

Abstract
Since the discovery of nitric oxide (NO), which is released from endothelial cells as the main mediator of vasodilation, its target, the soluble guanylyl cyclase (sGC), has become a focus of interest for the treatment of diseases associated with endothelial dysfunction. NO donors were developed to suppress NO deficiency; however, tolerance to organic nitrates was reported. Non-NO-based drugs targeting sGC were developed to overcome the problem of tolerance. In this review, we briefly describe the process of sGC activation by its main physiological activator NO and the advances in the development of drugs capable of activating sGC in a NO-independent manner. sGC stimulators, as some of these drugs are called, require the integrity of the reduced heme moiety of the prosthetic group within the sGC and therefore are called heme-dependent stimulators. Other drugs are able to activate sGC independent of heme moiety and are hence called heme-independent activators. Because pathologic conditions modulate sGC and oxidize the heme moiety, the heme-independent sGC activators could potentially become drugs of choice because of their higher affinity to the oxidized enzyme. However, these drugs are still undergoing clinical trials and are not available for clinical use.
Keywords: soluble guanylyl cyclase activators, nitric oxide, vascular smooth muscle, endothelial dysfunction, vascular disease
INTRODUCTION
The endothelium plays a role in the regulation of the vascular tone by releasing both relaxing and contracting agents. The basal release of nitric oxide (NO) by endothelium plays an important role in the maintenance of basal tone in resistance arteries and in tonic regulation of blood pressure and blood flow distribution.1,2 NO targets the soluble guanylyl cyclase (sGC) located in the smooth muscle cells and binds to its haem moiety leading to intracellular accumulation of the second messenger molecule cGMP, which in turn regulates numerous physiological events such as vessel tone and neurotransmission. In addition to the importance of NO-sGC-cGMP pathway for vasorelaxation, cardiovascular diseases are frequently associated with endothelial injury and, consequently, impairment of this pathway. Therefore, the NO-sGC-cGMP pathway became a target for treating cardiovascular diseases. Drugs releasing NO (or NO donors) such as sodium nitroprusside and nitroglycerin were developed to suppress the deficiency of endothelium-derived NO in patients with cardiovascular risks. However, patients taking NO donors become hyporesponsive and exhibit tolerance to organic nitrates.3,4 Tolerance to NO-releasing drugs are believed to be associated with the bioconversion of the molecules to NO, NO–superoxide interaction, and enzyme desensitization induced by exogenous NO. As a result, non-NO-based drugs activating the sGC were developed in an attempt to mimic NO effects without the development of tolerance by the patients. In this review, we briefly describe the physiological effects of sGC activation of smooth muscle and the evolution and development of drugs called NO-independent sGC activators/stimulators within the last decade.
Physiological Activation of Soluble Guanylyl Cyclase
sGC is a heterodimeric complex that consists of two subunits, α and β, and each one contains three common domains, as follows: 1) the N-terminal heme-binding domain that mediates the NO sensitivity of the enzyme; 2) a dimerization domain, which is found in the middle of the structure of each subunit and is required for basal or NO-stimulated sGC activity; and lastly 3) the C-terminal catalytic domain, which is the most highly conserved region between the subunits and is responsible for the conversion of GTP to the second messenger, cGMP.5 The mechanisms proposed for cGMP-mediated relaxations include: 1) inhibition of inositol-1,4,5-triphosphate generation; 2) enhanced cytosolic Ca2+ extrusion; 3) dephosphorylation of MLCK; 4) Ca2+ influx inhibition; 5) protein kinase G activation; 6) stimulation of membrane Ca2+ ATPase; and 7) potassium channels opening.5,6 The breakdown of cGMP to its inactive form 5′GMP is mediated by the PDE5, which is selective to cGMP and stops relaxation.7,8
NO, which is a heme-dependent sGC stimulator, stimulates sGC by binding to its prosthetic group containing the reduced Fe2+ haem moiety and removal or oxidation of the haem moiety leads to the NO-insensitive form of the enzyme.9 In the prosthetic group, the central ferrous iron is coordinated between four haem nitrogens and the axial ligand histidine-105, and forms a penta-coordinated histidyl–haem complex. NO binding results in the formation of a hexa-coordinated histidine-haem–NO intermediate, which is rapidly converted into a penta-coordinated nitrosyl–haem complex by cleavage of the haem–histidine bond, which is the molecular switch for sGC activation.10 However, in some cases, the redox state of the sGC can be changed by reactive oxygen and nitrogen species, which decrease its activity and expression.11
In the presence of an intact heme-moiety, the sGC is a constitutively active enzyme that basally releases cGMP. Heme-dependent compounds are unable to activate the sGC when it is on its inactive state (heme-free/oxidized enzyme). On the other hand, heme-independent compounds activate the sGC even if it is inactive. For these reasons, heme-dependent compounds are called sGC stimulators, whereas heme-independent compounds are called sGC activators. In summary, the physiological activation of sGC as well as sGC stimulation induced by heme-dependent compounds depends on the integrity of heme moiety of sGC, which needs to be in its reduced state, whereas heme-independent sGC activators are more potent in activating sGC when the heme moiety is oxidized (Fig. 1).
