

Abstract
PeptideAtlas is a multi-species compendium of peptides observed with tandem mass spectrometry methods. Raw mass spectrometer output files are collected from the community and reprocessed through a uniform analysis and validation pipeline that continues to advance. The results are loaded into a database and the information derived from the raw data is returned to the community via several web-based data exploration tools. The PeptideAtlas resource is useful for experiment planning, improving genome annotation, and other data mining projects. PeptideAtlas has become especially useful for planning targeted proteomics experiments.
Keywords: proteomics, data repository, proteome, database, SRM
1. Introduction
The advent of tandem mass spectrometry (MS/MS) has enabled the identification of a large numbers of proteins in a high throughput manner. A wide variety of instruments, sample preparation techniques, and data analysis methods have fostered an innovative research community, and a huge amount of data has been and continues to be generated at significant expense. It has long been recognized that public repositories of data would accelerate the advancement of proteomics [1] as it has done for other fields such as transcriptomics. Making the data easily accessible to the public fosters the validation of results, and more importantly the reuse of the data for purposes beyond the intents of the original researchers.
Making the raw mass spectrometer output files accessible to the community is important because the analysis techniques of proteomics continue to advance markedly over time. Modern analysis of older datasets yields many more identifications and information from the data due to better protein reference information and better informatics software. Indeed the newest spectral library searching techniques routinely identify 50% more spectra than sequence searching techniques, and different search engines are able to identify different peptides in the same datasets. One can expect that future workflows will apply several tools in parallel to achieve an analysis much closer to optimal.
The PeptideAtlas Project is a resource that accepts mass spectrometer output files in a variety of formats along with the metadata associated with the experiment. The raw data are reanalyzed using ever-improving techniques and coalesced into a compendium of identifications for each species. An important part of the resource is the tools that allow the research community to access the data in the PeptideAtlas database for experiment planning, validation of new datasets, and other data mining projects.
In addition to PeptideAtlas, several repositories for proteomics data have emerged over the last few years, including PRIDE (Proteomics Identifications Database) [2], OPD (Open Proteomics Database) [1], Tranche [3] and GPMDB (Global Proteome Machine Database) [4]. These repositories have different strengths and fill different niches. The strengths of PeptideAtlas are that only raw data are accepted and are processed through a uniform analysis and validation pipeline to insure high quality results with well-understood false discovery rates (FDR), and an advanced toolset for presenting the results in a manner conducive to experiment planning.
In the following sections, the PeptideAtlas Resource is described in detail. First a brief history of the early motivations and work is presented, followed by a description of the building of PeptideAtlas. Finally the many ways to use the PeptideAtlas is presented, ending with an outlook on the future of the resource.
2. History
With the increasing number of installed tandem mass spectrometers capable of generating large amount of MS/MS-based proteomics data, it became apparent that there was significant value in collecting and combining many of these datasets. Expected benefits from such work include high coverage of a proteome, sufficient data density for statistical arguments, and the possibility to contribute extensive observational data back to genome annotation projects.
The PeptideAtlas project thus began at the Seattle Proteomics Center as a compendium of peptides observed in a collection of human and Drosophila shotgun tandem mass spectrometry datasets acquired at the Institute for Systems Biology. Also available were the annotations describing in which samples the peptides and proteins were observed, which modified forms and how frequently the peptides were observed, and how these peptides mapped onto the genome [5].
Subsequently additional builds have been added for yeast [6], Streptococcus pyogenes [7], and Halobacterium salinarium [8]. In addition, several specialized builds for subproteomes were released for human plasma [9] and mouse plasma [10] samples. PeptideAtlas builds for several other species (mouse, E. coli, rat) and subproteomes (liver, pancreas, et al.) are expected to be released in 2009.
The tools have also evolved considerably in the past four years. In 2004, only basic query and browsing tools were available. As of this writing, there are a large number of tools that support new targeted proteomics strategies as well improvements to traditional approaches.
3. Building of the PeptideAtlas
The build process of the PeptideAtlas has evolved since it was initially described [11]. As illustrated in Fig 1, raw mass spectrometer output files for MS/MS experiments are collected from the community, processed through a consistent analysis pipeline, and then loaded into the PeptideAtlas database, thereby returning high-value information back to the community that provided the data. These different phases are described in great detail in the following subsections.
Figure 1.

