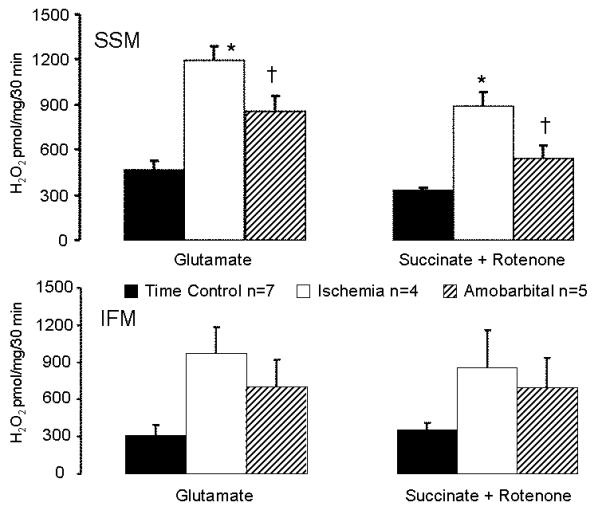
Compared to SSM from time control hearts, ischemic damage to electron transport chain increases net H2O2 production from SSM using glutamate as complex I substrate and succinate+rotenone as a complex II substrate. In contrast, protection of SSM by blockade of electron transport during ischemia by amobarbital decreases the H2O2 generation compared to the untreated ischemia (upper panel). Ischemia also tends to increase H2O2 generation in IFM compared to time control (lower panel). (Mean±SEM; *p<0.05 vs. time control; † p<0.05 vs. untreated-ischemia).
3.4 Blockade of electron transport during ischemia preserves cytochrome c content
The cytochrome c content in SSM decreased after 30 minutes of ischemia, whereas amobarbital treatment preserved cytochrome c content (Supplemental Table 2). Other cytochrome contents (c1, b, aa3) were unaffected. Ischemia did not alter cytochrome contents in IFM (Supplemental Table 2).
3.5 Blockade of electron transport during ischemia attenuates the opening of MPT
Ischemia significantly increased the calcium-induced swelling of SSM compared to time control. The SSM protected by amobarbital treatment during ischemia exhibited substantially decreased swelling compared to the SSM isolated from untreated ischemic hearts (Figure 3). Ischemia also led to calcium-induced swelling in IFM, but the rate and extent of swelling was less than in SSM (Figure 3).
Figure 3. Blockade of electron transport during ischemia prevents calcium-induced swelling in SSM following ischemia.
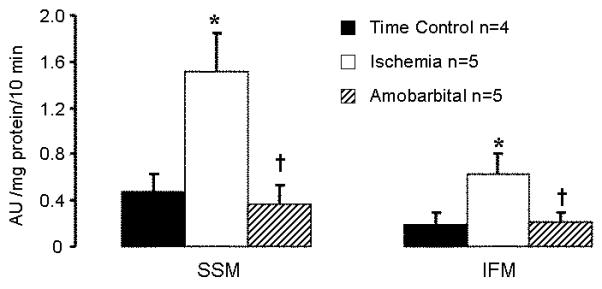
Ischemia leads to a significant increase in calcium-stimulated swelling in SSM. Blockade of electron transport protects SSM and limits the swelling compared to untreated mitochondria following ischemia. Ischemia also induces calcium stimulated swelling in IFM, albeit to a lesser degree. (Mean±SEM; *p<0.05 vs. time control; † p<0.05 vs. untreated ischemic group).
3.6 Inhibition of bcl-2 markedly induced MPT opening during oxidative stress
Mitochondrial swelling was used as an in vitro index of the susceptibility to opening of the permeability transition pore [38]. In the original tracing portion of Figure 4 (top), calcium (200 uM) stimulation led to mitochondrial swelling indicated by a decrease in absorbance at 520 nm (A520). H2O2 stimulation or HA14-1 treatment increased swelling in control SSM (Figure 4 top and bottom). In addition, H2O2 stimulation in the presence of HA14-1 dramatically increased swelling compared to H2O2 or HA14-1 treatment alone. Cyclosporine A prevented H2O2 or HA14-1 stimulated swelling, indicating that swelling occurred via MPT opening (Figure 4 top and bottom).