FIGURE 1.
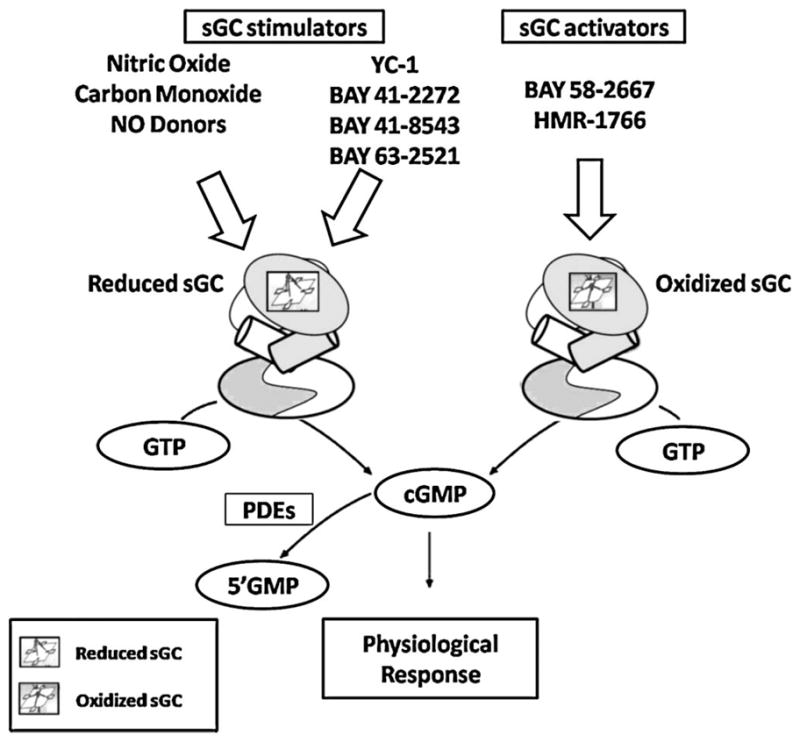
Soluble guanylyl cyclase activation by nitric oxide (NO), carbon monoxide (CO), NO donors, and NO-independent heme-dependent soluble guanylyl cyclase (sGC) stimulators requires integrity of heme-moiety of sGC, whereas NO- and heme- independent sGC activators are more potent to activate sGC if heme moiety is on its oxidized (planar) state.
Additionally, NO does not seem to be the only physiological molecule activating sGC. Despite the low potency, carbon monoxide (CO) also seems to activate sGC to cause vasorelaxation12 and to inhibit platelet aggregation.13
Based on the knowledge of the physiological stimulators of sGC, the indirect effects of synthetic sGC activators and stimulators were evaluated on the responses mediated by NO and CO as well as their direct effects on sGC.
HEME-DEPENDENT SOLUBLE GUANYLYL CYCLASE STIMULATORS: YC-1, BAY 41-2272, BAY 41-8543, AND BAY 63-2521
YC-1: The First Soluble Guanylyl Cyclase Stimulator
Tolerance to organic nitrates limited the therapeutic use of NO donors on NO-sGC-cGMP pathway activation in the treatment of cardiovascular diseases. Therefore, non-NO-based drugs activating sGC were developed. YC-1 (3-[5′-hydroxymethyl-2′-furyl]-1-benzylindazole), a chemically synthesized benzylindazole compound, was the first sGC activator developed and it was described to inhibit platelet aggregation by activating platelet sGC14 in an NO-independent mechanism.15 Although its action on sGC is still unclear, the effects of YC-1 were shown to be NO-independent but heme-dependent and capable to potentiate the effects of NO and CO.16 On the other hand, other authors suggested that YC-1 binds to a NO-independent regulatory site of sGC, an allosteric site on sGC that reduces ligand dissociation rates from the heme group without a direct change in the spectral characteristic of the heme moiety of sGC.17 Indeed, it was demonstrated that YC-1 has both heme-dependent and heme-independent properties.18
In vascular smooth muscle, YC-1 exhibited vasodilatory properties, causing endothelium-independent relaxation as well as increasing sGC activity and cGMP formation.19 In addition, YC-1 was also described to potentiate the vaso-relaxation and the reduction in the mean arterial pressure induced by CO20 and NO donor sodium nitroprusside,21 which might be of interest for treating pathologies associated with decreased NO bioavailability such as hypertension. Therefore, its effects were evaluated in animal models of hypertension. Acutely administered, YC-1 alone caused a transient decrease in the mean arterial pressure of hypertensive rats, whereas it strongly potentiated the effect of sodium nitroprusside inducing a sustained reduction in blood pressure.21 Corroborating this observation, YC-1 potentiated the effects of sodium nitroprusside in either vascular smooth muscle relaxation or sGC activity.19 However, its therapeutic relevance was not con-firmed because controversial effects of YC-1 were reported in animal models of hypertension. YC-1 equally relaxed the aorta of normotensive and hypertensive mice,22 whereas the ability of YC-1 to induce relaxation was smaller in spontaneously hypertensive rats, an effect attributed to the higher superoxide formation in spontaneously hypertensive rats, because this difference was blunted in the presence of superoxide dismutase.19 Moreover, YC-1 is less potent than NO donors in inducing vascular relaxation, which, therapeutically, raises the risk of toxicity. To date, chronic effects of this compound are unknown in both animals and humans as well as no clinical trials of YC-1 have been reported.