An overview of the build process of PeptideAtlas. Shotgun tandem mass spectrometry (MS/MS) experimental data are contributed by the community to the PeptideAtlas raw data repository, which is linked to other repositories via the ProteomExchange consortium. The raw data are processed through an evolving but consistent analysis and validation pipeline (Trans Proteomic Pipeline (TPP)) and loaded into the PeptideAtlas database, and made available to the community. Tranche, GPMDB (Global Proteome Machine Database), NIST (National Institute of Standards and Technology), and PRIDE (Protein Identifications Database) are the current major participants in the ProteomExchange consortium.
3.1. Acquiring Data and the Raw Data Repository
A key component of PeptideAtlas is a data repository in which raw data and search results are made available to the community. The PeptideAtlas data repository has had an important role in the advancement of research using high throughput technologies, acting as data provider to several projects, including the spectrum library building at the National Institute of Standards and Technology (NIST), the PepSeeker database [12], as well as large-scale genome annotation efforts [13] In addition to PeptideAtlas, several repositories for proteomics data have emerged over the last few years, including PRIDE, OPD, Tranche and GPMDB. These repositories have different strengths and fill different niches, but it is obvious that the highest benefit can be gained if all the repositories share data and metadata to allow users to access data from all the same experiments using the repository that best meets their requirements. PeptideAtlas is actively participating in the formation of the ProteomExchange consortium that attempts to facilitate this interoperability between the repositories.
However, most of the aforementioned repositories are largely passive—that is, results are stored and can be queried or downloaded, but the remaining untapped potential within the primary data is not extracted with continually advancing analysis tools. Typically, only a small fraction of acquired MS/MS spectra are confidently identified in the first attempt. Although many of the unidentified spectra are of inadequate quality to ever be identified, a considerable fraction of them can indeed be identified with more effort and newer techniques [14]. PeptideAtlas aims to be an active repository, in which only raw data are accepted and these raw data are periodically reprocessed with more advanced techniques for identification and statistical validation as these are becoming available. The results of this advancing analysis of the raw data are then made available back to the community in forms that enable additional research, specifically with tools that support the new targeted proteomics workflows.
3.2. Uniform Processing with Advanced Tools
Once raw mass spectrometer output files are available in the raw data repository, sequence database searching and automated validation of the results using the Trans Proteomic Pipeline (TPP) [15] is performed. This begins with conversion to a common mzXML file format, then sequence searching with either SEQUEST [16] or X!Tandem [17], followed by validation of the top hits with PeptideProphet [18], a program that models the correct and incorrect spectrum-peptide match populations and assigns a probability of being correct to each match.
All PeptideProphet results are then combined using ProteinProphet [19], a program that uses the spectrum-peptide match models from PeptideProphet to derive protein-level probabilities as well as to adjust the peptide-level probabilities based on the information available from the ensemble of experiments. Given a set of confidently identified spectra, the spectral library building tool SpectraST is used to create a consensus spectrum library comprising all observed peptide ions. As part of the library building process, many high scoring but incorrect identifications are rejected. Then all raw data are subjected to a second round of searching, this time by spectral library searching with SpectraST. This has the effect of identifying many more spectra from the available data, with a higher sensitivity and lower error [20]. Output of SpectraST is validated in the same manner as described above with PeptideProphet and ProteinProphet.
3.3. Populating the Database
All peptides are then mapped to a single reference Ensembl [21] build (if available for the species) and mapped to the genome. All this information is loaded into the PeptideAtlas database for browsing or downloading.
The information is loaded in as a discrete build within the PeptideAtlas database. A build represents a particular set of experiments that have been processed as described above at a certain point in time, and mapped to a specific build of the proteome/genome. This build version remains static thereafter. As additional data are acquired, old data reprocessed, or mappings to newer proteome/genome builds are performed, a new build becomes the default, but older builds remain available for comparison or historical reference.
The result of each build process is also made publicly available at the PeptideAtlas web site in several formats. The front-end web site software is distributed as part of the Systems Biology Experiment Analysis System (SBEAMS) framework [22]. A summary of the current state of the various PeptideAtlas builds is provided in Table 1.
Table 1.
