

Abstract
In idiopathic inflammatory myopathies (IIMs), extracellular inflammatory stimulation is considered to induce secondary intracellular inflammatory changes including expression of major histocompatibility complex class-I (MHC-I) and to produce self-sustaining loop of inflammation. We hypothesize that activation of calpain, a Ca2+-sensitive protease, bridges between these extracellular inflammatory stress and intracellular secondary inflammatory changes in muscle cells. In this study, we demonstrated that treatment of rat L6 myoblast cells with interferon-gamma (IFN-γ) caused expression of MHC-I and inflammation related transcription factors (phosphorylated-extracellular signal-regulated kinase 1/2 and nuclear factor-kappa B). We also demonstrated that treatment with tumor necrosis factor-alpha (TNF-α) induced apoptotic changes and activation of calpain and cyclooxygenase-2. Further, we found that post-treatment with calpeptin attenuated the intracellular changes induced by IFN-γ or TNF-α. Our results indicate that calpain inhibition attenuates apoptosis and secondary inflammatory changes induced by extracellular inflammatory stimulation in the muscle cells. These results suggest calpain as a potential therapeutic target for treatment of IIMs.
Keywords: Calpain, Immune inflammatory myopathy, Interferon-gamma, Myoblast cells, Tumor necrosis factor-alpha
INTRODUCTION
Idiopathic inflammatory myopathies (IIMs) are a constellation of disorders in which the immune system injures skeletal muscle. Various cytokines have been identified to involve in their pathogenesis (Greenberg 2007). Polymyositis (PM) and sporadic inclusion body myositis (s-IBM) are major disorders of IIMs. The hallmark of muscle pathology in PM and s-IBM is invariable expression of major histocompatibility complex class-I (MHC-I) antigens in non-necrotic muscle fibers, which is not usually observed in normal muscle fibers (Karpati et al. 1988). Cytokines including interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), and interleukin-1beta (IL-1β) upregulate MHC-I antigen in muscle cells (Michaelis et al. 1993; Nagaraju et al. 2000). In addition, overexpression of MHC-I leads to self-sustained autoimmune myositis (Nagaraju et al. 2000; Li et al. 2009). It is speculated that overexpression of MHC-I molecule in muscle cells leads to endoplasmic reticulum (ER) stress, which further leads to the activation of nuclear factor-kappa B (NF-κB), a pro-inflammatory transcription factor. NF-κB plays important roles in IIMs. NF-κB expression is observed in inflammatory cells and degenerating muscle cells invaded by mononuclear cells (Haslbeck et al. 2005) and regenerating muscle fibers (Monici et al. 2003; Haslbeck et al. 2005). In addition, activation of NF-κB causes profound muscle wasting due to acceleration of protein breakdown mediated by ubiquitin-dependent proteolysis (Cai et al. 2004). Activation of NF-κB leads to transcription of genes encoding cytokines and chemokines, resulting in a self-sustaining inflammatory response (Dalakas 2006). NF-κB is also involved in the regulation of cyclooxygenase-2 (COX-2), which is responsible for the formation of inflammatory mediators including prostaglandins (Crofford et al. 1997). In course of examination of muscle specimen obtained from patients with s-IBM, very strong focal immunoreactivity of extracellular signal-regulated kinase (ERK) in vacuolated fibers has been demonstrated (Nakano et al. 2001). These investigators suspect a defect in a chaperone-like molecule involved in its folding and nuclear transport.
IFN-γ and TNF-α are inflammatory cytokines, which are expressed in muscle specimen of IIM patients and are considered to play critical roles in the pathogenesis of IIMs (Dalakas 2006). We have identified that exposure of L6 rat myoblast cells to IFN-γ caused various intracellular changes, including apoptotic changes through activation of both ER stress and mitochondrial pathways, and expression of calpain, cathepsin D and amyloid protein (Nozaki et al. 2010). Since some of these changes are seen in IIMs, we think our in vitro model can be used for the study of IIM pathogenesis. We have also identified that calpeptin, a calpain inhibitor attenuates these changes (Nozaki et al. 2010). Based on these data, we propose that calpain induces various intracellular changes in muscle cells in response to extracellular inflammatory stimulation.
Calpain is an extralysosomal, intracellular protease. Ubiquitously expressed calpain exists in two forms: μcalpain and mcalpain, requiring μM and mM Ca2+ concentrations, respectively (Banik et al. 1992; Murachi 1984). Calpain activity is regulated by Ca2+, calpastatin (its endogenous inhibitor), lipids, and an activator protein (Chakrabarti et al. 1990a; Chakrabarti et al. 1990b; Coolican and Hathaway 1984; Murachi 1984). Calpain plays important roles in immune/inflammatory reactions. Calpain activates NF-κB through degradation of its inhibitor, inhibitor of kappa B (I-κB) (Schaecher et al. 2004). In multiple sclerosis, which is an autoimmune demyelinating disease of the central nervous system (CNS), and its animal model experimental allergic encephalomyelitis (EAE) (Waksman and Adams 1962), calpain plays critical roles in pathogenesis. These include Th1/Th2 cytokine dysregulation (Imam et al. 2007), T cell chemotaxis (Butler et al. 2009), demyelination (Shields et al. 1999), and inflammation and axonal damage (Guyton et al. 2010).
