

Abstract
Free radical-induced lipid peroxidation (LP) is critical in the evolution of secondary injury following traumatic brain injury (TBI). Previous studies in our laboratory demonstrated that U-83836E, a potent LP inhibitor, can reduce post-TBI LP along with an improved maintenance of mouse cortical mitochondrial bioenergetics and calcium (Ca++) buffering following severe (1.0 mm; 3.5 m/sec) controlled cortical impact TBI (CCI-TBI). Based upon this preservation of a major Ca++ homeostatic mechanism, we have now performed dose-response and therapeutic window analyses of the ability of U-83836e to reduce posttraumatic calpain-mediated cytoskeletal (α-spectrin) proteolysis in ipsilateral cortical homogenates at its 24 h post-TBI peak. In the dose-response analysis, mice were treated with a single i.v. dose of vehicle or U-83836e (0.1, 0.3, 1.3, 3.0, 10.0 or 30.0 mg/kg) at 15 min. after injury. U-83836e produced a dose-related attenuation of α-spectrin degradation with the maximal decrease being achieved at 3.0 mg/kg. Next, the therapeutic window was tested by delaying the single 3 mg/kg i.v. dose from 15 min. post-injury out to 1, 3, 6 or 12 h. No reduction in α-spectrin degradation was observed when the treatment delay was 1 h or longer. However, in a third experiment, we re-examined the window with repeated U-83836e dosing (3.0 mg/kg i.v. followed by 10 mg/kg i.p. maintenance doses at 1 and 3 h after the initial i.v. dose) which significantly reduced 24 h α-α-spectrin degradation even when treatment initiation was withheld until 12 h post-TBI. These results demonstrate the relationship between post-TBI LP, disruptions in neuronal Ca++ homeostasis and calpain-mediated cytoskeletal damage.
Keywords: U-83836E, lipid peroxidation, calpain, α-spectrin and traumatic brain injury
INTRODUCTION
The hallmark of the secondary phase of traumatic brain injury (TBI) is a triad of excitotoxicity, free radical-induced LP, and Ca++ dysregulation (Bullock and Fujisawa 1992; Tymianski and Tator 1996; Hall et al. 2010). Glutamate release after injury causes an influx of Ca++ into neuronal cells via activation of NMDA receptors (Arundine and Tymianski 2004). Activation of NMDA receptors has been shown to contribute to post-traumatic LP (Ozsuer et al. 2005) probably by causing an early increase of cytosolic Ca++ which ignites the production of free radicals by several mechanisms including the Ca++ induced activation of phospholipases and arachidonic acid cascade, conversion of xanthine dehydrogenase to xanthine oxidase, induction of nitric oxide synthases and mitochondrial leak (Hall and Springer 2004). The reactive species will attack cellular and mitochondrial membranes causing LP and protein oxidative damage (Beckman and Koppenol 1996; Violi et al. 1999; Singh et al. 2007). The inflicted oxidative damage causes further deterioration of Ca++ homeostasis (Hall et al. 1998) probably by targeting mitochondria and stimulating the formation of the mPTP (Castilho et al. 1995) which contributes to delayed Ca++ dysregulation (Jacquard et al. 2006). These events collectively culminate in a buildup of cytosolic Ca++ that will ultimately lead to neuronal degeneration though massive activation of cellular proteases like calpain (Kampfl et al. 1997).
Calpains are non-lysosomal Ca++-dependent cysteine proteases that function at neutral pH. However, Under physiological conditions, calpains exist as inactive proenzymes in the cytosol (Wang and Yuen 1994; Kawasaki and Kawashima 1996). Once activated by increased cytosolic Ca++ load after TBI, calpains degrade a large number of cellular proteins including cytoskeletal proteins such as α-spectrin (Roberts-Lewis and Siman 1993; Posmantur et al. 1996; Saatman et al. 1996a; Newcomb et al. 1997; Pike et al. 1998; Buki et al. 1999; Kupina et al. 2001; Kupina et al. 2002; Kupina et al. 2003; Deng et al. 2007) leading ultimately to post-traumatic neurodegeneration and neurological dysfunction (Saatman et al. 2000). α-Spectrin is an integral component of the cytoskeleton, especially in axons, dendrites and presynaptic terminals (Goodman et al. 1995). Calpain-mediated degradation of α-spectrin leads to the formation of breakdown products of two distinctive molecular weights;150 kDa and 145 kDa which are considered footprints of calpain activation (Roberts-Lewis and Siman 1993; Bartus et al. 1995; Pike et al. 2001) and are a reliable predictors of outcome after TBI (Pike et al. 2001). It should be pointed out that the 150 kD α-spectrin breakdown product is also generated by another cysteine protease caspase 3 (Wang 2000). However, our past work has indicated that calpain is the primary mediator of α-spectrin degradation in the mouse CCI-TBI model (Thompson et al. 2006; Deng et al. 2007). Similarly, studies by others in animal models of TBI (Liu et al. 1989; Pike et al. 1998; Pike et al. 2001; Aikman et al. 2006) and human TBI (Cardali and Maugeri 2006; Pineda et al. 2007; Brophy et al. 2009) have also shown that the contribution of calpain to post-TBI cytoskeletal degradation far exceeds that of caspase 3.
Studies done in a rat TBI model by (Buki et al. 1999) suggest that calpain-mediated α-spectrin degradation ultimately culminates in overt damage to the cytoskeleton, leading to irreversible damage of the axon and probably contributes to axotomy. Moreover, Inhibition of calpain–mediated proteolysis has proved to be a neuroprotective strategy since calpain inhibitors have been shown to salvage α-spectrin, attenuate axonal injury, and/or to improve motor and/or cognitive functions (Saatman et al. 1996b; Kampfl et al. 1997; Posmantur et al. 1997; Kupina et al. 2001; Ai et al. 2007). Recently published clinical studies have largely validated the use of α-spectrin degradation and immunoblot assessment of its proteolytic fragments in cerebrospinal fluid as biomarkers, the levels of which seem to correlate with TBI diagnosis, severity in terms of the Glasgow Coma Scale score and most importantly outcome (Mondello et al. 2010).
In the present report, we tested the hypothesis that LP plays a significant role in triggering posttraumatic calpain-mediated α-spectrin proteolysis, by determining whether pharmacologically scavenging lipid peroxyl radicals (LOO•) would attenuate calpain-mediated α-spectrin proteolysis after severe CCI-TBI. This was carried out with the use of U-83836E (Hall et al. 1991), which is a potent, selective and dual mechanism LOO• scavenger that we recently demonstrated to attenuate cortical mitochondrial oxidative damage and preserve mitochondrial functions following TBI including aerobic respiration and Ca++-buffering capacity (Mustafa et al. 2010).
