

Abstract
Cytokine signaling pathways are frequent targets of oncogenic mutations in acute myeloid leukemia, promoting proliferation and survival. We have previously shown that the transcription factor PLAGL2 promotes proliferation and cooperates with the leukemia fusion protein Cbfβ-SMMHC in acute myeloid leukemia development. Here we show that PLAGL2 upregulates expression of the thrombopoietin receptor Mpl, using 2 consensus sites in its proximal promoter. We also show that Mpl overexpression efficiently cooperates with Cbfβ-SMMHC in development of leukemia in mice. Finally, we demonstrate that PlagL2-expressing leukemic cells show hyper-activation of Jak2 and downstream STAT5, Akt and Erk1/2 pathways in response to Tpo ligand. These results show that PlagL2 expression activates expression of Mpl in hematopoietic progenitors, and that upregulation of wild type Mpl provides an oncogenic signal in cooperation with CBFβ-SMMHC in mice.
Keywords: acute myeloid leukemia, PlagL2, MPL, Cbfb-MYH11, gene regulation, transcription factors
Introduction
Acute myeloid leukemia (AML) results from the accumulation of genetic alterations in hematopoietic progenitors that alter survival and differentiation programs. The study of these alterations is key for understanding the mechanism of leukemia development. The chromosome 16 inversion inv(16)(p13;q22), called inv16, is a frequent mutation found in AML cases, which creates the leukemia fusion gene CBFB-MYH11 (1). The Cbfβ-SMMHC protein encoded by the fusion gene creates preleukemic myeloid progenitors unable to differentiate, which transform into full-blown leukemia in synergy with mutations that promote proliferation and survival (2, 3).
A fraction of inv16-AML samples also present activating mutations in genes encoding components of the receptor tyrosine kinase signaling pathways, including KIT and FLT3, and the their downstream GTPases NRAS and KRAS (4-7) (8). These mutations produce ligand-independent constitutively active signaling that enhances the expansion and survival of leukemic blasts. We have previously shown that the zinc finger PLAG transcription factors, Plag1 and PlagL2, have similar capacity to induce proliferation of hematopoietic progenitors and cooperate with Cbfβ-SMMHC in acute myeloid leukemia development (9, 10). The PLAG factors bind to the GRGGC(N)6-8RGGK consensus site in regulatory regions of target genes to activate transcription, such as the promoter 3 of the insulin-like growth factor 2 gene (11). The PLAG oncogenic activity has also been reported in chronic lymphocytic leukemia, breast cancer, and salivary gland tumors (12-14). However, little is known on how PLAG induces transformation in hematopoietic progenitors and leukemia blasts.
In this study, we use gene expression profile analysis of hematopoietic progenitors and leukemic cells expressing PLAGL2 to identify genes that are consistently deregulated by PLAGL2. We identify the thrombopoietin receptor Mpl as a downstream target, using gene profile analysis, and validated its expression levels using quantitative RT-PCR and flow cytometry. Furthermore, we identify two PLAG binding sites conserved in mammals in Mpl proximal promoter using luciferase reporter and electrophoretic mobility shift assays. We determined that Mpl is a key downstream mediator of PLAGL2 leukemogenesis as overexpression of wild type Mpl efficiently cooperates with CBFβ-SMMHC in leukemia development using transplantation assays. The leukemic cells expressing PLAGL2 exhibit sensitivity to TPO ligand as evidenced by increased phosphorylation of Jak2, Erk1/2, Akt, and Stat5. Together, these results demonstrate that PLAGL2 regulates Mpl expression, and that upregulation of wild type Mpl cooperates with CBFB-MYH11 in leukemia development in mice.
Materials and Methods
Microarray analyses
Sample preparation
For infected BM-HP cells, 129SvEv Cbfb+/56M;Mx1Cre mice were treated with 150 mg/kg 5-FU, and BM cells were harvested 5 days later. Cells were subjected to red-blood-cell lysis solution (Purgene, Gentra Systems, Minneapolis, MN), and spin-infected with 2 rounds of retrovirus (MIG or MIG-PLAGL2) as previously described (10). The GFP-positive cells were sorted by FACS and total RNA was immediately isolated (samples wt-MIG1, wt-MIG2, wt-P1, and wt-P2). The mouse AML cells were generated using Cbfb-MYH11 conditional knock-in mice as previously described (3). AML cells were isolated from spleen of 3 independent leukemic mice expressing MIG-PLAGL2 (samples AML-P1, AML-P2, and AML-P3) and from 3 independent leukemic mice with low Plagl2 levels (AML-x1, AML-x2, AML-x3). In all cases, RNA was extracted with Trizol (Invitrogen, Carlbad CA) and cleaned with RNA-Easy columns (Qiagen, Valencia CA). Complementary-DNA was synthesized using the Superscript II (Invitrogen, Carlbad CA), and Biotin-labeled cRNA was subsequently synthesized using RNA-transcript labeling kit (Affymetrix, Santa Clara, CA). Ten μg of labeled cRNA was fragmented and hybridized to Affymetrix Mouse Genome 430 2.0 Chip Arrays (Affymetrix, Santa Clara, CA).