In this investigation, we examined whether calpain induced intracellular changes including apoptosis and inflammation related transcription factor expression in response to extracellular inflammatory stimulation in muscle cells. We have presented data from our studies showing calpain activation and protein expression in rat myoblast cells following IFN-γ or TNF-α stimulation. We have also examined whether calpeptin (calpain inhibitor) attenuates these changes.
MATERIALS AND METHOS
Cell Culture and Treatment with IFN-γ and Calpeptin
The L6 rat myoblast cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were grown in 100-mm Petri dishes (Becton Dickinson, Franklin Lakes, NJ, USA) or in 75cm2 cell culture flasks (Corning Incorporated, Corning, NY, USA) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin (100 units/ml), streptomycin (100 μg/ml) (Sigma-Aldrich, St. Louis, MO, USA), grown to 50-60% confluency and washed twice with phosphate-buffered saline (PBS), 7.4. The medium was switched to 1% FBS-containing DMEM one overnight and then recombinant rat IFN-γ (500 units/ml) (R&D Systems, Minneapolis, MN, USA) or rat recombinant TNF-α (100 ng/ml) (Sigma-Aldrich, St. Louis, MO, USA) was added. The treatment of myoblast cells with IFN-γ was carried out for 6, 12, 24, or 48 hr at 37°C with 5% CO2 and full humidity. In order to observe the effect of calpain inhibition, various doses of calpeptin (1 and 5 μM) were added 5 min after IFN-γ (500 units/ml) or TNF-α (100 ng/ml) exposure. The treatment of myoblast cells with IFN-γ or TNF-α (100 ng/ml) or/and calpeptin was carried out for 24 hr at 37°C with 5% CO2 and full humidity. Cells were then used for determination of apoptotic changes and expression of specific proteins.
ApopTag Assay for Biochemical Evidence for Apoptosis
The L6 rat myoblast cells from each treatment were detached with a cell-scraper to harvest attached and detached cells together. The cells were centrifugated at 2000 rpm for 5 min to obtain a pellet. Then the cells were resuspended in PBS and sedimented onto the microscopic slides and fixed in ethanol. Finally, cells were subjected to ApopTag assay using a kit (Intergen, NY, USA) for biochemical detection of DNA fragmentation in apoptotic cells. The nuclei containing DNA fragments were stained dark brown with ApopTag assay and were not counterstained with methyl green, while normal nuclei were stained pale to medium green. After ApopTag assay, cells were counted to determine percentage of apoptosis. Cellular morphology was examined using light microscopy (Olympus, Tokyo, Japan) to assess apoptosis. At least 500 cells were counted in five randomly selected fields at 20 × magnification in each treatment and the percentage of apoptotic cells was calculated (three replicates per treatment).
Calpain Activity Assay
Calpain activity was examined using calpain activity assay kit (abcam, Cambridge, MA, USA). In brief, rat L6 myoblast cells were grown in 6-well cell culture plate (Corning, Corning, NY, USA). After the treatment with IFN-γ or TNF-α (100 ng/ml) or/and calpeptin as above, cells were counted and about 2×106 cells were centrifuged to make a pellet. Cells were resuspended in 100 μl Extraction Buffer (EB) and incubated on ice for 20 min. Samples were gently mixed by tapping several times during incubation and centrifuged for 1 min in a microcentrifuge (10,000 g). Supernatant was transferred to a fresh tube and put on ice. After determination of protein concentration, cell lysate (50-200 μg) was diluted to 85 μl using EB. Then, 10 μl of 10x reaction buffer and 5 μl of calpain substrate were added to each assay. After incubating samples at 37°C for 1 hr in the dark, samples were read in a fluorometer equipped with a 400-nm excitation filter and a 505-nm emission filter. The activity was expressed as Relative Fluorescent Unit (RFU) per milligram protein of each sample.
Protein Analysis with Western blotting
After cell pellets were obtained as above, they were homogenized on ice in a buffer (50 mM Tris-HCl, pH 7.4, 1 mM PMSF and 5 mM EDTA). Protein content was determined using standard Lowry protein assay. Then, each sample was diluted with the same volume of sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 5 mM β-mercaptoethanol, and 10% glycerol). Samples containing 10-15 μg of protein were separated on a 4-20% linear gradient SDS-PAGE (Laemmli 1970). After electrophoresis, proteins were transferred onto nitrocellulose membranes for immunoblot analysis (Towbin et al. 1979). Primary IgG antibodies against NF-κB, I-κB, and phosphorylated ERK (p-ERK) 1/2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and MHC-I (abcam, Cambridge, MA, USA) were used to probe the blots at 4°C overnight. Also, primary IgG antibody against β-actin (Sigma-Aldrich, St. Louis, MO, USA) was used to standardize the loading of cytosolic protein. Then, peroxidase conjugated goat anti-mouse or anti-rabbit IgG (MP Biomedicals, Solon, OH, USA) was applied as a secondary antibody at 37°C for 2 hr. For subsequent detection of specific proteins, the enhanced chemiluminescence (ECL) system was used (GE Health Care, Piscataway, NJ, USA). Blots were immediately processed to digitalize the image using FluorChem FC2 system (AlphaInnotech, San Leandro, CA, USA). Protein bands were quantified by using the public domain Image J software (http://rsb.info.nih.gov/ij/). The amount of protein was calculated as percent relative to control.