MATERIALS AND METHODS
Animals
The present studies employed 270 young adult male CF-1 mice (Charles River, Portage, MI, USA) weighing 29 to 32 g. All animals were fed ad libitum and housed in the Division of Laboratory Animal Resources (DLAR) sector of the University of Kentucky Medical Center, which is fully accredited by AALAC. Procedures follow protocols approved by the University of Kentucky’s Institutional Animal Care and Use Committee (IACUC).
Materials
Magnesium chloride (MgCl2), Mannitol, Ethylene-glycol tetraacetate (EGTA), Bovine serum albumin (BSA), N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), Potassium salt, Triton, Tris HCL, Sodium chloride, Dextrose, Sucrose, calcium chloride, EDTA, glycerol, Protease inhibitors (Complete Mini™ Protease Inhibitor Cocktail tablet).
Mouse Model of Focal (Controlled Cortical Impact - CCI) Traumatic Brain Injury
Mice were initially anesthetized in a Plexiglas chamber using 4.0% isoflurane, shaved, weighed and then placed in a stereotaxic frame (David Kopf, Tujunga, CA, USA). Throughout the injury procedure, mice were kept anesthetized by a constant flow of 3% isoflurane and oxygen both delivered via a nose cone. The head was positioned in the horizontal plane with the nose bar set at zero. A 2 cm sagittal incision is made in the scalp and the flap was retracted using hemostats to expose the skull. After that, a 4.0 mm craniotomy was made, using a dental bur (SS WHITE, Lakewood, NJ 08701) mounted on a cordless Dremel (Dremel, Racine, WI 53406), lateral to the sagittal suture and centered between bregma and lambda. The drilled skull cap at the craniotomy site was carefully removed to avoid inflicting damage to the dura.
A computer-controlled pneumatic impactor (Precision Systems Instrumentation TBI-030 Impactor, Fairfax Station, VA 22039) was used to generate the CCI-TBI. The impactor induced a non-penetrating, localized contusion of the cortex. The impactor tip was 3.0 mm in diameter with a slightly beveled edge. Injury severity is altered via independent adjustment of the impactor contact velocity and the depth of cortical deformation. In the present studies, a severe level of CCI-TBI was employed in which the contact velocity of the impactor was set at 3.5 m/sec, while the deformation depth was set at 1.0 mm as described previously (Scheff and Sullivan 1999; Singh et al. 2006b; Deng-Bryant et al. 2008; Mbye et al. 2008). After injury, the craniotomy was closed by placement of a 6.0 mm diameter disk made of dental acrylic that was cemented in place with cyanoacrylate to close the craniotomy. The mice were then placed in a Hova-Bator Incubator (model 1583; Randall Burkey Co, Boerne, TX, USA), set at 37°C, for at least 20 minutes to prevent post-traumatic hypothermia. Consciousness (i.e. return of right reflex and mobility) was regained within ten minutes after the injury. The survival time point for the injured mice was 24 h post-TBI which has been shown to be the time of the peak in posttraumatic calpain-mediated α-spectrin degradation in the mouse CCI-TBI model (Deng et al. 2007).
U-83836E Preparation and Dosing
U-83836E was purchased from the Biomol Company (Biomol International, LP, 5120 Butler Pike, PA 19462-1202 USA) and made up fresh daily in physiologic saline 0.9%). For i.v. injections, dilutions were made to deliver the assigned dose of U-83836E in an injection volume of ≈0.12 ml whereas for intraperitoneal injections dilutions were made to deliver 10 mg/kg in an injection volume of ≈0.30 ml.
Tissue Extraction and Protein Assay
At 24 h post-TBI, mice were overdosed with sodium pentobarbital (200mg/kg i.p.). Following decapitation, the contused ipsilateral cortical tissue was rapidly dissected on an ice-chilled stage as previously described (Deng et al. 2007). Immediately following dissection, samples were transferred into ice-cold Triton lysis buffer (1% triton, 20mM tris HCL, 150mM NaCl, 5mM EGTA, 10mM EDTA, 10% glycerol) with protease inhibitors (Complete Mini™ Protease Inhibitor Cocktail tablet). Samples were then briefly sonicated, and incubated on ice for 45 min. Following that, the samples were centrifuged at 14,000 rpm for 30 minutes at 4°C, and the supernatants were collected for protein assay. Protein concentration was determined by Bio-Rad DC Protein Assay, with sample solutions diluted to contain 1mg/ml of protein for immunoblotting.
Western-Blotting Analysis of Calpain-mediated Cytoskeletal Degradation
To measure calpain-mediated α-spectrin degradation products, aliquots of each sample (a total of 5 μg in a final volume of 15 μl) were run on a SDS/PAGE Precast gel (3–8 % Tris-Acetate Criterion™ XT Precast gel, Bio-Rad) and transferred onto a nitrocellulose membrane using a semi-dry electro-transferring unit set at 15 V for 15 min. Following transfer, the membranes were incubated in a TBS blocking solution with 5 % milk for 1 h at room temperature. For the detection of α-spectrin and its breakdown products, a mouse monoclonal anti-α-spectrin antibody (Affiniti FG6090) was used at a dilution of 1:5000 in TBST blocking solution with 5% milk with overnight incubation at 4°C. Positive bands were detected by a goat anti-mouse secondary antibody conjugated to an infrared dye (IRDye800CW, Rockland, Gilbertsville, PA, USA) at a dilution of 1:5000. All incubations and washing steps were performed according to the instructions given by the manufacturers. The intensity of the bands was visualized and quantified using a Li-Cor Odyssey Infrared Imager (Li-Cor; Lincoln, NE, USA). A loading control consisting of pooled samples containing intact α-spectrin and its 145 and 150 kD breakdown products from previous TBI experiments was included on each blot to enable normalization of the band densities across the multiple western blots needed to be run for the three experiments.
Sample Size and Statistical Analysis
Each experimental group contained 10 mice. We used Prizm 4.0 to perform a one-way analysis of variance (ANOVA). Where the ANOVA was statistically significant, it was followed by a Dunnett’s post-hoc analysis to determine the significance of differences between injured vehicle-treated vs. injured U-83836E-treated groups. A p<0.05 was required for statistical significance in all the experiments. All data are shown as mean ± standard deviation (SD).
Experimental Design
Three experiments are included in the present study. Experiment #1 was a dose-response experiment in which groups of 10 mice were treated with vehicle (0.9% saline or U-83836E (0.1, 0.3, 1.0, 3.0, 10.0 and 30.0 mg/kg i.v. in a single dose at 15 min. after CCI-TBI and ipsilateral cortical tissue α-spectrin degradation assessed at its 24 h posttraumatic peak (Deng et al. 2007). To make this ambitious dose-response more technically and statistically feasible, the experiment was done in two parts: Part 1 included the 0.1, 0.3 and 1.0 mg/kg i.v. doses which were examined against a vehicle-treated injured group and Part 2 included the 3.0, 10.0 and 30.0 mg/kg i.v. doses which were examined against a second vehicletreated group.