Data Analysis
To identify transcripts that were differentially expressed in MIG-PLAGL2, AML-PLAGL2 and AML samples, we used the GeneSpring (version 7.3; http://www.silicongenetics.com) and GENECLUSTER (version 2.0; http://www-genome.wi.mit.edu) software packages. The expression values of each probe set in each of the MIG-PLAGL2, AML-PLAGL2 and AML samples were normalized to that of the MIG control number one or number two samples. For a given transcript to be considered as differentially expressed in MIG-PLAGL2, AML-PLAGL2 and AML samples, it had to meet the following three criteria. First, the signal values in these samples and the MIG control sample had to exhibit a relative difference of at least a factor of 2 and an absolute difference of 100 units. Second, changes of comparable magnitude had to be reproduced in 2 out 2 independent experiments for the MIG-PLAG2 sample or 3 out 3 independent experiments for the AML-PLAGL2 and AML samples when compared to MIG controls. Third, when statistical group comparisons (Welch's approximate t test) were applied to the raw signal values of the MIG control samples and the MIG-PLAGL2, AML-PLAGL2 or AML samples, it yielded a significance value of p≤0.05. Transcripts differentially expressed in MIG-PLAGL2, AML-PLAGL2 or AML samples were subjected to hierarchical clustering analysis using the GeneSpring (version 7.3) software package. For this analysis, the signal value of each transcript in each of the samples was normalized to the corresponding value of the MIG control number one sample. The resulting ratios were subjected to two-dimensional clustering by using the average linkage algorithm and the standard correlation coefficient as a distance metric.
Reverse transcriptase and quantitative RT-PCR analyses
RNA from mouse BM and AML cells was extracted with Trizol (Invitrogen, Carlsbad CA). First-stand cDNA was generated by using 2 μg RNA, 1 U Superscript-II reverse transcriptase (Invitrogen, Carlsbad, CA), and 0.5 μM oligo-dT primer in a 20-μL reaction. SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) was used for qPCR according to the manufacturer's instructions. Mpl primers were Mplx1 and Mplx2 and β-actin primers were ActbF1 and ActbR1 (supplementary Table 1). QPCR was performed in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). Samples were normalized to β-actin transcript levels, and relative values were determined using standard curve method.
Retroviral production
The MIG-Mpl construct was generously provided by Harvey Lodish (Massachusetts Institutes of Technologies, Cambridge MA). The MIG-MplY mutant construct was generated by PCR using primers pMSCV1 and MplY112rev, cut with EcoR1 and Xho1, and cloned into MIG. Phoenix packaging cells (Gary Nolan, Stanford University, Stanford CA) were co-transfected with 2 μg retroviral constructs, 2 μg Psi-Eco packaging plasmid with Effectene (Qiagen, Valencia CA) according to manufacturer's protocol. Retrovirus supernatants were taken at 48, 56, and 72 hrs and titered in by GFP-FACS analysis.
Bone marrow transduction and transplantation (tBMT)
Endogenous Cbfb-MYH11 expression was induced in Cbfb56M/+Mx1Cre conditional knock-in mice (3) using the Mx1Cre transgenic system (15). Briefly, mice were injected with 3 doses every other day of polyinosinic-polycytidylic acid (pIpC; Sigma, St Louis, MO) at 3- to 5-weeks of age, treated with 150 mg/kg 5-fluorouracil (5FU), and BM cells were harvested 5 days later. Efficiency of Cre-mediated Cbfb-MYH11 induction was consistently above 90% (3). The BM progenitor cells were spin-infected twice with retrovirus supernatant, and 5×105 to 1×106 BM cells transplanted by intravenous injection (iv.) into 4- to 6-week old sublethally irradiated (650 rads) 129SvEv wild-type recipient mice. Mice were under daily observation for early signs of leukemia. These signs included limited motility, pale paws, and dehydration. At first signs of illness, peripheral blood was analyzed for the presence of immature cells. FACS analysis of peripheral blood was performed by using antibodies to cell-surface markers Gr-1, CD11b, B220, CD3, Ter119, and c-kit (BD Biosciences, San Diego CA). For flow cytometry analysis of Mpl receptor expression, a polyclonal rabbit anti-Mpl extracellular domain antibody (provided by Harvey Lodish, Whitehead Institute for Biomedical Research, Cambridge MA) and a PE-anti rabbit secondary antibody were used. For secondary transplantations, leukemic cells were harvested from the BM or spleen of affected mice in RPMI 1640 and 20% FBS (both from (Invitrogen, Carlsbad, CA), and 1×106 single-cell suspension aliquots were transplanted iv. into sub-lethally irradiated 4- to 6-week-old 129SvEv recipients.
Immunoblot analyses
Cryopreserved AML cells were thawed and immediately serum starved in RPMI media with 1% BSA for 60 min at 37°C (Invitrogen, Carlsbad CA). Cells were then incubated with RPMI media with 0.1% BSA and 0, 5, 25, or 50 ng/ml Tpo (Peprotech, Rockyhill NJ) for 10 min. Cells were washed with PBS and resuspended in RIPA buffer with protease-inhibitor-cocktail III (Calbiochem, Darmstadt Germany). Antibodies included anti-Jak2 (cat#sc-294; Santa Cruz Biotechnology Inc., Santa Cruz, CA), and anti-phospho-Jak2-Tyr221 (cat#3774), anti-phospho-Stat5-Tyr694 (cat#9351), anti-Stat5 (cat#9310), anti-phospho-Akt-Ser273 (cat#4058), anti-Akt (cat#9272), anti-phospho-Erk1/2 -Thr202/Tyr204 (cat#9101), and anti-Erk1,2 (cat#9102; all from Cell Signaling Technology, Danvers MA).
DNA Sequence Analysis
The search for PLAG consensus site GRGGC(6-8)RGGK was analyzed using the UCSC Genome Browser on Mouse July 2007 (NCBI37/mm9) Assembly (http://genome.ucsc.edu). Promoter regions were defined as 1000 base pairs upstream and 200 base pairs downstream of transcription start site, with this site as position 1000 (Supplementary Table 1). Evolutionary conservation of PLAG sites among 5 mammal species (human, orangutan, mouse, rat, dog and horse) was estimated. The sequence conservation analysis at the Mpl proximal promoter was performed using ClustalW-Alignment Analysis from MacVector9.5.2 (MacVector Inc.).