COX-2 Activity Assay
COX-2 activity assay kit (Cayman Chemical, Ann Arbor, MI, USA) was used according to the manufacturer’s protocol. Around 100 μl of supernatant (100 μg protein) was mixed with 280 μl of assay buffer, 20 μl of heme, and 20 μl of the COX-1 inhibitor SC-560 in triplicate in a 1.5-ml tube. The total volume was adjusted to 1 ml. The tube was shaken and incubated for 10 min at 25°C. Then, 40 μl of colorimetric substrate and 40 μl arachidonic acid solution were added sequentially, shaken, and incubated at 37°C for 10 min. The absorbance was read at 590 nm using spectrophotometer.
Statistical Analysis
Results were assessed using StatView software (Abacus Concepts, Berkeley, CA, USA) and compared using one-way analysis of variance (ANOVA) with Fisher’s protected least significant difference (PLSD) post-hoc test at a 95% confidence interval. Data were presented as mean ± standard deviation (SD) of separate experiments (n ≥ 3). Significant difference between control and IFN-γ or TNF-α was indicated by * (P < 0.05) or ** (P < 0.01). Significant difference between IFN-γ and IFN-γ + calpeptin, or TNF-α and TNF-α + calpeptin was indicated by # (P < 0.05) or ## (P < 0.01).
RESULTS
Induction of Time-Dependent Apoptosis in Muscle Cells Following IFN-γ Stimulation
To examine the time course of IFN-γ induced apoptosis in muscle cells, rat L6 myoblast cells were treated with IFN-γ (500 units/ml) for different times (Fig. 1). ApopTag assay was performed to detect induction of apoptosis in myoblast cells after stimulation with IFN-γ for 6, 12, 24, and 48 hr (Fig. 1A). Determination of amounts of apoptosis indicated that IFN-γ induced apoptosis in a time-dependent manner and significant apoptotic death occurred at 24 and 48 hr (p<0.05) in myoblast cells (Fig. 1B).
Fig. 1.
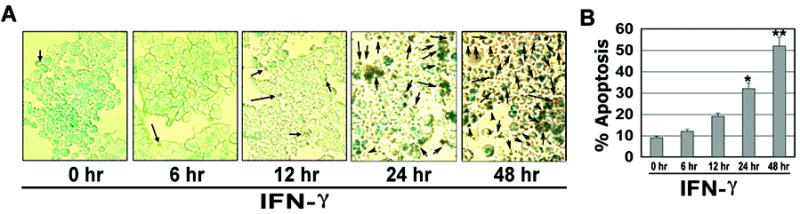
Time-dependent induction of apoptotosis in rat L6 myoblast cells after IFN-γ stimulation. Cells were treated with IFN-γ (500 units/ml) for 6, 12, 24 and 48 hr. (A) Photomicrographs showing representative cells from each treatment following ApopTag assay. The arrows indicate apoptotic cells. (B) Determination of percentage of apoptosis based on ApopTag assay (n = 3).
Time-Dependent Increases in Capain Activity and Expression of MHC-I and Transcription Factors in Muscle Cells Following IFN-γ Stimulation
In order to examine whether extracellular inflammatory stimulation affects intracellular calpain activity and expression of MHC-I and transcription factors in muscle cells, we treated L6 rat myoblast cells with IFN-γ (500 units/ml) for various time periods (Fig. 2). Treatment of myoblast cells with IFN-γ increased calpain activity in a time-dependent manner and significant increases (P < 0.05) in calpain activity occurred at 12, 24, and 48 hr (Fig. 2A). We also performed Western blotting following treatment of myoblast cells with IFN-γ (500 units/ml) and found that IFN-γ significantly increased (P < 0.05) expression of 41 kD MHC-I at 24 hr (Fig. 2B, C). Then, we examined expression of the inflammation related transcription factors (44/42 kD p-ERK 1/2 and 65 kD NF-κB). Our results showed significant increases (p<0.05) in expression of 44/42 kD p-ERK 1/2 at 12, 24, and 48 hr (Fig. 2B, C). Expression 42 kD β-actin was monitored to ensure that equal amounts of cytosolic protein were loaded in all lanes (Fig. 2B, D). There were increases in expression of 65 kD NF-κB relative to expression of its inhibitor 37 kD I-κB (Fig. 2D) resulting in significant increases (P < 0.01) in the NF-κB:I-κB ratio at 24 and 48 hr (Fig. 2E).
Fig. 2.