Summary of public PeptideAtlas builds
| Build | # Exps | # MS Runs | Searched Spectra | IDs P>0.9 | Distinct Peptides | Distinct Proteins |
|---|---|---|---|---|---|---|
| Human All | 219 | 54 k | 49 M | 5.6 M | 97 k | 12141 |
| Human Plasma | 76 | 48 k | 16 M | 1.8 M | 18 k | 2486 |
| Drosophila | 43 | 1769 | 7.5 M | 498 k | 72 k | 9124 |
| Drosophila PhosphoPep | 4 | 448 | 0.9 M | 170 k | 10 k | 4583 |
| Yeast | 53 | 2957 | 6.5 M | 1.1 M | 36 k | 4336 |
| Mouse | 59 | 3097 | 10 M | 1.4 M | 51 k | 7686 |
| Halobacterium | 88 | 497 | 0.5 M | 76 k | 12 k | 1518 |
| S. pyogenes | 5 | 64 | 215 k | 52 k | 7 k | 1068 |
4. Using PeptideAtlas
A crucial aspect to the success of the PeptideAtlas Project is the tools available for accessing the information therein. The following subsections highlight some of the most visible and useful features of the PeptideAtlas.
4.1. Build Overviews
As described above a build represents a particular set of experiments that have been processed as described above at a certain point in time, and mapped to a specific build of the proteome/genome. For each build there is a summary page that provides such information as the build date, the number of experiments included, the number of spectra searched to create the build, and the resulting number of identifications.
This is followed by some tables and charts that demonstrate the individual contributions of the experiments to the build. Experiments are usually listed in approximate chronological addition to the PeptideAtlas and therefore the charts track the growth of the atlas build over time.
4.2. Protein Views
For each protein in the reference proteome for a given build, a dynamic protein view page summarizes the information available for that protein. The page is segmented into several collapsible sections that can be easily minimized when they are not of relevance to the user. Minimized sections persist over multiple page views.
The top section provides basic information about the protein including all the aliases and related names and accessions available in the database, as well as the total number of spectra and distinct peptides that map to the protein.
The following two sections summarize the peptide coverage of the protein. A graphical diagram similar to genome browser views summarizes all the peptides that map either uniquely or redundantly to the proteins plus information on segments unlikely to be observed with mass spectrometers, as well as signal peptides and transmembrane information where available. The actual protein sequence is displayed with amino acids occurring in observed peptides highlighted.
This is followed by a section listing all the peptides observed and mapping to this protein. The table listing includes many attributes of the peptides, including the number of times they were observed with what best probabilities, theoretically calculated hydrophobicities, and the samples in which the peptides were observed. Empirical Observability Score (EOS) and Suitability score metrics are listed as well. The EOS reflects a likelihood that if the protein is detectable in the sample, it is detected via that peptides. The Suitability Score represents a ranking of how suitable the peptide is as a reference or proteotypic peptide. The score includes information about the total number of observations, the EOS, the best probability of identification, and includes penalties if the peptides are not fully tryptic, contain missed cleavages, or undesirable residues that impact a peptides suitability for targeting (such as methionine which is variably oxidized).
Below this is a section about theoretical peptides for the protein. Each protein is digested in silico and both the PeptideSieve [23] and DetectabilityPredictor [24] software tools are used to predict which peptides might be most suitable for targeting. This can be compared with the empirical evidence for many proteins. For low abundance or otherwise hard-to-detect proteins, these theoretical predictions are useful.
Finally, the last section provides a summary of the samples in which the protein was observed. This is also potentially quite useful for planning future experiments.
4.3. Peptide Views
For each peptide observed in the data for a given build, a dynamic peptide view page summarizes the information available for that peptide. The page is segmented into several collapsible sections as described above. The first section provides a number of attributes for the peptide including predicted hydrophobicity and pI, as well as the number of spectra supporting the identifications.
The following section diplays the peptide-to-protein and chromosomal mapping information. Since the peptide-to-protein mapping can be multiplex and confusing, this section tries to simplify the mapping information. If the peptide can map to multiple isoforms of the same gene, this is noted, and when a peptide spans an intron in the genome, the chromosomal coordinates reflect this. In order to better visualize a complex mapping relationship, a hyperlink to a secondary page displays all the proteins to which a peptide maps aligned together with an overlap of which peptides are observed for each isoform or different protein.
The next section lists all of the different observed peptide ions, i.e. the different charge states or mass modifications that were observed. For each peptide ion, the predicted monoisotopic precursor m/z is listed along with the number of observations, number of experiments, and hyperlinks to visualize the consensus spectra for each peptide ion.