4.0 Discussion
Blockade of electron transport during ischemia decreases ROS production and the probability of MPT opening. Ischemia decreases bcl-2 content whereas protection of the electron transport chain by amobarbital inhibition preserves the bcl-2 content, providing strong support that the electron transport chain contributes to the previously observed [41,42] bcl-2 depletion during ischemia. The decreased bcl-2 content facilitates MPT opening in the presence of oxidative stress. The current results indicate that the ischemia-induced decrease in bcl-2 content in combination with increased ROS generation from the damaged electron transport chain will trigger MPT opening and increase cardiac injury during early reperfusion.
Mitochondria are the key mediators of cardiomyocyte survival and death during ischemiareperfusion, mediated in large part via MPT opening [28,32]. The exact components of the MPT are not clear, although cyclophilin D in the mitochondrial matrix regulates MPT opening [43]. Although MPT may open during cardiac ischemia [44-46], MPT opening mostly occurs during reperfusion [14,15,17,28,32,47-49]. Electron microscopy discloses that mitochondria remain intact in the isolated rabbit heart at the end of ischemia [3], supporting that MPT does not open during ischemia in situ [17]. The burst of ROS generation and rapid normalization of intracellular pH during early reperfusion are key factors that induce MPT [28,29]. Ischemic damage to electron transport increases ROS generation in isolated mitochondria [10]. In contrast, reversible blockade of electron transport only during ischemia decreases ROS generation from mitochondria isolated following reperfusion [4]. Moreover, reversible blockade of electron transport decreases in situ ROS generation during reperfusion [31]. Thus, the ETC is the source of ROS generation, and ischemia-damaged mitochondria release ROS during reperfusion [11]. In the present study, we found that the ischemia-damaged electron transport chain increased ROS generation accompanied by mitochondrial swelling. In isolated mitochondria, oxidative stress induced by exogenous H2O2 increased mitochondrial swelling. These results suggest that the ischemia-damaged ETC increases MPT opening by inducing ROS generation.
Although SSM were isolated at the end of ischemia, the swelling assay was conducted under the conditions of physiological pH, oxygen content, substrate, and high concentration of calcium. This condition is similar to the intracellular environment encountered by mitochondria during early reperfusion. In the same milieu, SSM isolated from ischemic hearts yet without ischemic damage as a result of amobarbital treatment did not exhibit significant swelling, indicating that damage to the ETC from ischemia was required for MPT opening. Our results support that ischemic damage to the ETC is a mechanism of MPT opening during reperfusion.
Bcl-2 is an anti-apoptotic protein located on the outer mitochondrial membrane and endoplasmic reticulum [13,50]. Over-expression of bcl-2 decreases ischemia-reperfusion injury in isolated hearts [42], whereas functional inhibition of bcl-2 with HA14-1, a small molecule that prevents bcl-2 inhibition of pro-apoptotic proteins [40], blocked cardioprotection [34]. Inhibition of bcl-2 with HA14-1 stimulates mitochondrial swelling [34], indicating that manipulation of bcl-2 function impacts MPT opening [13,14]. In the current study, ischemic damage to the ETC decreased bcl-2 content and favored MPT opening, whereas blockade of electron transport during ischemia prevented the loss of bcl-2 in association with the decreased mitochondrial swelling. In isolated mitochondria, inhibition of bcl-2 increased calcium-stimulated mitochondrial swelling, and swelling was further enhanced in the presence of oxidative stress. At the onset of reperfusion, re-oxygenation leads to ROS generation from the ischemia-damaged ETC [4,31,51]. This oxidative stress occurs in the setting of decreased mitochondrial bcl-2 content and favors MPT opening, eventually leading to the ischemia-reperfusion injury [28]. Thus, preservation of bcl-2 content via reversible blockade of electron transport contributes to reduced myocardial injury during ischemia-reperfusion by inhibition of MPT opening.