Experiment #2 was a therapeutic window analysis of the effects of the single 3.0 mg/kg i.v. dose of U-83836E (selected from Experiment #1) on 24 h α-spectrin degradation in which the time of drug administration was delayed to 1, 3, 6 or 12 h post-injury.
Experiment #3 was a second therapeutic window analysis in which the initial 3.0 mg/kg i.v. dose of U-83836E was followed by two additional 10.0 mg/kg i.p. doses administered at 1 and 3 h after the i.v. dose. In this experiment 7 treatment delays were included that were split between two parts: Part 1 included treatment delays of 1, 3 and 6 h post-injury vs. a vehicle-treated group and Part 2 included treatment delays of 1, 9, 12 and 18 h vs. a second vehicle-treated group.
RESULTS
Dose-Response Analysis of the Effect of U-83836E on α-Spectrin Degradation
Figure 1 shows the results of the two part Experiment #1 dose-response analysis examining ipsilateral cortical α-spectrin fragments level following CCI-TBI. Part 1 included the U-83836E i.v.-treated groups; 0.1 mg/kg, 0.3 mg/kg, and 1.0 mg/kg while Part 2 included the U-83836E i.v.-treated groups; 3.0 mg/kg, 10.0 mg/kg, and 30.0 mg/kg (n=10/dose group). Each part included its own vehicle-treated injured group. The six different single i.v. dosages were administrated (tail vein) to the mice 15 min after CCI-TBI. All mice were euthanized 24 h after injury when α-spectrin degradation is at its peak (Deng et al. 2007). In Part 1, the ANOVA did not show a significant difference across experimental groups (145 kDa; F(3,36)= 1.732, p>0.05. 150 kDa; F(3,36)= 1.045, p>0.05) which precluded any subsequent post-hoc analysis. However, in Part 2, which involved the higher doses, the ANOVA did show a significant difference across the experimental groups (145 kDa; F(3,36)= 9.895, p<0.01. 150 kDa; F(3,36)= 7.909, p<0.01). Subsequent Dunnett’s post-hoc analysis revealed that the thee U-83836E doses (3.0, 10.0 and 30 mg/kg i.v.) significantly reduced both the 150 and 145 kD SBDPs compared with the vehicle-treated injured mice. However, there were no significant differences among different dosage groups. Therefore, the lowest maximally effective dose (3.0 mg/kg i.v.) was chosen for the subsequent Experiments #2 and #3. While not shown, U-83836E did not effect the levels of α-spectrin or it’s degradation products in sham, non-injured mouse brain tissue.
Figure 1.
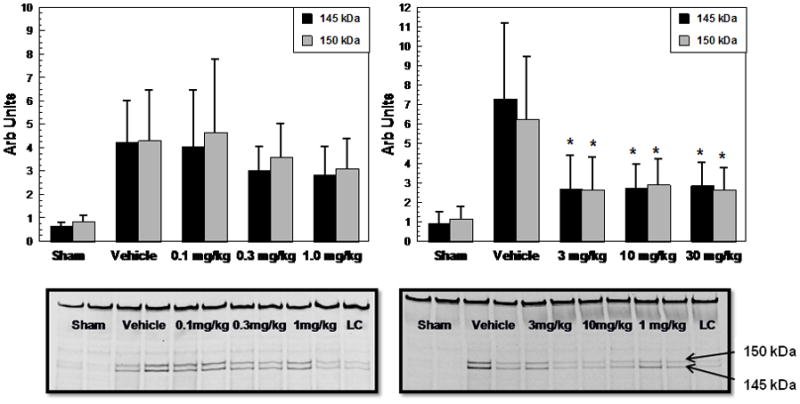
U-83836E dose-response analysis of the effects of a single i.v. dose administered at 15 min after CCI-TBI in male mice on α-spectrin degradation in injured cortical tissue as assessed by quantitative western blotting of the calpain-specific 145 kDa and the mixed calpain/caspase 3-generated 150 kDa α-spectrin breakdown products at their 24 h peak. This was a two part study in which Part 1 involved testing the three lower doses whereas in Part 2 the higher three doses were examined. The three highest doses; 3.0, 10.0 and 30.0 mg/kg doses significantly reduced both SBDPs compared to their paired vehicle-treated group. Representative western blots are displayed below the graphs (LC stands for loading control). N = 10 animals/group; values = mean (±SD); one-way ANOVA followed by Dunnett’s post hoc test: * p < 0.05 vs. vehicle.
Therapeutic Window Analysis of a Single Dose of U-83836E Measured by Calpain-Mediated α-Spectrin Breakdown
A therapeutic window study (Experiment 2) was next conducted in which the onset of U-83836E treatment (3.0 mg/kg i.v. was delayed from the 15 min post-injury tested in Experiment #1 to either 1, 3, 6, or 12 h (Figure 2). The vehicle treated group displayed high levels of SBDPs at 24 h post-injury as in Parts 1 and 2 of Experiment #1. Although there was a slight decline of both SBDPs among U-83836E treatment groups compared with the vehicle-treated group, the overall one way ANOVA was not significant across the various treatment groups (145 kDa; F(4,45)=2.006, p>0.1. 150 kDa; F(4,45)= 1.668, p>0.1). Thus, in the case of single U-83836E i.v. dose administration, the therapeutic window is less than 1 h.
Figure 2.
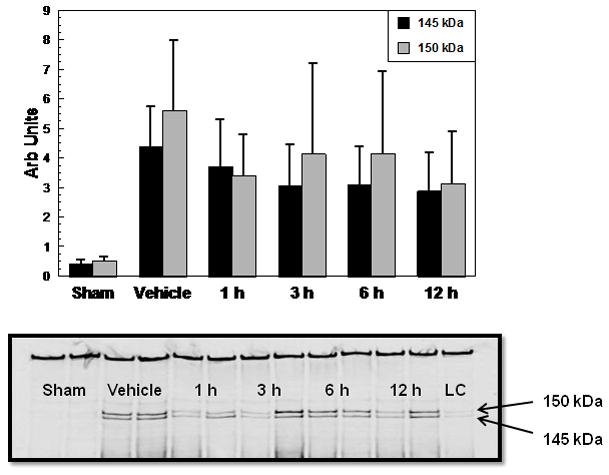
Therapeutic window analysis of the effects of a single 3.0 mg/kg i.v. dose of U-83836E, administered at 1, 3, 6 or 12 h after CCI-TBI in male mice on α-spectrin degradation in injured cortical tissue as assessed by quantitative western blotting of the calpain-specific 145 kDa and the mixed calpain/caspase 3-generated 150 kDa α-spectrin breakdown products at their 24 h peak. Values = mean (±SD) (N=10 mice/group) following administration of a single dose of U-83836E at either 1, 3, 6, or 12 h post-injury. A one-way ANOVA did not reveal a statistically significant difference across experimental groups in regards to the 145 and 150 kD SBDPs. Representative western blots are displayed below the graphs (LC stands for loading control).