Luciferase assays
A 300 bp fragment of the Mpl proximal promoter was amplified with primers MPLFLPromkpnF and MPLFLpromBglR using PFU (Stratagene, La Jolla CA), and the amplicon was digested with KpnI and BglII and cloned into pGL3-Basic (Promega, Madison WI). Mutants were constructed by overlapping PCR with primers Mpls1F and Mpls1R for “site-1”, Mplps2F and Mplps2R for “site-2”, and Mplps3F and Mplps3R for “site-3” (Supplementary Table 1). The mutant primers were used with primers MPLFLpromkpnF and MPLFLpromBglR to amplify overlapping fragments that were annealed, digested with KpnI and BglII and cloned into the PGL3-basic vector. Constructs were transfected into NIH3T3 cells along with MhCD4 or MhCD4-PLAGL2 and PRL-TK with Effectene (Quiagen Valencia CA). Cells were analyzed for luciferase levels with the Dual-Luciferase Reporter Assay System (Promega, Madison WI).
Electrophoretic mobility shift assay
The electrophoretic mobility shift assay was performed as previously described (16). Briefly, 3T3 cells transfected with MIG-PlagL2 were used for isolation of nuclear proteins and added to protein-DNA binding reactions with double stranded DNA oligos representing PlagL2 binding sites in the Mpl promoter. The sequence for DNA oligos P1ss and P1as, P2ss and P2as, P3ss and P3as, mutP1ss and mutP1as, mutP2ss and mutP2as, and mutP3ss and mutP3as are shown in Supplementary Table 1.
The P1ss, P2ss and P3ss oligos were radioactively labeled with 32P using OptiKinase (USB, Cleveland OH), and the excess 32P-γATP was removed by purifying the labeled oligos using G-25 Mini Quick Spin Oligo columns (Roche, Basel, Switzerland). The oligos were annealed with equal amounts of antisense oligos (P1as, P2as, P3as respectively) to obtain a double stranded labeled oligo with a PlagL2 binding site. 100 pg of the oligo were used for each binding reaction. Cold competitors and mutant competitors were obtained the same way, but without labeling the oligos. When using cold competitors (wild type or mutant), a 1:200 molar ratio of labeled and cold oligo was used. Constant DNA amount was maintained by adding scrambled oligos Sss and Sas. The protein-DNA binding reactions were loaded onto a 4% polyacrylamide gel, ran until unlabeled 32P-γATP went out into buffer, dried and exposed for 24 hrs.
Cytology Analysis
Cells from peripheral blood smears were stained with Wright-Giemsa. Pictures were taken using a Zeiss Axioskop 40 microscope using 100x lenses and a Zeiss AxioCam MRc camera and software.
Results
Mpl is upregulated by the transcription factor PLAGL2 in hematopoietic progenitors and AML blasts
We have previously shown that the transcription factor PLAGL2 induces AML in synergy with Cbfβ-SMMHC in mice (10). In order to identify PLAGL2 target genes that participate in leukemia development, we defined a group of genes consistently deregulated by PLAGL2 by assessing those genes consistently deregulated in bone marrow (BM) hematopoietic progenitors (HP) and AML blasts expressing PLAGL2. The HPs from Cbfb+/MYH11 conditional knock in mice expressing Cbfb-MYH11 were transduced with a pMSCV-IRES-GFP (MIG) or pMSCV-PLAGL2-IRES-GFP (PL2-MIG) retrovirus (Supplementary Figure 1a). The RNA from GFP sorted cells was used in a gene profile analysis using 2 independent samples per group, and represent the “early deregulated gene-set”. In addition, the expression profiles of 6 mouse AML samples expressing Cbfb-MYH11, 3 expressing (P+1AML to-P+3AML) and 3 not expressing PLAGL2 (P-1AML to-P-3AML), were analyzed (experimental design in Supplementary Figure 1b). Two-dimensional hierarchical clustering of the 10 samples was performed using GeneSpring® software (Supplementary Figure 1c). Thirty-three probes identified 23 genes consistently upregulated over 2-fold in PLAGL2 expressing samples (Figure 1A). These included upregulation of transcripts for the thrombopoietin receptor Mpl, the G-protein couple receptors Gpr56, Prokr1 and Gpr124, the endothelial protein-C receptor, the vitamin-D receptor, endoglin, and collagenase genes Col18a1 and Col4a2. In addition, the transcripts for the extracellular matrix proteins tenascin-XB and Spondin-2, the ubiquitin ligase Cullin-7, the actin-associated Cyb5r3, and Janus kinase Jak3 were consistently increased by PLAGL2. Consistent with the reported transcription activation role of PLAGL2, this analysis did not identify down-regulated genes (17, 18). The presence of PLAG consensus GRGGC(8-6)RGGK sites (11) was searched in a 1,200 base pairs window (1000 base pairs upstream and 200 base pairs downstream of the gene transcription start), with the start site as position 1,000 (Supplementary Table 2). The conservation of these sites in 5 mammal species was analyzed using the UCSC Genome Sequence. From the 23 identified genes, 3 genes (Mpl, Ccdc8 and Eng) showed high conservation in all sites, while 4 genes (Hif3a and 3 genes with unknown function) have at least 1 conserved site.
Figure 1. Identification of PLAGL2 target genes in leukemia development using microarray analysis.