Damage to the ETC occurs mainly during ischemia [3,27,52] and persists during reperfusion [5,26]. The unexpected finding that interventions that protect myocardium during reperfusion, including postconditioning [26,47] and brief inhibition of electron transport [23,25] are effective without the recovery of oxidative phosphorylation [26] point to ETC dependent processes other than respiration as key effectors of mitochondrial-driven cardiac injury. In the present study, enhanced net production of oxidants, shift of the mitochondrial outer membrane proteome to a pro-apoptotic ensemble via depletion of bcl-2, and enhanced susceptibility to mitochondrial permeability transition are mechanisms that transduce the electron transport chain damage caused by ischemia into cardiomyocyte death during reperfusion.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs and 2PO1AG15885 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5.0 References
- 1.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–89. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 2.Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol. 2001;280:H2770–8. doi: 10.1152/ajpheart.2001.280.6.H2770. [DOI] [PubMed] [Google Scholar]
- 3.Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol. 1997;273:H1544–54. doi: 10.1152/ajpheart.1997.273.3.H1544. [DOI] [PubMed] [Google Scholar]
- 4.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 2006;319:1405–12. doi: 10.1124/jpet.106.110262. [DOI] [PubMed] [Google Scholar]
- 5.Lesnefsky EJ, Chen Q, Slabe TJ, Stoll MS, Minkler PE, Hassan MO, Tandler B, Hoppel CL. Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: role of cardiolipin. Am J Physiol Heart Circ Physiol. 2004;287:H258–67. doi: 10.1152/ajpheart.00348.2003. [DOI] [PubMed] [Google Scholar]
- 6.Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol. 2003;284:H549–58. doi: 10.1152/ajpheart.00708.2002. Epub 2002 Oct 10. [DOI] [PubMed] [Google Scholar]
- 7.Gross GJ, Auchampach JA. Reperfusion injury: does it exist? J Mol Cell Cardiol. 2007;42:12–8. doi: 10.1016/j.yjmcc.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 9.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–70. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–6. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- 11.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turrens JF, Beconi M, Barilla J, Chavez UB, McCord JM. Mitochondrial generation of oxygen radicals during reoxygenation of ischemic tissues. Free Radic Res Commun. 1991;12-13:681–9. doi: 10.3109/10715769109145847. [DOI] [PubMed] [Google Scholar]
- 13.Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol. 2007;292:C45–51. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 14.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–85. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 16.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 17.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307:93–8. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Q, Hoppel CL, Lesnefsky EJ. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J Pharmacol Exp Ther. 2006;316:200–7. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 19.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during Ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–7. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 20.Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH:ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002;544:687–93. doi: 10.1113/jphysiol.2002.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ismaeil MS, Tkachenko I, Gamperl AK, Hickey RF, Cason BA. Mechanisms of isoflurane-induced myocardial preconditioning in rabbits. Anesthesiology. 1999;90:812–21. doi: 10.1097/00000542-199903000-00024. [DOI] [PubMed] [Google Scholar]
- 22.Sgobbo P, Pacelli C, Grattagliano I, Villani G, Cocco T. Carvedilol inhibits mitochondrial complex I and induces resistance to H2O2 -mediated oxidative insult in H9C2 myocardial cells. Biochim Biophys Acta. 2007;1767:222–32. doi: 10.1016/j.bbabio.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 23.Ambrosio G, et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem. 1993;268:18532–41. [PubMed] [Google Scholar]
- 24.Park JW, Chun YS, Kim YH, Kim CH, Kim MS. Ischemic preconditioning reduces Op6 generation and prevents respiratory impairment in the mitochondria of post-ischemic reperfused heart of rat. Life Sci. 1997;60:2207–19. doi: 10.1016/s0024-3205(97)00236-1. [DOI] [PubMed] [Google Scholar]