Therapeutic Window Analysis of a Multiple Dose Regimen of U-83836E Measured by Calpain-Mediated α-Spectrin Breakdown
We next investigated the therapeutic window of a U-83836E treatment regimen involving an initial 3mg/kg i.v dose followed by two consecutive doses of 10mg/kg i.p, 1 h after the first dose and 2 h after the second dose, respectively (Experiment #3). The onset of U-83836E treatment was delayed from the 15 min post-injury time point tested earlier to either 1, 3, 6, 9, 12, or 18 h (Figure 3). Due to the large number of animals needed for this study, it was divided into two parts as already noted in the Materials and Methods. Part 1 included groups that were treated with U-83836E beginning at 1, 3 or 6 h whereas Part 2 included groups that were treated with U-83836E beginning at 1, 9, 12 or 18 h (n=10). Each part of the therapeutic window experiment included a a separate vehicle-treated injured group and a separate 1 hr treatment delay group to determine the consistency of the injury effect as assessed in the vehicle-treated groups and consistency of the U-83836E effect between the two parts of the experiment. As expected, the vehicle-treated groups showed high levels of SBDPs. In Part 1, involving a maximum treatment delay up to 6 h post-TBI, there was a significant difference across all treatment groups for both the 145 and 150 kD α-spectrin fragments (145 kDa; F(4,45)=21.78, p<0.0001, 150 kDa; F(4,45)=23.91, p<0.0001). Subsequent post-hoc Dunnett’s testing showed that for both fragments, the U-83836E treated groups were all significantly different than the corresponding vehicle treated injured group. In Part 2 involving a repeat of the 1 h treatment delay as well as groups in which treatment was delayed either 9, 12 or 18 h, the ANOVA also showed a significant difference across treatment groups (145 kDa; F(4,45)=21.78, p<0.005, 150 kDa; F(4,45)=6.38, p<0.05). The post-hoc Dunnett’s testing showed for both fragments that there was a statistically significant difference between the U-83836E and vehicle-treated groups out to, and including, a 12 h treatment delay. Even with a delay of 18 h post-injury, the 150 kD fragment was significantly suppressed compared to the vehicle-treated 150 kD fragment levels. However, U-83836E did not have an effect on the 24 h calpain-specific 145 kDa SBDP (p>0.05) when treatment initiation was withheld to 18 h post-TBI.
Figure 3.
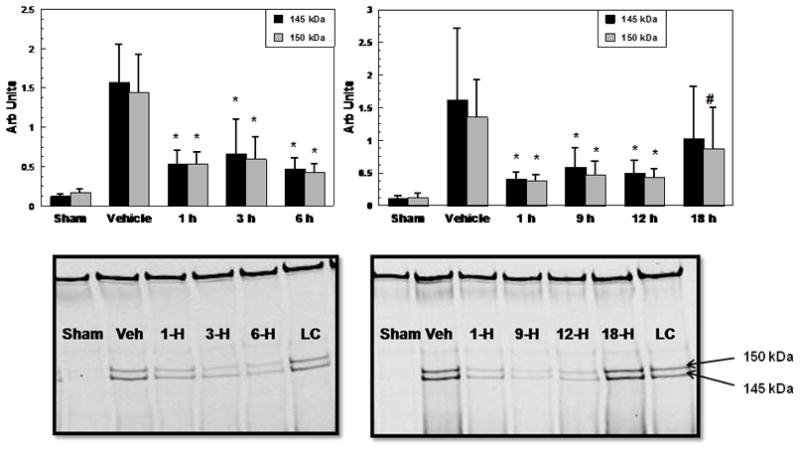
Therapeutic window analysis of a multiple dose regimen of U-83836E initiated at either 1, 3, 6, 9, 12, or 18 h post-injury as measured by calpain-mediated α-spectrin proteolysis 24 h following severe CCI. The initial dose was 3mg/kg i.v followed one hour later by a 10mg/kg i.p maintenance dose. Two hours after the second dose, a third dose of 10mg/kg i.p was also administered. This was a two part study in which Part 1 involved testing of treatment delays of 1, 3 and 6 h whereas Part 2 involved a repeat of the 1 h treatment delay as well as 9, 12 and 18 h delays. Values = mean (±SD) for the 145 and 150 kDa SBDPs with N = 10 mice/group. One-way ANOVAs revealed a statistically significant difference across experimental groups in both Part 1 and Part 2. Subsequent post hoc analysis revealed that the mice treated with multiple doses of U-83836E at, 1, 3, 6, 9, or 12 h post-injury all had a significantly (p<0.05) lower mean levels of the 145 and 150 kDa SBDPs compared with the paired vehicle-treated groups. However, even the group treated with multiple doses of U-83836E beginning at 18 h after injury did display a lower level of the 150 kDa SBDP, but not that of the calpain-specific 145 kDa fragment compared to the vehicle-treated group. Statistical differences (one-way ANOVA and Dunnett’s post hoc test), *p<0.01 vs. vehicle, #p<0.05 vs. vehicle, n=10. Representative western blots are displayed below the graphs (LC stands for loading control)
DISCUSSION
In our previous investigation, we demonstrated that U-83836E treatment can attenuate post-traumatic LP in cerebral cortical tissue or mitochondria together with a preservation of aerobic respiratory function and Ca++-buffering capacity (Mustafa et al. 2010). Consistent with that overall effect, and the preservation of Ca++-buffering capacity in particular, the current results have demonstrated that the LOO• scavenger U-83836E produces a dose-related attenuation of α-spectrin degradation after TBI. As discussed earlier, posttraumatic α-spectrin degradation is produced by both calpain and another cysteine protease caspase 3. The 150 kDa α-spectrin fragment is produced by both proteases whereas the 145 kDa fragment is calpain-specific (Wang 2000). However, since we detected very little of the caspase 3-specific 120 kDa fragment (Wang 2000) in these or other TBI experiments (Kupina et al. 2001; Kupina et al. 2002; Kupina et al. 2003; Thompson et al. 2006; Deng et al. 2007; Deng-Bryant et al. 2008), we believe that the leading source of posttraumatic α-spectrin degradation. From this, we conclude that even though U-83836E does not directly interact with calpain, it nevertheless attenuates the activation of this Ca++-activated protease by preserving neuronal intracellular Ca++ homeostatic mechanisms which are known to be compromised by posttraumatic LP (Hall et al. 2010) and thereby decreases post-traumatic Ca++ overload and it’s activation of neuronal calpain. Calpain-mediated α-spectrin proteolysis peaks at 24 h after CCI-TBI in mice (Deng et al. 2007), and it has been repeatedly implicated in posttraumatic neurodegeneration (Posmantur et al. 1994; Kampfl et al. 1996; Posmantur et al. 1996; Kampfl et al. 1997; Buki and Povlishock 2006; Thompson et al. 2006; Deng et al. 2007; Saatman et al. 2010). Posttraumatic activation of calpains is mediated via the evolution of intracellular Ca++ dysregulation after injury (Kampfl et al. 1997). The early phase of Ca++ dysregulation is an acute manifestation of glutamate-mediated excitotoxicity which triggers an influx of Ca++ via activation of NMDA receptors (Hayes et al. 1988; Faden et al. 1989; Randall and Thayer 1992). This initial increase in intracellular Ca++ triggers a “burst” of highly reactive free radicals (Hall et al. 1993; Lewen et al. 2000) contributed to by multiple mechanisms including membrane phospholipase and arachidonic acid cascade activation, increased biogenic amine release and turnover and mitochondrial ”leak” (Hall and Braughler 1993). The radical burst initiates LP oxidative damage which can progressively increase exponentially as it propagates through cell membranes (Hall and Braughler 1993).