(A) Cluster of genes specifically up-regulated in PLAGL2+ cells from two-dimensional hierarchical clustering analysis. Hematopoietic progenitor samples infected with MIG (MIG1-HP and MIG2-HP), HPs infected with PLAGL2-MIG (PL2+1HP and PL2+2MIG), AML samples expressing PLAGL2 (PL2+1, PL2+2, and PL2+ AMLs), and AML samples not expressing PLAGL2 (PL2−1, PL2−2, PL2−3 AMLs). All samples express Cbfb-MYH11. (B) Relative Mpl transcript levels of Cbfb-MYH11/hematopoietic progenitor (HP) transduced with MIG (lane 1) or MIG-PLAGL2 (lane 2) retrovirus, and of PLAGL2-negative (PL2−; lanes 3-6) and PLAGL2-positive (PL2+; lanes 7-10) Cbfb-MYH11 AML samples using quantitative RT-PCR analysis. (C) Expression of Mpl receptor in AML cells with non-detectable PLAGL2 expression (left panel) or expressing PLAGL2 (right panel) using FACS analysis. Percentage of cells expressing Mpl is shown relative to negative control.
Considering that PLAGL2 induces proliferation and replating capacity of hematopoietic progenitors (10), and the high conservation of the three PLAG sites in its promoter region, we focused our study on the cytokine thrombopoietin receptor Mpl. Quantitative RT-PCR (qRT-PCR) analysis revealed that the level of Mpl transcript was increased over 100-fold 48 hours after transduction (Figure 1B, lanes 1 and 2). A similar increase (30- to 500-fold) was observed in AML cells expressing PLAGL2 when compared to AML cells not expressing PLAGL2 (Figure 1B, lanes 3 to 10). In addition, the levels of Mpl receptor in membrane were markedly increased in AML cells expressing PLAGL2 using flow cytometry (Figure 1C).
PLAGL2 induces expression of the Mpl proximal promoter
The Mpl gene consists of 12 exons highly conserved in mammals and its expression is regulated from a proximal promoter spanning approximately 200 bp upstream of the translation start site (Figure 2A, top). This region is highly conserved in the mammalian genomes (30 way multiz-alignment and conservation; USCS-Mouse genome Browser; Figure 2A, middle). The Mpl proximal promoter (−189 to +3 from translation start site) is highly conserved in mammals, including human, monkey, mouse, dog, horse, and cow genomes (ClustalW sequence alignment; Figure 2A, bottom). This region has 3 PLAG consensus binding-sites. To test whether PLAGL2 could directly activate Mpl expression, we tested PLAGL2 responsiveness of a luciferase reporter carrying the Mpl proximal promoter in NIH3T3 cells. PLAGL2 activated the reporter over 30-fold relative to control (Figure 2B, lanes 1 and 2). The introduction of previously described (11) point mutations in PLAG binding sites 1 (m1) and 2 (m2) resulted in a 2- and 5-fold reduction in luciferase activity respectively (Figure 2B, columns 3-6), indicating that these sites are critical for PLAGL2 induced Mpl transcription. Conversely, mutation of the third PLAG site (m3) did not change significantly the PLAGL2 responsiveness, suggesting that this site may not participate in the Mpl promoter activity (Figure 2B, columns 7 and 8). Importantly, mutation of all three PLAG binding sites totally obliterated PLAGL2 responsiveness of the reporter, confirming that binding of PLAGL2 to the promoter is necessary for transcriptional activation (Figure 2B, columns 9 and 10). To validate PLAGL2 binding to these sites, we performed electrophoretic mobility shift assays using oligonucleotides covering each of the three Mpl binding sites (o), and a combination of labeled oligonucleotides with cold competitor (c), or mutated competitor (m) oligonucleotides (Figure 2C). These assays show that P1 and P2 sites bind to PLAGL2 with specificity, but P3 site binding is poorly competed with cold competitor. These results suggest that PLAGL2 can directly activate the Mpl proximal promoter using the P1 and P2 binding sites.
Figure 2. Mpl expression is upregulated by PLAGL2 in hematopoietic cells.
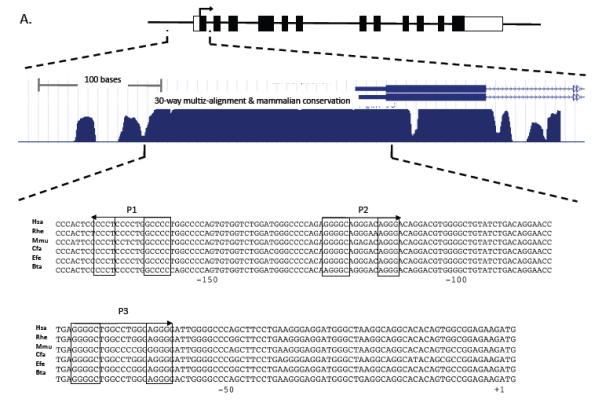

(A) Top: depiction of the Mpl gene structure, including untranslated exon sequences (blank boxes), coding regions (black boxes), translation start (arrow). Middle: zoom-in of the proximal promoter, exon 1 (black box) and section of intron 1 (arrowed line), and 30-way multiz-alignment & mammalian conservation analysis of the mouse genome segment chr4:118,129,933-118,130,333; using the UCSC Genome Browser on Mouse July 2007 (NCBI37/mm9) Assembly. Bottom: ClustalW sequence alignment of the Mpl proximal promoter (−189 to +3 from translation start site) including human (Hsa), monkeys (Rhe), mouse (Mmu), dog (Cfa), horse (Efe), and cow (Bta). The PLAGL2 consensus binding sites GRGGC(6-8)RGGK (P1 and P2) and a third site (P3) with an (N)9 linker are shown in boxes. Arrows indicate orientation of binding site. (B) Analysis of PLAGL2 activation of the Mpl proximal promoter using luciferase reporters in NIH3T3 cells. The luciferase reporters with 300 bp of the Mpl proximal promoter (wt) and respective mutants ablating one or more sites (m1, m2, m3, m1, 2, 3) are illustrated on the left panel. Fold activation relative to wt reporter and no PLAGL2 (MIG + wt) of each construct with (PL2) or without (MIG) PLAGL2 is shown on right panel. (C) Electrophoretic mobility shift assay of PLAGL2 consensus sites P1, P2, and P3 in the Mpl proximal promoter, using labeled wild type oligo (o), unlabeled oligo (c), or labeled oligo with point mutation in the core box of the Consensus PLAGL2 site (m). Arrow indicates PLAGL2:DNA binding.