Below this is a listing of every spectrum that supports the identification of this peptide, along with individual attributes of the identifications such as probability of being correctly identified. Each individual spectrum is available for viewing.
Finally at the bottom is a listing of all the experiments that included the peptide along with some simple charts that depict the relative number of spectral counts in each of the experiments.
4.4. Queries
The previously described peptide and proteins views are useful for exploring proteomes one protein at a time. However, they are impractical for extracting lists of interesting results for many proteins and peptides. For this reason, there are several query pages that can return many peptides or proteins at once. These pages allow users to specify a list of constraints for the desired output and receive a list of either proteins, peptides, or transitions (see section 4.6) based on the specified constraints. The lists may be browsed interactively via the embedded hyperlinks, or downloaded in XML or tab-separated-value formats, or even right into tools like Excel.
4.5. Proteotypic Peptides
For targeted proteomics strategies, it is important to determine which peptides are the optimal ones to target; these are termed proteotypic peptides [25]. A proteotypic peptide is one that is easily observable with current mass spectrometry technology and one that maps uniquely to a single protein or isoform. Such peptides make optimum targets and PeptideAtlas provides tools that make the extraction of such proteotypic peptides easy via the query form described above.
The relationships between proteins and constituent peptides can be quite complex in higher eukaryotes and difficult to grasp using ordinary tabular views. We therefore provide a mechanism to visualize peptide and proteins within a PeptideAtlas build using the Cytoscape network visualization software [26]. On the protein view web page, below the list of constituent peptides, there is a button to launch Cytoscape. The information on the current page, including the protein and peptides is combined into a Cytoscape-compatible format. Proteins and peptides are nodes in the network; peptides that map to the protein are connected with an edge. Additionally, the network is then grown to include all proteins and peptides that have any relationship to peptides or proteins already in the network. This final dataset is then packaged up in a jar file and sent to the client with the application via Java Web Start. The user sees a new Java window appear as shown and further described in Figure 2.
Figure 2.
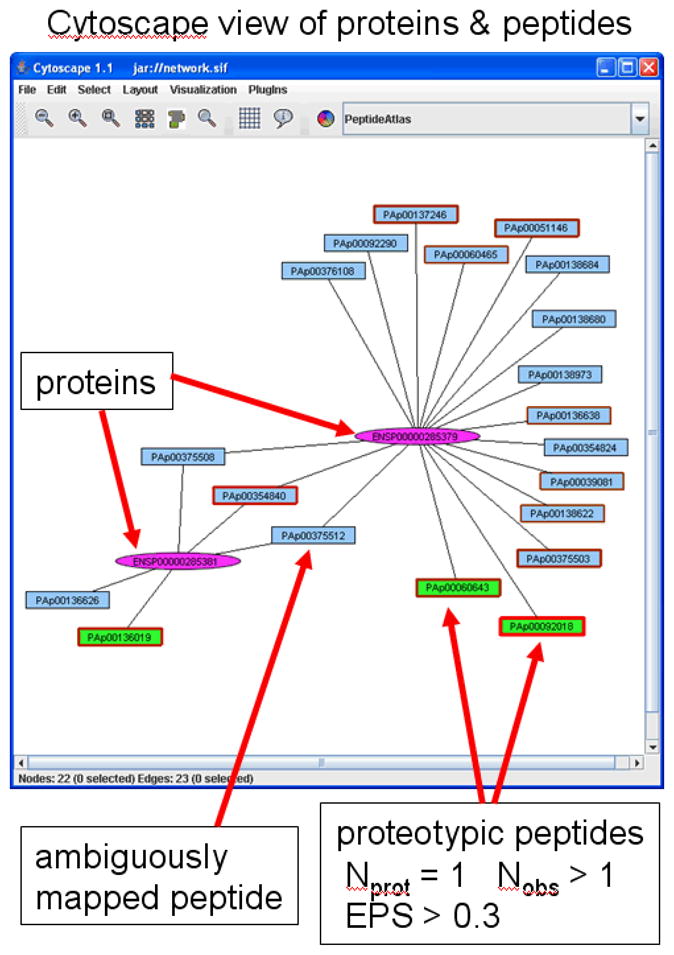
Cytoscape visualization of a simple set of proteins and peptides. The 2 proteins are drawn as purple oval nodes. Peptides are drawn as rectangular nodes. Edges indicate the mapping of peptides to proteins. Peptides that have only one edge are uniquely mapping; peptides with two or more edges are ambiguously mapped. Peptide rectangle borders become thicker and redder with greater numbers of observations. Proteotypic peptides (uniquely mapping, multiply observed, and having EOS > 0.3) are shaded in green.