The ensuing LP damage exacerbates the loss of intracellular Ca++ regulation by several mechanisms as illustrated in Figure 4. First of all, it has been shown that the accumulation of LP products such as 4-hydroxynonenal (4-HNE) impairs glutamate transport mechanisms (Keller et al. 1997; Pedersen et al. 1999) which prolongs the extracellular duration of synaptically-released glutamate and thus the duration of its activation of NMDA receptors and inward Ca++ influxes. Secondly, LP damages membrane phospholipid architecture (Hall et al. 2010) which can accentuate cell membrane ionic permeabilities, and most importantly that of Ca++. This is evident by the occurrence of Ca++ influxes in cultured cells following oxidant treatment which has been shown to be ameliorated by U-83836E treatment (Kimura et al. 1992; Munns and Leach 1995). Thirdly, LP damage targets the Ca++ ATPase within the cell membrane impairing its ability to pump Ca++ either into the endoplasmic reticulum or through the plasma membrane (Rohn et al. 1993, 1996; Durmaz et al. 2003). Fourth, LP can potentially disrupt Ca++ homeostasis by mobilizing Ca++ from endoplasmic reticular stores (Racay et al. 1997). Fifth, LP in mitochondrial membranes aggravates mitochondrial dysfunction (Gadelha et al. 1997; Kowaltowski and Vercesi 1999; Sullivan et al. 1999; Singh et al. 2006b; Mbye et al. 2008) including a compromise of the ability of mitochondria to buffer cytosolic Ca++ (Singh et al. 2006b; Mustafa et al. 2010). If oxidative damage to the mitochondrion is severe enough, it can trigger formation of the mitochondrial permeability transition pore (mPTP) leading to mitochondrial permeability transition (MPT), collapse of the mitochondrial membrane potential and a dramatic release of mitochondrial matrix Ca++ into the cytoplasm (Gadelha et al. 1997; Nicholls and Budd 2000). These events culminate in a dramatic increase of cytosolic Ca++ causing massive activation of calpain and caspase 3. Such immense protease activation following TBI is revealed by the evolution of high levels of cytoskeletal SBDPs after TBI (Kampfl et al. 1997; Posmantur et al. 1997; Pike et al. 1998; Pike et al. 2001). Thus, it is our hypothesis that LP is a major contributor to the delayed Ca++ dysregulation following TBI. Consequently, inhibiting LP by scavenging LOO• will protect cellular and mitochondrial membranes and hence maintain Ca++ homeostasis sufficiently to prevent the delayed posttraumatic phase of Ca++ dysregulation. Consistent with this theory, a single i.v. dose of U-83836E administered at 15 min. post-injury was able to decrease α-spectrin degradation at its peak at 24 h after injury.
Figure 4.
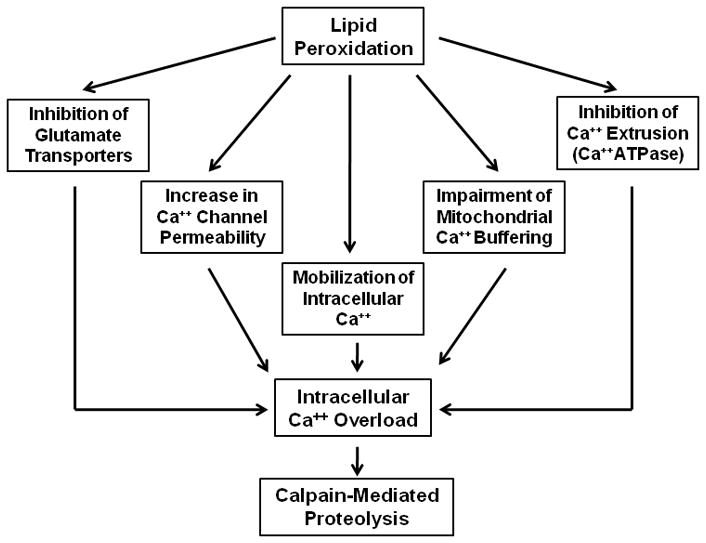
Mechanisms by which free radical-induced lipid peroxidation contributes to intracellular Ca++ overload and calpain activation after TBI (see DISCUSSION for details).
The fact that delaying the single dose treatment beyond 15 min did not have a significant effect is attributed to the nature of LP process. Lipid peroxidation can be initiated by a variety of free radicals generated by the reactive nitrogen species peroxynitrite or by iron-dependent mechanisms (Hall et al. 2010). However, once initiated, branching and propagation of LP reactions damage is dependent on LOO• which is generated within the process of LP (Spiteller 2006; Hall et al. 2010). As more time passes after injury before the start of antioxidant treatment, then more and more LOO• radicals are generated and the propagation of LP becomes amplified and therefore more difficult to stop. As a result, when we delayed the treatment from 15 min to 1 h, the single dose was not sufficient to stop the LP which was already well along. In contrast, when we employed the multiple dosing paradigm to increase the total amount of U-83836E administered and prolong its antioxidant action, it significantly attenuated the levels of SBDPs even though the treatment was delayed up to 12 h after injury. The ability of the drug to attenuate calpain-mediated α-spectrin proteolysis even when the treatment is delayed for 12 h is indicative that both branching and propagation reactions of LP and their contribution to Ca++ dysregulation are still active at least out to 12 h following TBI. This is supported by our previously reported data which shows that in addition to the early increase in calpain-mediated α-spectrin proteolysis which begins during the first hr and plateaus at 6 h, there is a significant secondary increase in α-spectrin degradation between 12 and 24 h following CCI-TBI followed by a progressive decline thereafter (Deng et al. 2007).