- 25.Stewart S, Lesnefsky EJ, Chen Q. Reversible blockade of electron transport with amobarbital at the onset of reperfusion attenuates cardiac injury. Transl Res. 2009;153:224–31. doi: 10.1016/j.trsl.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Paillard M, Gomez L, Augeul L, Loufouat J, Lesnefsky EJ, Ovize M. Postconditioning inhibits mPTP opening independent of oxidative phosphorylation and membrane potential. J Mol Cell Cardiol. 2009;46:902–9. doi: 10.1016/j.yjmcc.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 27.Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol. 2007;292:C137–47. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- 28.Hausenloy DJ, Ong SB, Yellon DM. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol. 2009;104:189–202. doi: 10.1007/s00395-009-0010-x. [DOI] [PubMed] [Google Scholar]
- 29.Cohen MV, Yang XM, Downey JM. The pH hypothesis of postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation. 2007;115:1895–903. doi: 10.1161/CIRCULATIONAHA.106.675710. [DOI] [PubMed] [Google Scholar]
- 30.Chen Q, Lesnefsky EJ. Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic Biol Med. 2006;40:976–82. doi: 10.1016/j.freeradbiomed.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 31.Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ, Camara AK. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc Res. 2008;77:406–15. doi: 10.1016/j.cardiores.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77:334–43. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 33.McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2004;286:H1923–35. doi: 10.1152/ajpheart.00935.2003. Epub 2004 Jan 8. [DOI] [PubMed] [Google Scholar]
- 34.Obame FN, Zini R, Souktani R, Berdeaux A, Morin D. Peripheral benzodiazepine receptor-induced myocardial protection is mediated by inhibition of mitochondrial membrane permeabilization. J Pharmacol Exp Ther. 2007;323:336–45. doi: 10.1124/jpet.107.124255. [DOI] [PubMed] [Google Scholar]
- 35.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–9. [PubMed] [Google Scholar]
- 36.Williams JN., Jr. A Method For The Simultaneous Quantitative Estimation Of Cytochromes A, B, C1, And C In Mitochondria. Arch Biochem Biophys. 1964;107:537–43. doi: 10.1016/0003-9861(64)90313-3. [DOI] [PubMed] [Google Scholar]
- 37.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: Central role of complex III. J Biol Chem. 2003;278:36027–31. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 38.Wang G, et al. Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2005;288:H1290–5. doi: 10.1152/ajpheart.00796.2004. [DOI] [PubMed] [Google Scholar]
- 39.Milanesi E, et al. The mitochondrial effects of small organic ligands of BCL-2: sensitization of BCL-2-overexpressing cells to apoptosis by a pyrimidine-2,4,6-trione derivative. J Biol Chem. 2006;281:10066–72. doi: 10.1074/jbc.M513708200. [DOI] [PubMed] [Google Scholar]
- 40.An J, Chen Y, Huang Z. Critical upstream signals of cytochrome C release induced by a novel Bcl-2 inhibitor. J Biol Chem. 2004;279:19133–40. doi: 10.1074/jbc.M400295200. [DOI] [PubMed] [Google Scholar]
- 41.Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280:H2313–20. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 42.Imahashi K, Schneider MD, Steenbergen C, Murphy E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ Res. 2004;95:734–41. doi: 10.1161/01.RES.0000143898.67182.4c. [DOI] [PubMed] [Google Scholar]
- 43.Baines CP, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 44.Borutaite V, Jekabsone A, Morkuniene R, Brown GC. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J Mol Cell Cardiol. 2003;35:357–66. doi: 10.1016/s0022-2828(03)00005-1. [DOI] [PubMed] [Google Scholar]
- 45.Borutaite V, Morkuniene R, Arandarcikaite O, Jekabsone A, Barauskaite J, Brown GC. Nitric oxide protects the heart from ischemia-induced apoptosis and mitochondrial damage via protein kinase G mediated blockage of permeability transition and cytochrome c release. J Biomed Sci. 2009;16:70. doi: 10.1186/1423-0127-16-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104:1240–52. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3beta by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117:2761–8. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- 48.Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–31. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem. 1997;174:167–72. [PubMed] [Google Scholar]
- 50.Rudner J, Lepple-Wienhues A, Budach W, Berschauer J, Friedrich B, Wesselborg S, Schulze-Osthoff K, Belka C. Wild-type, mitochondrial and ER-restricted Bcl-2 inhibit DNA damage-induced apoptosis but do not affect death receptor-induced apoptosis. J Cell Sci. 2001;114:4161–72. doi: 10.1242/jcs.114.23.4161. [DOI] [PubMed] [Google Scholar]
- 51.Chen Q, Lesnefsky EJ. Ischemic damage to the mitochondrial electron transport chain favors opening of the permeability transition pore. FASEB J. 2008;22:E345. abstract 750.6. [Google Scholar]
- 52.Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, Turkaly PJ, Hoppel CL. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys. 2001;385:117–28. doi: 10.1006/abbi.2000.2066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.