Overexpression of Mpl induces AML in cooperation with CBF fusion genes
Since PLAGL2 induces expression of Mpl in HPs and AML cells, we asked whether Mpl expression is a key event triggered by PLAGL2 in leukemia development. We tested whether co-expression of Cbfb-MYH11 and Mpl is sufficient for AML development in a BM transplantation assay (Figure 3A-B, and “Materials and Methods”). All of the mice transplanted with BM cells expressing Cbfb-MYH11/MIG-Mpl (n=14) readily developed leukemia between 4 and 10 weeks post transplantation (Figure 3C, blue line). Control groups remained healthy over 5 months, including Cbfb-MYH11/MIG (n=10) or MIG-Mpl (n=8) transplanted mice (Figure 3C, black line). Secondary transplantation of AML cells into sublethally irradiated recipients succumbed with leukemia with a median latency of 4 weeks (Figure 3C, green line; n=11) confirming the leukemic nature of the disease. The pathology of disease in Cbfb-MYH11/Mpl AML mimics that of Cbfb-MYH11/MIG-PLAGL2 leukemia previously described (9, 10, 19). Briefly, we found a significant increase of GFP(+), Kit(+) immature cells in peripheral blood (Figure 3D), confirming the presence of the MIG-Mpl retrovirus, and were lineage negative (Lin: CD3, Gr-1, B220, Ter119, and Mac-1). These cells were blast-like cells with dark and pigmented cytoplasm, and monocytic-like cells with lobulated nucleus (Figure 3E, red arrow and asterisks respectively). Moribund mice also displayed infiltration of leukemic cells into the spleen, causing a significant enlargement (Figure 3F). The MPL mutation tyrosine to phenylalanine at amino acid 112 (Y112F) in the carboxyl-terminus largely reduces downstream signaling through the PI3k/Akt, Ras/Erk and Stat3/5 pathways, and is critical for proliferation of hematopoietic cell lines and for transformation of FRE-rat fibroblasts (20-22). To test if downstream MPL signaling was critical for leukemogenesis, we performed BM transplantation assays with BM expressing Cbfb-MYH11 and transduced with MIG-MplY112F retrovirus. Recipient mice transplanted with BM cells expressing Cbfb-MYH11 and MIG-MplY112F mice developed AML with decreased penetrance (3/8) and latency (range 12 and 16 weeks; Figure 3C, red line). These data suggest that activation of signaling networks by upregulation of Mpl receptor is critical in leukemia development.
Figure 3. Mpl expression cooperates with Cbfβ-SMMHC in leukemia development.

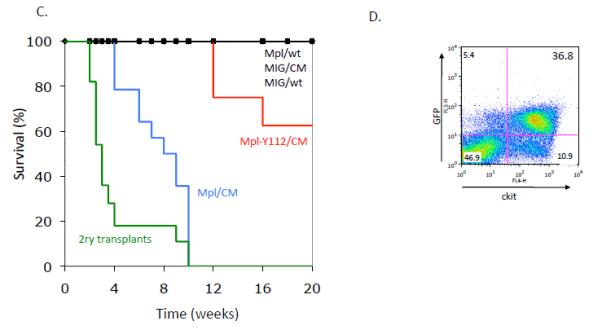

Schematic representation of viral vectors (A) and experimental strategy (B) used in BM transplantation assay. Hematopoietic progenitors from wild type and Cbfb+/56M;Mx1Cre mice (expressing CBFb-MYH11) were infected with MIG, MIG-Mpl, or MIG-Mpl-Y112F retrovirus and transplanted into sub-lethally irradiated recipient mice. (C) Kaplan Meier survival curves for mice transplanted with wild type (wt) or Cbfb+/56M;Mx1Cre (CM) cells infected with MIG-Mpl (Mpl), MIG-MplY112, or MIG retroviruses. (D) Representative FACS analysis of peripheral blood white blood cells from a leukemic mouse expressing Cbfβ-SMMHC and MIG-Mpl (GFP+cKit+ cells). (E) Representative cell morphology of 2 AML samples from peripheral blood of CM/Mpl leukemic mice, depicting balstlike (red arrow) and monocyticlike (asterisks), with magnification 100X. (F) Spleen weight of leukemic (n=11) and control (n=6) mice.
CBFb-MYH11-AML cells expressing PlagL2 are hypersensitive to Tpo ligand
The TPO/MPL signaling pathway regulates proliferation and survival of hematopoietic progenitor cells. In contrast to other members of the cytokine-receptor superfamily, MPL lacks intrinsic tyrosine kinase activity. Ligand stimulation of MPL results in the phosphorylation of JAK2 kinase, a required step for phosphorylation of cytoplasmic residues of MPL, which then triggers activation of AKT/mTOR, STAT3/5 and RAS/ERK cascades (23). We analyzed the phosphorylation status of Jak2 in CBFb-MYH11-AML cells expressing PLAGL2 (PL2+) or not (PL2−) cultured in presence of Tpo after serum starvation. We found increased phospho-Jak2 levels in presence of Tpo only in samples expressing PLAGL2 (Figure 4A). In addition, the phosphorylation of downstream signals Akt1, Stat5, and Erk1/2 was increased accordingly in response to Tpo in these AML samples (Figure 4B). Of note, phopspho-Akt levels were significantly increased upon Top induction (Figure 4C). These results demonstrate that Cbfb-MYH11-AML cells expressing PLAGL2 have an activated Mpl signaling pathway in response to Tpo ligand.
Figure 4. AML cells expressing Cbfb-MYH11 and PLAGL2 are hypersensitive to Tpo ligand.