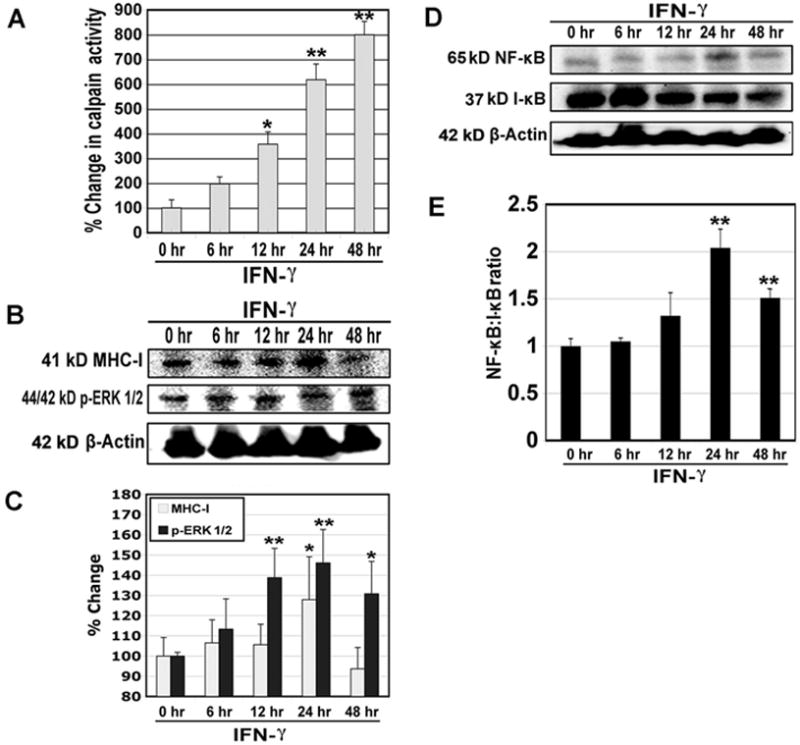
Changes in calpain activity and expression of MHC-I, p-ERK 1/2, NF-κB, and I-κB in rat L6 myoblast cells following IFN-γ stimulation. Cells were treated with IFN-γ (500 units/ml) for 6, 12, 24 and 48 hr. (A) Determination of percent changes in calpain activity (n = 6). (B) Representative Western blots to show levels of 41 kD MHC-I, 44/42 kD p-ERK 1/2, and 42 kD β-actin. (C) Determination of percent changes in 41 kD MHC-I (n = 5) and 44/42 kD p-ERK 1/2 (n = 3) expression based on Western blotting. (D) Representative Western blots to show levels of 65 kD NF-κB, 37 kD I-κB, and 42 kD β-actin. (E) Determination of NF-κB:I-κB ratio based on Western blotting (n = 3).
Calpeptin Attenuated Apoptosis in Muscle Cells Following IFN-γ Stimulation
In order to confirm our hypothesis that an increase in calpain activity was associated with induction of apoptosis, we examined the effect of the calpain inhibitor calpeptin on apoptotic changes following IFN-γ stimulation in rat L6 myoblast cells (Fig. 3). Morphological and biochemical features of apoptosis were examined using ApopTag assay (Fig. 3A). The post-treatment with calpeptin (1 and 5 μM) in the presence of IFN-γ (500 units/ml) significantly decreased (P < 0.05) apoptotic death in myoblast cells at 24 hr, when compared with IFN-γ treatment alone (Fig. 3B).
Fig. 3.
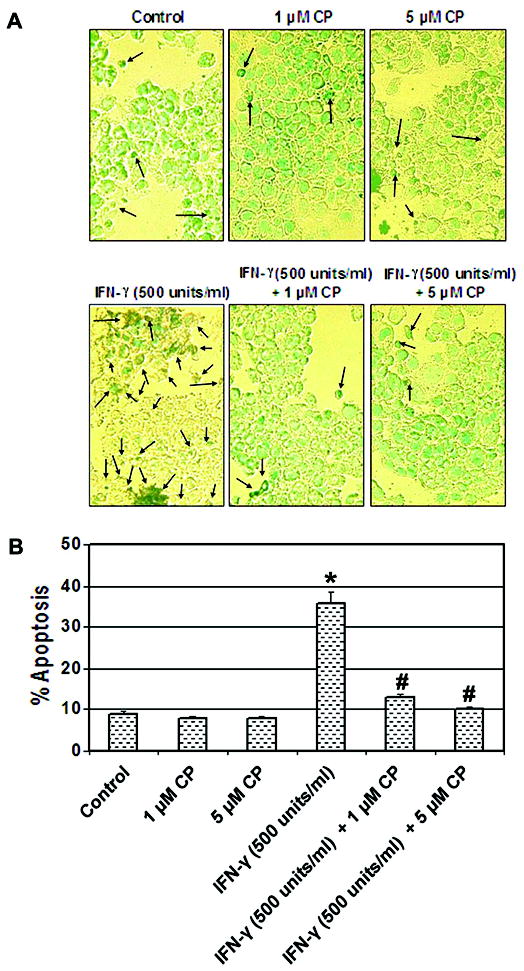
Attenuation of biochemical features of apoptosis by the calpain inhibitor. Cells were treated with IFN-γ (500 units/ml) for 24 hr. Calpeptin (1 and 5 μM) was added 5 min after the IFN-γ addition. (A) Photomicrographs showing representative cells from each treatment following ApopTag assay. The arrows indicate apoptotic cells. (B) Determination of percentage of apoptosis based on ApopTag assay (n = 3).