4.6. Selecting Transitions
The emerging targeted proteomics workflows such as selected reaction monitoring (SRM; also called MRM) are gaining popularity. In this workflow, the mass spectrometer is configured to monitor unique ion signatures, called transitions, of predetermined peptides in order to achieve a detection or confident upper limit for desired peptides to the exclusion of all other peptides. A transition is merely the set of precursor m/z and one or more product ion m/z values. However, the selection of appropriate transitions can be a difficult task. PeptideAtlas has several tools to aid in the selection of transitions, the signatures of peptides needed for SRM [27]. As described above, the individual and consensus spectra are all available within the PeptideAtlas interface and can be used to select transitions either by hand or in batch queries. The ViewMRMList query allows users to specify a list of input proteins and the desired attributes of the transitions, and the result is a tab-separated-value list of candidate transitions for followup.
One problem with predicting candidate transitions from PeptideAtlas is that most of the spectra in the atlas are from ion trap instruments, simply because that is what is predominantly submitted. However, the relative intensities of the fragment ions in triple quadrupole mass spectrometers, the one typically used for SRM, can be quite different from that of ion traps, and thus the predicted transitions do need to be validated.
However, a special build of the PeptideAtlas, called MRMAtlas, is built using MS/MS spectra only from triple-quad instruments. Only a relatively small number of such spectra are available in the MRMAtlas, and only for certain species. However, the data that are contained therein provide the best available transitions for the proteins represented in these special builds.
4.7. PeptideAtlas results in other resources
Besides the interfaces at http://www.peptideatlas.org described so far, the builds from PeptideAtlas can be accessed via several other sites on the World Wide Web. At the Ensembl genome browser site, one can overlay PeptideAtlas peptides onto the genome exploration interface by selecting PeptideAtlas in the DAS (distributed annotation server) sources section. Indeed PeptideAtlas builds are available as DAS sources on our DAS server, and therefore any application that can access genome annotation information via DAS can access PeptideAtlas builds.
Also, we have adapted our interfaces so that they may be easily indexed by the very popular Google search engine. If one performs a Google search for any of the peptides contained in the public PeptideAtlas builds, the top hit will usually be to a PeptideAtlas page that summarizes attributes of the desired peptides, including in which builds the peptide occurs.
As a final example, the iSPIDER resource [28] allows its users to search for proteomics identifications across multiple proteomics databases including PeptideAtlas. When a protein name or accession is entered into iSPIDER, it dynamically queries several repositories including PeptideAtlas and summarizes the results to the user.
5. Conclusion
This chapter has provided an overview of the PeptideAtlas proteomics data resource and repository, including a description of its history, the build process, and the many tools that can be used to access the information in PeptideAtlas. Although it has many uses from improving genome annotation to complex data mining projects, PeptideAtlas is also a very valuable resource for the design of experiments for emerging targeted proteomics workflows. Work is underway to make PeptideAtlas an even more valuable resource for SRM experiments. Soon users will be able to shop for the best available transitions for their favorite list of proteins based on the various data types in PeptideAtlas, including community-submitted validated transitions, transitions based on MRMAtlas observations, transitions based on main PeptideAtlas builds, and finally if insufficient information if available from the previous sources, transitions will be predicted based on the best available theoretical prediction software.
PeptideAtlas is designed as an engine to turn the community’s data into information that everyone can use to enable future work. It relies critically on the availability of raw data, which is now starting to become common. As better and more extensive datasets are processed through PeptideAtlas with ever-improving analysis tools, the resource will serve everyone designing future proteomics experiments.
Acknowledgments
The PeptideAtlas Project has involved a great many contributors. The author would like to thank the following for their contributions to the design and implementation of PeptideAtlas: Dave Campbell, Nichole King, Luis Mendoza, David Shteynberg, Natalie Tasman, Abhishek Pratap, Pat Moss, Jimmy Eng, Ning Zhang, Frank Desiere, Terry Farrah, Zhi Sun, Michael Johnson, and Ruedi Aebersold.