(A) Phosphorylation analysis of Mpl target Jak2 protein in CBFb-MYH11 AML samples expressing PLAGL2 (PL2+ AML) or not (PL2− AML), after serum starvation and incubation with Tpo ligand. (B) Expression of phospho-Stat5, Stat5, phospho-Erk1/2, Erk1/2, phospho-Akt, Akt, and B-actin by immunoblot analysis after serum starvation and incubation with Tpo ligand. (C) Densitometric quantification of phospho-protein levels shown in B.
Discussion
The leukemias with core-binding factor inv(16)(p13;q22) frequently present oncogenic mutations in receptor tyrosine-kinase pathway proteins KIT, FLT3 and RAS, which provide proliferation and survival capacity to hematopoietic blasts (8). We have previously shown that the related transcription factors PLAG1 and PLAGL2 cooperate with CBFβ-SMMHC in leukemia development (10). This study shows that the transcription factor PLAGL2 activates Mpl receptor, inducing activation of the Erk, Stat, Akt pathways.
The members of the PLAG zinc finger transcription factors PLAG1 and PLAGL2 have been shown to activate growth factor associated genes, including IGFII and CLF1, in epithelial cells (24). We found that the PLAG factors promote proliferation of hematopoietic progenitors and participate in AML development (9, 10). Here we designed a gene expression profiling approach to identify genes consistently deregulated by PLAGL2 in hematopoietic progenitors as an “early” readout of PLAG function, and in leukemic cells expressing PLAGL2 as a “leukemic” readout. In this analysis, 23 genes were upregulated over 2 fold while no repressed genes were found, highlighting the transcription activation role of PLAG proteins. The sustained upregulation of Mpl levels in membrane by PLAGL2 strongly suggests that Mpl is a major player in PLAG mediated oncogenicity. Consistent with these findings, mutations in the MPL and TPO genes that promote translation and protein stability have been found in the inherited myeloproliferative syndrome familial essential thrombocythemia (25, 26). In addition, somatic activating mutations in MPL are present in a fraction of patients with myelofibrosis with myeloid metaplasia and essential thrombocythemia (27, 28). Future studies should evaluate the levels of PLAGL2 and MPL in myeloproliferative disorders that lack oncogenic mutations in MPL.
Mpl expression is regulated by the ETS, RUNX and GATA factors in megakaryocytes (29-31). The Tpo/Mpl pathway is a major regulator of hematopoietic stem cells (32-34). However, the mechanism of regulation of Mpl expression in HSCs is not well understood. We identified two PLAG consensus sites within a 180 base pair evolutionary conserved region upstream of the Mpl translation initiation site. These sites can be bound by PLAGL2 which upregulates transcription, strongly implicating PlagL2 as regulator of Mpl transcription. Importantly, RUNX and ETS binding sites are also located within the Mpl proximal promoter, suggesting that Mpl expression could be regulated by the combinatorial role of multiple transcription factors in megakaryocytes, stem cells and leukemic cells. Future studies should unravel the specific PLAG, RUNX and ETS factors that interact to regulate Mpl expression in different cell types. In addition to the oncogenic role of PLAG1 and PLAGL2, PLAGL1 (the third member of the PLAG family) has been implicated as a tumor suppressor in multiple types of cancer, including breast and ovarian cancer and neck squamous cell carcinomas (reviewed in (35)), and has recently been found repressed in diffuse large B-cell lymphoma (36). Since PLAGL1 binds to the first GC-block, it is also possible that MPL expression in hematopoietic progenitors and leukemia may be affected by PLAGL1-loss of expression. Coexpression analysis of available gene expression datasets of human AML (www.oncomine.org) suggest no correlation between PlagL2 and Mpl transcript levels. However, further study of the three PLAG proteins and other factors as modulators of Mpl signaling in human AML is warranted.
The present study demonstrates that PLAGL2 activates expression of Mpl, using two PLAG-consensus binding sites in its proximal promoter, and activates its downstream signaling in hematopoietic progenitors and leukemic cells. This activation is at least one of PLAGL2 induced oncogenic signals that promotes leukemia development in cooperation with CBFβ-SMMHC in mice.
Supplementary Material
Representation of microarray strategy to identify PLAGL2 target genes in leukemia development. (A) Representation of pMSCV-IRES-GFP (MIG) and MIG expressing PLAGL2 (MIG-PL2) retroviruses. (B) Bone marrow derived hematopoietic progenitors (HP) expressing CBFb-MYH11 are transduced with MIG or MIG-PL2 retrovirus, GFP-sorted, and used for Affymetrix-based microarray analysis (n=2 per group). Leukemic cells expressing CBFb-MYH11 (PL2−AML blasts) and leukemic cells expressing CBFb-MYH11 and PLAGL2+ (PL2+ AML) (n=3 per group) are used in Affymetrix-based microarray analysis.
(C) Two-dimensional hierarchical clustering analysis of probe sets that are differentially expressed in hematopoietic progenitor cells expressing Cbfb-MYH11 and PLAGL2 (PL2− HP), and leukemic samples expressing Cbfb-MYH11 and PLAGL2 (PL2+ AML) or expressing Cbfb-MYH11 but not PLAGL2 (PL2− AML) compared to a MIG-infected progenitor sample (MIG-HP) expressing Cbfb-MYH11. Triangle marks position of data shown in Figure 1.
Expression pattern of Mpl probe in gene profiled samples. (A) Scatter plot comparison of the raw signals of the entire probe sets of mouse genome from the HPs expressing PLAGL2 sample (Y axis, PL2+1 HP) and the HPs not expressing PLAGL2 sample (X-axis, MIG1-HP). Up-regulated (red) and down-regulated (green) probe-sets over 2 fold are shown. Arrow indicates the position of Mpl probe set. (B) The normalized expression patterns of probe sets upregulated in PLAGL2 samples in a sub-tree containing the Mpl probe set. The profile of the Mpl probe set is indicated by black arrow.