Calpeptin Attenuated Calpain Activity and Expression of MHC-I and Transcription Factors in Muscle Cells Following IFN-γ Stimulation
We treated rat L6 myoblast cells with calpeptin following extracellular inflammatory stimulation and examined whether calpeptin could attenuate calpain activity and expression of MHC-I and inflammatory transcription factors (Fig. 4). The post-treatment of myoblast cells with calpeptin (1 and 5 μM) in the presence of IFN-γ (500 units/ml) for 24 hr significantly decreased (P < 0.01) calpain activity, when compared with IFN-γ treatment alone (Fig. 4A). We also performed Western blotting to examine whether calpain inhibition affected intracellular expression of MHC-I and inflammation related transcription factors in muscle cells following extracellular inflammatory stimulation (Fig. 4B). The post-treatment of myoblast cells with calpeptin (1 and 5 μM) in the presence of IFN-γ (500 units/ml) for 24 hr significantly decreased (P < 0.05) expression of 41 kD MHC-I and 44/42 kD p-ERK 1/2, when compared with IFN-γ treatment alone (Fig. 4B, C). We also observed that post-treatment with calpeptin for 24 hr decreased the expression of 65 kD NF-κB (Fig. 4B) resulting in a significant decrease (P < 0.01) in NF-κB:I-κB ratio in the muscle cells (Fig. 4B,D).
Fig. 4.
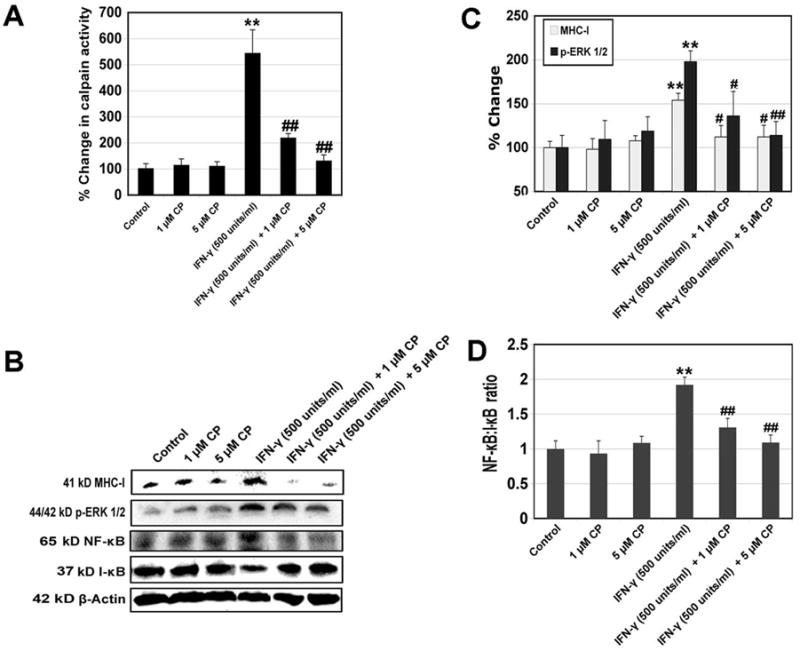
Alterations in calpain activity and expression of MHC-I, p-ERK 1/2, NF-κB, and I-κB by the calpain inhibitor. Cells were treated with IFN-γ (500 units/ml) for 24 hr. Calpeptin (1 and 5μM) was added 5 min after the IFN-γ addition. (A) Determination of percent changes in calpain activity (n = 3). (B) Representative Western blots to show levels of 41 kD MHC-I, 44/42 kD p-ERK 1/2, 65 kD NF-κB, 37 kD I-κB, and 42 kD β-actin. (C) Determination of percent changes in 41 kD MHC-I (n = 4) and 44/42 kD p-ERK 1/2 (n = 5) expression based on Western blotting. (D) Determination of NF-κB:I-κB ratio based on Western blotting (n = 3).
Induction of Apoptosis and Activities of Calpain and COX-2 in Muscle Cells by TNF-α and Their Attenuation by Calpeptin
In order to examine whether another cytokine could induce apoptosis and intracellular inflammatory changes in muscle cells, we treated rat L6 myoblast cells with TNF-α (100 ng/ml) for 24 hr. ApopTag assay showed induction of apoptosis in TNF-α treated cells (Fig. 5A). Amount of apoptosis and activities of calpain and COX-2 were significantly increased (P < 0.01) in TNF-α treated cells compared with control cells (Fig. 5B). We also examined whether calpeptin attenuated apoptosis and intracellular inflammatory changes. The post-treatment of myoblast cells with calpeptin (1 and 5 μM) in the presence of TNF-α (100 ng/ml) for 24 hr significantly decreased (P < 0.01) the apoptotic changes and activities of calpain and COX-2 when compared with TNF-α treatment alone (Fig. 5)
Fig. 5.