The author has been funded in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, under contract No. N01-HV-28179, and from PM50 GMO76547/Center for Systems Biology.
References
- 1.Prince JT, et al. The need for a public proteomics repository. Nature Biotechnology. 2004;22:471–472. doi: 10.1038/nbt0404-471. [DOI] [PubMed] [Google Scholar]
- 2.Martens L, et al. PRIDE: the proteomics identifications database. Proteomics. 2005;5(13):3537–45. doi: 10.1002/pmic.200401303. [DOI] [PubMed] [Google Scholar]
- 3.Falkner JA, Andrews PC. Tranche: Secure Decentralized Data Storage for the Proteomics Community. Journal of Biomolecular Techniques. 2007;18(1):3. [Google Scholar]
- 4.Craig R, Cortens JP, Beavis RC. Open source system for analyzing, validating, and storing protein identification data. J Proteome Res. 2004;3(6):1234–42. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- 5.Desiere F, et al. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 2004;6(1):R9. doi: 10.1186/gb-2004-6-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King NL, et al. Analysis of the Saccharomyces cerevisiae proteome with PeptideAtlas. Genome Biol. 2006;7(11):R106. doi: 10.1186/gb-2006-7-11-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lange V, et al. Targeted quantitative analysis of Streptococcus pyogenes virulence factors by multiple reaction monitoring. Mol Cell Proteomics. 2008;7(8):1489–500. doi: 10.1074/mcp.M800032-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van PT, et al. Halobacterium salinarum NRC-1 PeptideAtlas: toward strategies for targeted proteomics and improved proteome coverage. J Proteome Res. 2008;7(9):3755–64. doi: 10.1021/pr800031f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deutsch EW, et al. Human Plasma PeptideAtlas. Proteomics. 2005;5(13) doi: 10.1002/pmic.200500160. in press. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Q, et al. A mouse plasma peptide atlas as a resource for disease proteomics. Genome Biol. 2008;9(6):R93. doi: 10.1186/gb-2008-9-6-r93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desiere F, et al. The PeptideAtlas project. Nucleic Acids Res. 2006;34(Database issue):D655–8. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLaughlin T, et al. PepSeeker: a database of proteome peptide identifications for investigating fragmentation patterns. Nucleic Acids Res. 2006;34(Database issue):D649–54. doi: 10.1093/nar/gkj066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanner S, et al. Improving gene annotation using peptide mass spectrometry. Genome Res. 2007;17(2):231–9. doi: 10.1101/gr.5646507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nesvizhskii AI, et al. Dynamic spectrum quality assessment and iterative computational analysis of shotgun proteomic data: toward more efficient identification of post-translational modifications, sequence polymorphisms, and novel peptides. Mol Cell Proteomics. 2006;5(4):652–70. doi: 10.1074/mcp.M500319-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Keller A, et al. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol. 2005;1:2005 0017. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eng J, McCormack AL, Yates JR. An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 17.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–7. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 18.Keller A, et al. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 19.Nesvizhskii AI, et al. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 20.Lam H, et al. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics. 2007;7(5):655–67. doi: 10.1002/pmic.200600625. [DOI] [PubMed] [Google Scholar]
- 21.Hubbard TJ, et al. Ensembl 2007. Nucleic Acids Res. 2007;35(Database issue):D610–7. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marzolf B, et al. SBEAMS-Microarray: database software supporting genomic expression analyses for systems biology. BMC Bioinformatics. 2006;7:286. doi: 10.1186/1471-2105-7-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mallick P, et al. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol. 2007;25(1):125–31. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 24.Tang H, et al. A computational approach toward label-free protein quantification using predicted peptide detectability. Bioinformatics. 2006;22(14):e481–8. doi: 10.1093/bioinformatics/btl237. [DOI] [PubMed] [Google Scholar]
- 25.Kuster B, et al. Scoring proteomes with proteotypic peptide probes. Nat Rev Mol Cell Biol. 2005;6(7):577–83. doi: 10.1038/nrm1683. [DOI] [PubMed] [Google Scholar]
- 26.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008;9(5):429–34. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siepen JA, et al. ISPIDER Central: an integrated database web-server for proteomics. Nucleic Acids Res. 2008;36(Web Server issue):W485–90. doi: 10.1093/nar/gkn196. [DOI] [PMC free article] [PubMed] [Google Scholar]