Acknowledgments
We wish to thank Julie Zhu and Stephen Baker for the statistical analyses of the expression data. This work was supported by National Institutes of Health Grant CA09683 to L.H.C, and Core resources were supported by the Diabetes Endocrinology Research Center Grant DK32520. L.H.C is recipient of a Scholar Award from the Leukemia & Lymphoma Society.
Footnotes
The authors declare no conflict of interest in relation to the work described in this manuscript.
References
- 1.Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278(5340):1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 2.Castilla LH, Wijmenga C, Wang Q, Stacy T, Speck NA, Eckhaus M, et al. Failure of embryonic hematopoiesis and lethal hemorrhages in mouse embryos heterozygous for a knocked-in leukemia gene CBFB-MYH11. Cell. 1996;87(4):687–696. doi: 10.1016/s0092-8674(00)81388-4. [DOI] [PubMed] [Google Scholar]
- 3.Kuo YH, Landrette SF, Heilman SA, Perrat PN, Garrett L, Liu PP, et al. Cbf beta-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell. 2006 Jan;9(1):57–68. doi: 10.1016/j.ccr.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 4.Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML) Leukemia. 2006 Jun;20(6):965–970. doi: 10.1038/sj.leu.2404188. [DOI] [PubMed] [Google Scholar]
- 5.Care RS, Valk PJ, Goodeve AC, Abu-Duhier FM, Geertsma-Kleinekoort WM, Wilson GA, et al. Incidence and prognosis of c-KIT and FLT3 mutations in core binding factor (CBF) acute myeloid leukaemias. Br J Haematol. 2003 Jun;121(5):775–777. doi: 10.1046/j.1365-2141.2003.04362.x. [DOI] [PubMed] [Google Scholar]
- 6.Schessl C, Rawat VP, Cusan M, Deshpande A, Kohl TM, Rosten PM, et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J Clin Invest. 2005 Aug;115(8):2159–2168. doi: 10.1172/JCI24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004 Apr 15;350(16):1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- 8.Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002 Jul;2(7):502–513. doi: 10.1038/nrc840. [DOI] [PubMed] [Google Scholar]
- 9.Castilla LH, Perrat P, Martinez NJ, Landrette SF, Keys R, Oikemus S, et al. Identification of genes that synergize with Cbfb-MYH11 in the pathogenesis of acute myeloid leukemia. Proc Natl Acad Sci U S A. 2004 Apr 6;101(14):4924–4929. doi: 10.1073/pnas.0400930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landrette SF, Kuo YH, Hensen K, Barjesteh van Waalwijk van Doorn-Khosrovani S, Perrat PN, Van de Ven WJ, et al. Plag1 and Plagl2 are oncogenes that induce acute myeloid leukemia in cooperation with Cbfb-MYH11. Blood. 2005 Apr 1;105(7):2900–2907. doi: 10.1182/blood-2004-09-3630. [DOI] [PubMed] [Google Scholar]
- 11.Voz ML, Agten NS, Van de Ven WJ, Kas K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000;60(1):106–113. [PubMed] [Google Scholar]
- 12.Declercq J, Skaland I, Van Dyck F, Janssen EA, Baak JP, Drijkoningen M, et al. Adenomyoepitheliomatous lesions of the mammary glands in transgenic mice with targeted PLAG1 overexpression. Int J Cancer. 2008 Oct 1;123(7):1593–1600. doi: 10.1002/ijc.23586. [DOI] [PubMed] [Google Scholar]
- 13.Kas K, Voz ML, Roijer E, Astrom AK, Meyen E, Stenman G, et al. Promoter swapping between the genes for a novel zinc finger protein and beta-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat Genet. 1997 Feb;15(2):170–174. doi: 10.1038/ng0297-170. [DOI] [PubMed] [Google Scholar]
- 14.Pallasch CP, Patz M, Park YJ, Hagist S, Eggle D, Claus R, et al. miRNA deregulation by epigenetic silencing disrupts suppression of the oncogene PLAG1 in chronic lymphocytic leukemia. Blood. 2009 Oct 8;114(15):3255–3264. doi: 10.1182/blood-2009-06-229898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 16.Javed A, Zaidi SK, Gutierrez SE, Lengner CJ, Harrington KS, Hovhannisyan H, et al. Protein-deoxyribonucleic acid interactions linked to gene expression: electrophoretic mobility shift assay. Methods Mol Biol. 2004;285:45–55. doi: 10.1385/1-59259-822-6:045. [DOI] [PubMed] [Google Scholar]
- 17.Hensen K, Van Valckenborgh IC, Kas K, Van de Ven WJ, Voz ML. The tumorigenic diversity of the three PLAG family members is associated with different DNA binding capacities. Cancer Res. 2002 Mar 1;62(5):1510–1517. [PubMed] [Google Scholar]
- 18.Kas K, Voz ML, Hensen K, Meyen E, Van de Ven WJ. Transcriptional activation capacity of the novel PLAG family of zinc finger proteins. J Biol Chem. 1998;273(36):23026–23032. doi: 10.1074/jbc.273.36.23026. [DOI] [PubMed] [Google Scholar]
- 19.Castilla LH, Garrett L, Adya N, Orlic D, Dutra A, Anderson S, et al. The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia. Nat Genet. 1999;23(2):144–146. doi: 10.1038/13776. [DOI] [PubMed] [Google Scholar]
- 20.Challier C, Cocault L, Flon M, Pauchard M, Porteu F, Gisselbrecht S, et al. A new feature of Mpl receptor: ligand-induced transforming activity in FRE rat fibroblasts. Oncogene. 2000 Apr 13;19(16):2033–2042. doi: 10.1038/sj.onc.1203472. [DOI] [PubMed] [Google Scholar]