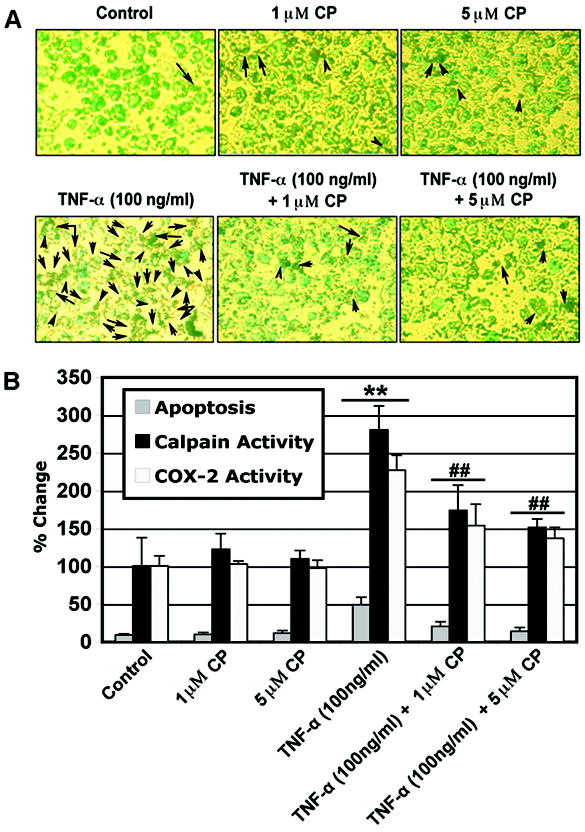
Induction of apoptosis and changes in calpain and COX-2 activities by TNF-α and their attenuation by calpain inhibitor. Cells were treated with TNF-α (100 ng/ml) for 24 hr. Calpeptin (1 and 5 μM) was added 5 min after the TNF-α addition. (A) Photomicrographs showing representative cells from each treatment following ApopTag assay. The arrows indicate apoptotic cells. (B) Determination of apoptosis based on ApopTag assay and percent changes in calpain and COX-2 activities (n = 3).
DISCUSSION
In this study, we have demonstrated that IFN-γ increased the expression of MHC-1 and p-ERK 1/2 and NF-κB:I-κB ratio and that TNF-α increased COX-2 activity in rat L6 myoblast cells. The expression of these proteins is associated with the immune/inflammatory process in the muscle cells. The overexpression of MHC-I molecules is an early event in many autoimmune disease (Nagaraju et al. 2000). Because NF-κB is a major transcription factor modulating the cellular immune and inflammatory processes, it is considered to be essential for the development of autoimmunity (Creus et al. 2009; Monici et al. 2003). ERK belongs to the mitogen-activated protein kinase family and plays a central role in transducing extracellular signals to the nucleus (Nakano et al. 2001). ERK is known to be involved in the regulation of interleukin-6 (IL-6), IL-12, IL-23 and tumor necrosis factor-alpha (TNF-α) synthesis (Thalhamer et al. 2008). In our recent study, we identified the induction of apoptosis by IFN-γ in rat L6 myoblast cells (Nozaki et al. 2010). In this study, we demonstrated that TNF-α also induced apoptosis in rat L6 myoblast cells. Interestingly, increases in the expression of MHC-I and p-ERK 1/2, and NF-κB:I-κB ratio reached maximum levels in muscle cells at 24 hr after the treatment with IFN-γ, while apoptotic death reached its maximum at 48 hr. Based on these results, we propose that extracellular inflammatory stimulation induces secondary inflammatory changes and makes a sustained loop of inflammation leading to apoptotic death in the muscle cells.
We recently identified that IFN-γ induced apoptosis with calpain activation and calpain inhibition with calpeptin attenuated the apoptotic changes in rat L6 myoblast cells (Nozaki et al. 2010). In this study, we demonstrated that TNF-α also induced apoptosis with an increase in calpain activity in rat L6 myoblast cells. Calpain inhibition with calpeptin attenuated apoptosis and COX-2 activity in muscle cells exposed to TNF-α and expression of MHC-I and inflammation related transcription factors in muscle cells exposed to IFN-γ. Intracellular Ca2+ homeostasis is maintained primarily by ER. In response to a variety of external stimuli, Ca2+ is released from the lumen of the ER into the cytoplasm (Corbett and Michalak 2000). Skeletal muscle has a highly specialized ER, the sarcoplasmic reticulum, where Ca2+ binding proteins play a pivotal role in signals critical to muscle contraction and function (Li et al. 2009). Previously, we showed that IFN-γ induced apoptosis through the activation of caspase-12, an ER stress associated pro-apoptotic protease in myoblast cells (Nozaki et al. 2010). Hence, it is possible that extracellular IFN-γ and TNF-α stimulate ER and induces Ca2+ release to activate calpain and produce further intracellular changes including secondary inflammation and apoptosis in the muscle cells. While IFN-γ and TNF-α have been found to promote Ca2+ influx in neural cells in vitro (Das et al. 2006; Das et al. 2010), TNF-α may also have similar effect on Ca2+ homeostasis. Increased Ca2+ will lead to activation of calpain, a Ca2+-dependent protease. Increased calpain activity has been found to cause apoptosis in neuron and oligodendrocyte and also axon-myelin degeneration in the inflammation, neurodegenerative disease, and EAE (Guyton et al. 2010). However, it is not clear whether cytokines stimulate ER directly. Since TNF-α is known to produce oxidants in skeletal muscle cells (Reid and Li 2001), it is possible that cytokines induce oxidative stress, which triggers above changes in muscle cells. The contribution of other proteases should also be considered for the expression of inflammation related transcription factors, MHC-I, and activation of COX-2 in response to extracelluar inflammatory stress. Indeed, we recently identified increased expression of caspase-3, caspase-12, and cathepsin D in rat L6 myoblast cells in response to IFN-γ stimulation (Nozaki et al. 2010). It is noteworthy that calpeptin inhibits tyrosine phosphatases besides calpain (Schoenwaelder and Burridge 1999).