As explained above, and illustrated in Figure 4, there are several mechanisms by which LP contributes to posttraumatic intracellular Ca++ dysregulation, and a cell-permeable LP inhibitor like U-83836E could potentially counteract each of these. Having said that, we demonstrated in our previous experiments that administration of a 3 mg/kg i.v. dose to mice at 15 min. after CCI-TBI was able to significantly preserve the bioenergetics and Ca++ buffering capacity of mitochondria isolated from the injured cortex (Mustafa et al. 2010). In the same study, the mitochondrial functional protection was accompanied by a significant reduction in the levels of the mitochondrial protein-bound LP product 4-HNE. In other work in our laboratory, it has been shown that direct application of 4-HNE to isolated cortical mitochondria potently impairs their bioenergetic function (Vaishnav et al. 2010). Thus, it seems certain that U-83836E is attenuating calpain-mediated cytoskeletal damage at least in part via mitochondrial functional protection. Further support for this notion comes from the fact that the peak of mitochondrial functional collapse after CCI-TBI does not occur until 12 h post-injury (Singh et al. 2006a) which coincides with our present observation of a U-83836E repeated dose therapeutic window of 12 h. In other words, the 12 h window is demonstrable because the absolute window for mitochondrial functional preservation is also 12 h. Secondary to the 12 h bottoming out of cortical mitochondrial function, we have documented a delayed secondary spike in α-spectrin degradation between 12 and 24 h post-injury in the CCI-TBI model (Deng et al. 2007). The coincidence of these events provides an explanation for how a 12 h therapeutic window in regards to stopping cytoskeletal proteolysis could be feasible. Nevertheless, the requirement for U-83836E to be effective when delayed until 12 h is that intensive (i.e. repeated) dosing be applied to stop the probably intense LP that is well developed by 12 h after injury.
Lastly, it should be pointed out that this is not our first demonstration that a 12 h therapeutic window in regards to pharmacological inhibition of post-TBI calpain-mediated cytoskeletal damage can occur or that such a prolonged window is associated with a compound that acts to protect mitochondria. In recent work, we examined the cytoskeletal and neuroprotective effects of the novel compound NIM-811 (Mbye et al. 2008; Mbye et al. 2009). NIM811 is a non-immunosuppressive cyclosporine A analog, and like cyclosporine A, it appears to protect mitochondria by binding to cyclophilin D and preventing the formation of the mPTP (Waldmeier et al. 2002). Identical to our present therapeutic window data with U-83836E, we observed that NIM811 can attenuate post-TBI calpain-mediated α-spectrin degradation if administered as late as 12 h after injury (Mbye et al. 2009) and that this is paralleled by a significant reduction in mitochondrial LP-related oxidative damage (Mbye et al. 2008) and ultimately a decrease in cortical neurodegeneration and motor dysfunction (Mbye et al. 2009). This further emphasizes the contribution of mitochondrial dysfunction to overall posttraumatic Ca++ dysregulation as well as the conclusion that it is amenable to pharmacological intervention as late as 12 h post-TBI.
Concerning U-83836E, our ongoing studies include a careful assessment of whether this potent and selective LOO• scavenging LP inhibitor, like NIM811 (Mbye et al. 2009), is also able to attenuate post-TBI cortical neuronal damage and improve motor function. However, extensive work in animal models has shown a connection between the brain tissue or CSF levels of calpain-generated α-spectrin fragments and neuronal damage in preclinical TBI models (Posmantur et al. 1996; Saatman et al. 1996a; Newcomb et al. 1997; Buki et al. 1999; Kupina et al. 2002) and the neuroprotective effects of compounds that attenuate α-α-spectrin degradation by either directly or indirectly decreasing calpain activation (Posmantur et al. 1997; Kupina et al. 2002; Deng-Bryant et al. 2008; Mbye et al. 2009). Furthermore, the value of CSF α-spectrin proteolytic fragment levels as an outcome predictor has recently been extended to human TBI (Mondello et al. 2010).
Acknowledgments
These studies were supported by NIH grants 1P30 NS051220 and 1 P01 NS058484 and funding from the Kentucky Spinal Cord & Head Injury Research Trust.
References
- Ai J, Liu E, Wang J, Chen Y, Yu J, Baker AJ. Calpain inhibitor MDL-28170 reduces the functional and structural deterioration of corpus callosum following fluid percussion injury. J Neurotrauma. 2007;24:960–978. doi: 10.1089/neu.2006.0224. [DOI] [PubMed] [Google Scholar]
- Aikman J, O’Steen B, Silver X, Torres R, Boslaugh S, Blackband S, Padgett K, Wang KK, Hayes R, Pineda J. Alpha-II-spectrin after controlled cortical impact in the immature rat brain. Developmental neuroscience. 2006;28:457–465. doi: 10.1159/000094171. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus RT, Elliott PJ, Hayward NJ, Dean RL, Harbeson S, Straub JA, Li Z, Powers JC. Calpain as a novel target for treating acute neurodegenerative disorders. Neurological research. 1995;17:249–258. doi: 10.1080/01616412.1995.11740322. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Brophy GM, Pineda JA, Papa L, Lewis SB, Valadka AB, Hannay HJ, Heaton SC, Demery JA, Liu MC, Tepas JJ, 3rd, Gabrielli A, Robicsek S, Wang KK, Robertson CS, Hayes RL. alphaII-Spectrin breakdown product cerebrospinal fluid exposure metrics suggest differences in cellular injury mechanisms after severe traumatic brain injury. Journal of neurotrauma. 2009;26:471–479. doi: 10.1089/neu.2008.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buki A, Povlishock JT. All roads lead to disconnection?--Traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148:181–193. doi: 10.1007/s00701-005-0674-4. discussion 193–184. [DOI] [PubMed] [Google Scholar]
- Buki A, Siman R, Trojanowski JQ, Povlishock JT. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365–375. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- Bullock R, Fujisawa H. The role of glutamate antagonists for the treatment of CNS injury. J Neurotrauma. 1992;9(Suppl 2):S443–462. [PubMed] [Google Scholar]
- Cardali S, Maugeri R. Detection of alphaII-spectrin and breakdown products in humans after severe traumatic brain injury. J Neurosurg Sci. 2006;50:25–31. [PubMed] [Google Scholar]
- Castilho RF, Kowaltowski AJ, Meinicke AR, Bechara EJ, Vercesi AE. Permeabilization of the inner mitochondrial membrane by Ca2+ ions is stimulated by t-butyl hydroperoxide and mediated by reactive oxygen species generated by mitochondria. Free Radic Biol Med. 1995;18:479–486. doi: 10.1016/0891-5849(94)00166-h. [DOI] [PubMed] [Google Scholar]
- Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab. 2008;28:1114–1126. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- Deng Y, Thompson BM, Gao X, Hall ED. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol. 2007;205:154–165. doi: 10.1016/j.expneurol.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmaz R, Kanbak G, Akyuz F, Isiksoy S, Yucel F, Inal M, Tel E. Lazaroid attenuates edema by stabilizing ATPase in the traumatized rat brain. Can J Neurol Sci. 2003;30:143–149. doi: 10.1017/s0317167100053415. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Gadelha FR, Thomson L, Fagian MM, Costa AD, Radi R, Vercesi AE. Ca2+-independent permeabilization of the inner mitochondrial membrane by peroxynitrite is mediated by membrane protein thiol cross-linking and lipid peroxidation. Arch Biochem Biophys. 1997;345:243–250. doi: 10.1006/abbi.1997.0259. [DOI] [PubMed] [Google Scholar]
- Goodman SR, Zimmer WE, Clark MB, Zagon IS, Barker JE, Bloom ML. Brain spectrin: of mice and men. Brain Res Bull. 1995;36:593–606. doi: 10.1016/0361-9230(94)00264-2. [DOI] [PubMed] [Google Scholar]
- Hall ED, Braughler JM. Free radicals in CNS injury. Res Publ Assoc Res Nerv Ment Dis. 1993;71:81–105. [PubMed] [Google Scholar]
- Hall ED, Springer JE. Neuroprotection and acute spinal cord injury: a reappraisal. NeuroRx. 2004;1:80–100. doi: 10.1602/neurorx.1.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Yonkers PA. Brain hydroxyl radical generation in acute experimental head injury. Journal of neurochemistry. 1993;60:588–594. doi: 10.1111/j.1471-4159.1993.tb03189.x. [DOI] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG. Antioxidant Therapies for Traumatic Brain Injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Oostveen JA, Fleck TJ, Gurney ME. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. Journal of neuroscience research. 1998;53:66–77. doi: 10.1002/(SICI)1097-4547(19980701)53:1<66::AID-JNR7>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Hall ED, Braughler JM, Yonkers PA, Smith SL, Linseman KL, Means ED, Scherch HM, Von Voigtlander PF, Lahti RA, Jacobsen EJ. U-78517F: a potent inhibitor of lipid peroxidation with activity in experimental brain injury and ischemia. J Pharmacol Exp Ther. 1991;258:688–694. [PubMed] [Google Scholar]
- Hayes RL, Jenkins LW, Lyeth BG, Balster RL, Robinson SE, Clifton GL, Stubbins JF, Young HF. Pretreatment with phencyclidine, an N-methyl-D-aspartate antagonist, attenuates long-term behavioral deficits in the rat produced by traumatic brain injury. Journal of neurotrauma. 1988;5:259–274. doi: 10.1089/neu.1988.5.259. [DOI] [PubMed] [Google Scholar]
- Jacquard C, Trioulier Y, Cosker F, Escartin C, Bizat N, Hantraye P, Cancela JM, Bonvento G, Brouillet E. Brain mitochondrial defects amplify intracellular [Ca2+] rise and neurodegeneration but not Ca2+ entry during NMDA receptor activation. FASEB J. 2006;20:1021–1023. doi: 10.1096/fj.05-5085fje. [DOI] [PubMed] [Google Scholar]
- Kampfl A, Posmantur RM, Zhao X, Schmutzhard E, Clifton GL, Hayes RL. Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: implications for pathology and therapy: a review and update. J Neurotrauma. 1997;14:121–134. doi: 10.1089/neu.1997.14.121. [DOI] [PubMed] [Google Scholar]
- Kampfl A, Posmantur R, Nixon R, Grynspan F, Zhao X, Liu SJ, Newcomb JK, Clifton GL, Hayes RL. mu-calpain activation and calpain-mediated cytoskeletal proteolysis following traumatic brain injury. J Neurochem. 1996;67:1575–1583. doi: 10.1046/j.1471-4159.1996.67041575.x. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Kawashima S. Regulation of the calpain-calpastatin system by membranes (review) Molecular membrane biology. 1996;13:217–224. doi: 10.3109/09687689609160599. [DOI] [PubMed] [Google Scholar]
- Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Waeg G, Mattson MP. 4-Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997;80:685–696. doi: 10.1016/s0306-4522(97)00065-1. [DOI] [PubMed] [Google Scholar]
- Kimura M, Maeda K, Hayashi S. Cytosolic calcium increase in coronary endothelial cells after H2O2 exposure and the inhibitory effect of U78517F. Br J Pharmacol. 1992;107:488–493. doi: 10.1111/j.1476-5381.1992.tb12772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, Vercesi AE. Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med. 1999;26:463–471. doi: 10.1016/s0891-5849(98)00216-0. [DOI] [PubMed] [Google Scholar]
- Kupina NC, Detloff MR, Dutta S, Hall ED. Neuroimmunophilin ligand V-10,367 is neuroprotective after 24-hour delayed administration in a mouse model of diffuse traumatic brain injury. J Cereb Blood Flow Metab. 2002;22:1212–1221. doi: 10.1097/01.wbc.0000037994.34930.bc. [DOI] [PubMed] [Google Scholar]
- Kupina NC, Detloff MR, Bobrowski WF, Snyder BJ, Hall ED. Cytoskeletal protein degradation and neurodegeneration evolves differently in males and females following experimental head injury. Experimental neurology. 2003;180:55–73. doi: 10.1016/s0014-4886(02)00048-1. [DOI] [PubMed] [Google Scholar]
- Kupina NC, Nath R, Bernath EE, Inoue J, Mitsuyoshi A, Yuen PW, Wang KK, Hall ED. The novel calpain inhibitor SJA6017 improves functional outcome after delayed administration in a mouse model of diffuse brain injury. Journal of neurotrauma. 2001;18:1229–1240. doi: 10.1089/089771501317095269. [DOI] [PubMed] [Google Scholar]
- Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. Journal of neurotrauma. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- Liu TH, Beckman JS, Freeman BA, Hogan EL, Hsu CY. Polyethylene glycol-conjugated superoxide dismutase and catalase reduce ischemic brain injury. Am J Physiol. 1989;256:H589–593. doi: 10.1152/ajpheart.1989.256.2.H589. [DOI] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Exp Neurol. 2008;209:243–253. doi: 10.1016/j.expneurol.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Carrico KM, Saatman KE, Hall ED. Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:87–97. doi: 10.1038/jcbfm.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondello S, Robicsek SA, Gabrielli A, Brophy GM, Papa L, Tepas J, Robertson C, Buki A, Scharf D, Jixiang M, Akinyi L, Muller U, Wang KK, Hayes RL. AlphaII-spectrin breakdown products (SBDPs): diagnosis and outcome in severe traumatic brain injury patients. Journal of neurotrauma. 2010;27:1203–1213. doi: 10.1089/neu.2010.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munns PL, Leach KL. Two novel antioxidants, U74006F and U78517F, inhibit oxidant-stimulated calcium influx. Free Radic Biol Med. 1995;18:467–478. doi: 10.1016/0891-5849(94)00163-e. [DOI] [PubMed] [Google Scholar]
- Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. Journal of neurochemistry. 2010;114:271–280. doi: 10.1111/j.1471-4159.2010.06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb JK, Kampfl A, Posmantur RM, Zhao X, Pike BR, Liu SJ, Clifton GL, Hayes RL. Immunohistochemical study of calpain-mediated breakdown products to alpha-spectrin following controlled cortical impact injury in the rat. Journal of neurotrauma. 1997;14:369–383. doi: 10.1089/neu.1997.14.369. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Ozsuer H, Gorgulu A, Kiris T, Cobanoglu S. The effects of memantine on lipid peroxidation following closed-head trauma in rats. Neurosurg Rev. 2005;28:143–147. doi: 10.1007/s10143-004-0374-1. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Cashman NR, Mattson MP. The lipid peroxidation product 4-hydroxynonenal impairs glutamate and glucose transport and choline acetyltransferase activity in NSC-19 motor neuron cells. Exp Neurol. 1999;155:1–10. doi: 10.1006/exnr.1998.6890. [DOI] [PubMed] [Google Scholar]
- Pike BR, Zhao X, Newcomb JK, Posmantur RM, Wang KK, Hayes RL. Regional calpain and caspase-3 proteolysis of alpha-spectrin after traumatic brain injury. Neuroreport. 1998;9:2437–2442. doi: 10.1097/00001756-199808030-00002. [DOI] [PubMed] [Google Scholar]
- Pike BR, Flint J, Dutta S, Johnson E, Wang KK, Hayes RL. Accumulation of non-erythroid alpha II-spectrin and calpain-cleaved alpha II-spectrin breakdown products in cerebrospinal fluid after traumatic brain injury in rats. J Neurochem. 2001;78:1297–1306. doi: 10.1046/j.1471-4159.2001.00510.x. [DOI] [PubMed] [Google Scholar]
- Pineda JA, Lewis SB, Valadka AB, Papa L, Hannay HJ, Heaton SC, Demery JA, Liu MC, Aikman JM, Akle V, Brophy GM, Tepas JJ, Wang KK, Robertson CS, Hayes RL. Clinical significance of alphaII-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. Journal of neurotrauma. 2007;24:354–366. doi: 10.1089/neu.2006.003789. [DOI] [PubMed] [Google Scholar]
- Posmantur R, Hayes RL, Dixon CE, Taft WC. Neurofilament 68 and neurofilament 200 protein levels decrease after traumatic brain injury. Journal of neurotrauma. 1994;11:533–545. doi: 10.1089/neu.1994.11.533. [DOI] [PubMed] [Google Scholar]
- Posmantur R, Kampfl A, Siman R, Liu J, Zhao X, Clifton GL, Hayes RL. A calpain inhibitor attenuates cortical cytoskeletal protein loss after experimental traumatic brain injury in the rat. Neuroscience. 1997;77:875–888. doi: 10.1016/s0306-4522(96)00483-6. [DOI] [PubMed] [Google Scholar]
- Posmantur RM, Kampfl A, Liu SJ, Heck K, Taft WC, Clifton GL, Hayes RL. Cytoskeletal derangements of cortical neuronal processes three hours after traumatic brain injury in rats: an immunofluorescence study. J Neuropathol Exp Neurol. 1996;55:68–80. doi: 10.1097/00005072-199601000-00007. [DOI] [PubMed] [Google Scholar]
- Racay P, Kaplan P, Mezesova V, Lehotsky J. Lipid peroxidation both inhibits Ca(2+)-ATPase and increases Ca2+ permeability of endoplasmic reticulum membrane. Biochem Mol Biol Int. 1997;41:647–655. doi: 10.1080/15216549700201691. [DOI] [PubMed] [Google Scholar]
- Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Lewis JM, Siman R. Spectrin proteolysis in the hippocampus: a biochemical marker for neuronal injury and neuroprotection. Ann N Y Acad Sci. 1993;679:78–86. doi: 10.1111/j.1749-6632.1993.tb18290.x. [DOI] [PubMed] [Google Scholar]
- Rohn TT, Hinds TR, Vincenzi FF. Ion transport ATPases as targets for free radical damage. Protection by an aminosteroid of the Ca2+ pump ATPase and Na+/K+ pump ATPase of human red blood cell membranes. Biochem Pharmacol. 1993;46:525–534. doi: 10.1016/0006-2952(93)90530-a. [DOI] [PubMed] [Google Scholar]
- Rohn TT, Hinds TR, Vincenzi FF. Inhibition of Ca2+-pump ATPase and the Na+/K+-pump ATPase by iron-generated free radicals. Protection by 6,7-dimethyl-2,4-DI-1-pyrrolidinyl-7H-pyrrolo[2,3-d] pyrimidine sulfate (U-89843D), a potent, novel, antioxidant/free radical scavenger. Biochem Pharmacol. 1996;51:471–476. doi: 10.1016/0006-2952(95)02222-8. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Creed J, Raghupathi R. Calpain as a Therapeutic Target in Traumatic Brain Injury. Neurotherapeutics. 2010;7:31–42. doi: 10.1016/j.nurt.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Zhang C, Bartus RT, McIntosh TK. Behavioral efficacy of posttraumatic calpain inhibition is not accompanied by reduced spectrin proteolysis, cortical lesion, or apoptosis. J Cereb Blood Flow Metab. 2000;20:66–73. doi: 10.1097/00004647-200001000-00010. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996a;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Murai H, Bartus RT, Smith DH, Hayward NJ, Perri BR, McIntosh TK. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci U S A. 1996b;93:3428–3433. doi: 10.1073/pnas.93.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16:783–792. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Hall ED. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J Neurosci Res. 2007;85:2216–2223. doi: 10.1002/jnr.21360. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006a;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006b;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Spiteller G. Peroxyl radicals: inductors of neurodegenerative and other inflammatory diseases. Their origin and how they transform cholesterol, phospholipids, plasmalogens, polyunsaturated fatty acids, sugars, and proteins into deleterious products. Free Radic Biol Med. 2006;41:362–387. doi: 10.1016/j.freeradbiomed.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Experimental neurology. 2006;201:253–265. doi: 10.1016/j.expneurol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Tator CH. Normal and abnormal calcium homeostasis in neurons: a basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery. 1996;38:1176–1195. doi: 10.1097/00006123-199606000-00028. [DOI] [PubMed] [Google Scholar]
- Vaishnav RA, Singh IN, Miller DM, Hall ED. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. Journal of neurotrauma. 2010;27:1311–1320. doi: 10.1089/neu.2009.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violi F, Marino R, Milite MT, Loffredo L. Nitric oxide and its role in lipid peroxidation. Diabetes Metab Res Rev. 1999;15:283–288. doi: 10.1002/(sici)1520-7560(199907/08)15:4<283::aid-dmrr42>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- Wang KK. Calpain and caspase: can you tell the difference?, by kevin K.W. WangVol. 23, pp. 20–26. Trends Neurosci. 2000;23:59. doi: 10.1016/s0166-2236(99)01536-2. [DOI] [PubMed] [Google Scholar]
- Wang KK, Yuen PW. Calpain inhibition: an overview of its therapeutic potential. Trends in pharmacological sciences. 1994;15:412–419. doi: 10.1016/0165-6147(94)90090-6. [DOI] [PubMed] [Google Scholar]