- 21.Bouscary D, Lecoq-Lafon C, Chretien S, Zompi S, Fichelson S, Muller O, et al. Role of Gab proteins in phosphatidylinositol 3-kinase activation by thrombopoietin (Tpo) Oncogene. 2001 Apr 26;20(18):2197–2204. doi: 10.1038/sj.onc.1204317. [DOI] [PubMed] [Google Scholar]
- 22.Drachman JG, Griffin JD, Kaushansky K. The c-Mpl ligand (thrombopoietin) stimulates tyrosine phosphorylation of Jak2, Shc, and c-Mpl. J Biol Chem. 1995 Mar 10;270(10):4979–4982. doi: 10.1074/jbc.270.10.4979. [DOI] [PubMed] [Google Scholar]
- 23.Kaushansky K. Lineage-specific hematopoietic growth factors. N Engl J Med. 2006 May 11;354(19):2034–2045. doi: 10.1056/NEJMra052706. [DOI] [PubMed] [Google Scholar]
- 24.Voz ML, Mathys J, Hensen K, Pendeville H, Van Valckenborgh I, Van Huffel C, et al. Microarray screening for target genes of the proto-oncogene PLAG1. Oncogene. 2004 Jan 8;23(1):179–191. doi: 10.1038/sj.onc.1207013. [DOI] [PubMed] [Google Scholar]
- 25.Kondo T, Okabe M, Sanada M, Kurosawa M, Suzuki S, Kobayashi M, et al. Familial essential thrombocythemia associated with one-base deletion in the 5′-untranslated region of the thrombopoietin gene. Blood. 1998 Aug 15;92(4):1091–1096. [PubMed] [Google Scholar]
- 26.Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood. 2004 Jun 1;103(11):4198–4200. doi: 10.1182/blood-2003-10-3471. [DOI] [PubMed] [Google Scholar]
- 27.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006 Jul 25; doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 28.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006 Jul 18;3(7):e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deveaux S, Filipe A, Lemarchandel V, Ghysdael J, Romeo PH, Mignotte V. Analysis of the thrombopoietin receptor (MPL) promoter implicates GATA and Ets proteins in the coregulation of megakaryocyte-specific genes. Blood. 1996 Jun 1;87(11):4678–4685. [PubMed] [Google Scholar]
- 30.Huang H, Yu M, Akie TE, Moran TB, Woo AJ, Tu N, et al. Differentiation-dependent interactions between RUNX-1 and FLI-1 during megakaryocyte development. Mol Cell Biol. 2009 Aug;29(15):4103–4115. doi: 10.1128/MCB.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackers P, Szalai G, Moussa O, Watson DK. Ets-dependent regulation of target gene expression during megakaryopoiesis. J Biol Chem. 2004 Dec 10;279(50):52183–52190. doi: 10.1074/jbc.M407489200. [DOI] [PubMed] [Google Scholar]
- 32.Alexander WS, Roberts AW, Nicola NA, Li R, Metcalf D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood. 1996 Mar 15;87(6):2162–2170. [PubMed] [Google Scholar]
- 33.Kimura S, Roberts AW, Metcalf D, Alexander WS. Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proc Natl Acad Sci U S A. 1998 Feb 3;95(3):1195–1200. doi: 10.1073/pnas.95.3.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007 Dec 13;1(6):685–697. doi: 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Abdollahi A. LOT1 (ZAC1/PLAGL1) and its family members: mechanisms and functions. J Cell Physiol. 2007 Jan;210(1):16–25. doi: 10.1002/jcp.20835. [DOI] [PubMed] [Google Scholar]
- 36.Valleley EM, Cordery SF, Carr IM, MacLennan KA, Bonthron DT. Loss of expression of ZAC/PLAGL1 in diffuse large B-cell lymphoma is independent of promoter hypermethylation. Genes Chromosomes Cancer. May;49(5):480–486. doi: 10.1002/gcc.20758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representation of microarray strategy to identify PLAGL2 target genes in leukemia development. (A) Representation of pMSCV-IRES-GFP (MIG) and MIG expressing PLAGL2 (MIG-PL2) retroviruses. (B) Bone marrow derived hematopoietic progenitors (HP) expressing CBFb-MYH11 are transduced with MIG or MIG-PL2 retrovirus, GFP-sorted, and used for Affymetrix-based microarray analysis (n=2 per group). Leukemic cells expressing CBFb-MYH11 (PL2−AML blasts) and leukemic cells expressing CBFb-MYH11 and PLAGL2+ (PL2+ AML) (n=3 per group) are used in Affymetrix-based microarray analysis.
(C) Two-dimensional hierarchical clustering analysis of probe sets that are differentially expressed in hematopoietic progenitor cells expressing Cbfb-MYH11 and PLAGL2 (PL2− HP), and leukemic samples expressing Cbfb-MYH11 and PLAGL2 (PL2+ AML) or expressing Cbfb-MYH11 but not PLAGL2 (PL2− AML) compared to a MIG-infected progenitor sample (MIG-HP) expressing Cbfb-MYH11. Triangle marks position of data shown in Figure 1.
Expression pattern of Mpl probe in gene profiled samples. (A) Scatter plot comparison of the raw signals of the entire probe sets of mouse genome from the HPs expressing PLAGL2 sample (Y axis, PL2+1 HP) and the HPs not expressing PLAGL2 sample (X-axis, MIG1-HP). Up-regulated (red) and down-regulated (green) probe-sets over 2 fold are shown. Arrow indicates the position of Mpl probe set. (B) The normalized expression patterns of probe sets upregulated in PLAGL2 samples in a sub-tree containing the Mpl probe set. The profile of the Mpl probe set is indicated by black arrow.