Calpain is involved in the pathogenesis of neurological diseases including Alzheimer’s disease (Saito et al. 1993), Parkinson’s disease (Samantaray et al. 2008), MS, and EAE (Shields et al. 1999; Guyton et al. 2010), spinal cord injury (SCI) (Sribnick et al. 2010), and traumatic brain injury (Samantaray et al. 2008). In addition, its mutation has been linked to limb-girdle muscular dystrophy type 2A (Richard et al. 1995) and diabetes mellitus type 2 (Horikawa et al. 2000). Since muscle weakness or fatigue is a common feature in progressive cases of MS and SCI where calpain activity is increased, it is possible that calpain mediated muscle cell dysfunction or death may be a contributing factor to these disorders. In conclusion, we have identified that extracelluar inflammatory stimulation induces apoptosis with overexpression of MHC-I and inflammation related transcription factors in the muscle cells. These changes are associated with increase in calpain activity and are therefore attenuated by the calpain inhibitor. Since expression of MHC-I, NF-κB, and p-ERK is associated with the pathogenesis of IIMs, calpain can be a potential target in their therapy. Experiments using primary culture of myoblast cells may be useful to confirm our findings.
Acknowledgments
Grant Funding: NIH-NINDS (NS-31622, NS-38146, and NS-41088)
References
- Banik NL, Chakrabarti AK, Konat GW, Gantt-Wilford G, Hogan EL. Calcium-activated neutral proteinase (calpain) activity in C6 cell line: compartmentation of μ and m calpain. J Neurosci Res. 1992;31:708–714. doi: 10.1002/jnr.490310414. [DOI] [PubMed] [Google Scholar]
- Butler JT, Samantaray S, Beeson CC, Ray SK, Banik NL. Involvement of calpain in the process of Jurkat T cell chemotaxis. J Neurosci Res. 2009;87:626–635. doi: 10.1002/jnr.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Chakrabarti AK, Dasgupta S, Banik NL, Hogan EL. Ganglioside-modulated proteolysis by Ca2+-activated neutral proteinase (CANP): a role of glycoconjugates in CANP regulation. J Neurochem. 1990a;54:1816–1819. doi: 10.1111/j.1471-4159.1990.tb01241.x. [DOI] [PubMed] [Google Scholar]
- Chakrabarti AK, Dasgupta S, Banik NL, Hogan EL. Regulation of the calcium-activated neutral proteinase (CANP) of bovine brain by myelin lipids. Biochim Biophys Acta. 1990b;1038:195–198. doi: 10.1016/0167-4838(90)90204-s. [DOI] [PubMed] [Google Scholar]
- Coolican SA, Hathaway DR. Effect of L-alpha-phosphatidylinositol on a vascular smooth muscle Ca2+-dependent protease. Reduction of the Ca2+ requirement for autolysis. J Biol Chem. 1984;259:11627–11630. [PubMed] [Google Scholar]
- Corbett EF, Michalak M. Calcium, a signaling molecule in the endoplasmic reticulum? Trends in biochemical sciences. 2000;25:307–311. doi: 10.1016/s0968-0004(00)01588-7. [DOI] [PubMed] [Google Scholar]
- Creus KK, De Paepe B, De Bleecker JL. Idiopathic inflammatory myopathies and the classical NF-κB complex: current insights and implications for therapy. Autoimmunity reviews. 2009;8:627–631. doi: 10.1016/j.autrev.2009.02.026. [DOI] [PubMed] [Google Scholar]
- Crofford LJ, Tan B, McCarthy CJ, Hla T. Involvement of nuclear factor kappa B in the regulation of cyclooxygenase-2 expression by interleukin-1 in rheumatoid synoviocytes. Arthritis and rheumatism. 1997;40:226–236. doi: 10.1002/art.1780400207. [DOI] [PubMed] [Google Scholar]
- Dalakas MC. Mechanisms of disease: signaling pathways and immunobiology of inflammatory myopathies. Nat Clin Pract Rheumatol. 2006;2:219–227. doi: 10.1038/ncprheum0140. [DOI] [PubMed] [Google Scholar]
- Das A, Garner DP, Del Re AM, Woodward JJ, Kumar DM, Agarwal N, Banik NL, Ray SK. Calpeptin provides functional neuroprotection to rat retinal ganglion cells following Ca2+ influx. Brain Res. 2006;1084:146–157. doi: 10.1016/j.brainres.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Das A, McDowell M, Pava MJ, Smith JA, Reiter RJ, Woodward JJ, Varma AK, Ray SK, Banik NL. The inhibition of apoptosis by melatonin in VSC4.1 motoneurons exposed to oxidative stress, glutamate excitotoxicity, or TNF-α toxicity involves membrane melatonin receptors. J Pineal Res. 2010;48:157–169. doi: 10.1111/j.1600-079X.2009.00739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SA. Proposed immunologic models of the inflammatory myopathies and potential therapeutic implications. Neurology. 2007;69:2008–2019. doi: 10.1212/01.WNL.0000291619.17160.b8. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Das A, Samantaray S, Wallace GCt, Butler JT, Ray SK, Banik NL. Calpeptin attenuated inflammation, cell death, and axonal damage in animal model of multiple sclerosis. J Neurosci Res. 2010;88:2398–2408. doi: 10.1002/jnr.22408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck KM, Friess U, Schleicher ED, Bierhaus A, Nawroth PP, Kirchner A, Pauli E, Neundorfer B, Heuss D. The RAGE pathway in inflammatory myopathies and Limb Girdle muscular dystrophy. Acta Neuropathol. 2005;110:247–254. doi: 10.1007/s00401-005-1043-3. [DOI] [PubMed] [Google Scholar]
- Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, del Bosque-Plata L, Oda Y, Yoshiuchi I, Colilla S, Polonsky KS, Wei S, Concannon P, Iwasaki N, Schulze J, Baier LJ, Bogardus C, Groop L, Boerwinkle E, Hanis CL, Bell GI. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet. 2000;26:163–175. doi: 10.1038/79876. [DOI] [PubMed] [Google Scholar]
- Imam SA, Guyton MK, Haque A, Vandenbark A, Tyor WR, Ray SK, Banik NL. Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. J Neuroimmunol. 2007;190:139–145. doi: 10.1016/j.jneuroim.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpati G, Pouliot Y, Carpenter S. Expression of immunoreactive major histocompatibility complex products in human skeletal muscles. Ann Neurol. 1988;23:64–72. doi: 10.1002/ana.410230111. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Li CK, Knopp P, Moncrieffe H, Singh B, Shah S, Nagaraju K, Varsani H, Gao B, Wedderburn LR. Overexpression of MHC class I heavy chain protein in young skeletal muscle leads to severe myositis: implications for juvenile myositis. Am J Pathol. 2009;175:1030–1040. doi: 10.2353/ajpath.2009.090196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis D, Goebels N, Hohlfeld R. Constitutive and cytokine-induced expression of human leukocyte antigens and cell adhesion molecules by human myotubes. Am J Pathol. 1993;143:1142–1149. [PMC free article] [PubMed] [Google Scholar]
- Monici MC, Aguennouz M, Mazzeo A, Messina C, Vita G. Activation of nuclear factor-kappaB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology. 2003;60:993–997. doi: 10.1212/01.wnl.0000049913.27181.51. [DOI] [PubMed] [Google Scholar]
- Murachi T. Calcium-dependent proteinases and specific inhibitors: calpain and calpastatin. Biochem Soc Symp. 1984;49:149–167. [PubMed] [Google Scholar]
- Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, Danning C, Wada R, Thompson C, Bahtiyar G, Craft J, Hooft Van Huijsduijnen R, Plotz P. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci U S A. 2000;97:9209–9214. doi: 10.1073/pnas.97.16.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S, Shinde A, Kawashima S, Nakamura S, Akiguchi I, Kimura J. Inclusion body myositis: expression of extracellular signal-regulated kinase and its substrate. Neurology. 2001;56:87–93. doi: 10.1212/wnl.56.1.87. [DOI] [PubMed] [Google Scholar]
- Nozaki K, Das A, Ray SK, Banik NL. Calpain inhibition attenuates intracellular changes in muscle cells in response to extracellular inflammatory stimulation. Exp Neurol. 2010;225:430–435. doi: 10.1016/j.expneurol.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MB, Li YP. Cytokines and oxidative signalling in skeletal muscle. Acta Physiol Scand. 2001;171:225–232. doi: 10.1046/j.1365-201x.2001.00824.x. [DOI] [PubMed] [Google Scholar]
- Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C, Pasturaud P, Roudaut C, et al. Mutations in the proteolytic enzyme calpain 3 cause Limb-Girdle muscular dystrophy type 2A. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samantaray S, Ray SK, Banik NL. Calpain as a potential therapeutic target in Parkinson’s disease. CNS Neurol Disord Drug Targets. 2008;7:305–312. doi: 10.2174/187152708784936680. [DOI] [PubMed] [Google Scholar]
- Schaecher K, Goust JM, Banik NL. The effects of calpain inhibition on IkB alpha degradation after activation of PBMCs: identification of the calpain cleavage sites. Neurochem Res. 2004;29:1443–1451. doi: 10.1023/b:nere.0000026410.56000.dd. [DOI] [PubMed] [Google Scholar]
- Schoenwaelder SM, Burridge K. Evidence for a calpeptin-sensitive protein-tyrosine phosphatase upstream of the small GTPase Rho. A novel role for the calpain inhibitor calpeptin in the inhibition of protein-tyrosine phosphatases. J Biol Chem. 1999;274:14359–14367. doi: 10.1074/jbc.274.20.14359. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Saido TC, Banik NL. A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc Natl Acad Sci USA. 1999;96:11486–11491. doi: 10.1073/pnas.96.20.11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sribnick EA, Samantaray S, Das A, Smith J, Matzelle DD, Ray SK, Banik NL. Postinjury estrogen treatment of chronic spinal cord injury improves locomotor function in rats. J Neurosci Res. 2010;88:1738–1750. doi: 10.1002/jnr.22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford) 2008;47:409–414. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waksman BH, Adams RD. A histologic study of the early lesion in experimental allergic encephalomyelitis in the guinea pig and rabbit. Am J Pathol. 1962;41:135–162. [PMC free article] [PubMed] [Google Scholar]