BAY 41-2272, BAY 41-8543, AND BAY 63-2521
Based on the molecular structure of YC-1, a series of pyrazolopyridinylpyrimidines derivatives were synthesized to find a more potent drug to activate sGC.23 One of the first selected compounds was 5-cyclopropyl-2-(1-[2-fluorobenzyl]-1H-pyrazolo [3,4-b]pyridine-3-yl)pyrimidin-4-ylamine, a pyrazolopyridine named BAY 41-2272, two orders of magnitude more potent than YC-1.23,24 Together with BAY 41-2272, BAY 41-8543 was released as a heme-dependent drug and the most potent sGC activator, causing vascular relaxation and reducing coronary perfusion pressure and collagen-mediated platelet aggregation.25,26 Chemical structures of YC-1, BAY 41-2272, and BAY 41-8543 are presented in Figure 2(A, B, and C, respectively) . Nevertheless, only few studies investigated the effects of BAY 41-8543.25,26 On the other hand, BAY 41-2272 was extensively investigated and it was shown to have similar effects to YC-1, including anticoagulant,27 antiplatelet,28 and anti-inflammatory activities,29 chemotaxis inhibition,30 vasodilatory properties,23,31,32 pro erectile activity33,34 and angiogenesis.35 Similar to YC-1, BAY 41-2272 potentiated the relaxant effects of NO in the corpus cavernosum,33,34 aorta,23,31 penile urethra,36 and anococcygeus muscle.37 BAY 41-2272 binds to the cysteine 238 and cysteine 243 spanning region of the α1 subunit of the enzyme, and its mechanism of action was described to be heme-dependent in the isolated enzyme,23 although in smooth muscles, heme oxidation by ODQ did not blunt the relaxant effect of BAY 41-2272.31,33 The intriguing and remaining effects of BAY 41-2272 after sGC blockade led to the investigation of the underlying mechanisms of BAY 41-2272 after enzyme oxidation and the proposed mechanisms include inhibition of PDE538 and calcium entry blockade. This last effect was reproducible in the basilar artery,39 urinary bladder,40 and penile urethra.36 However, PDE5 inhibition occurred only in high concentrations and therefore this inhibitory effect is considered irrelevant because these concentrations would not be reached in therapeutic doses. In the cardiovascular system, BAY 41-2272 diminished blood pressure in normotensive mice41 and in vessels BAY 41-2272 was described to cause a potent and long-lasting relaxation of aorta,23,31 basilar,39 and pulmonary42 arteries in a synergistic fashion with NO and exhibited a higher potency than YC-1 in the relaxation of rat mesenteric arteries.32 Interestingly, BAY 41-2272 maintains its activity in tissues that have developed tolerance to organic nitrates.25 BAY 41-2242 has been shown to be effective in the treatment of acute43 and chronic44 pulmonary hypertension administered either intravenously or inhaled. However, its effects in arterial hypertension remain controversial. In mesenteric arteries of spontaneously hypertensive rats, the augmented oxidative stress decreased the relaxation induced by BAY 41-2272.45 Additionally, BAY 41-2272 prevented the increase in blood pressure and heart abnormalities caused by chronic NO inhibition46 and cardiovascular remodeling in angiotensin II-induced hypertension.47 It is important to clarify that in both studies, although administered chronically, BAY 41-2272 was given before the development of high blood pressure, whereas the decreased relaxation was observed in mesenteric arteries after hypertension was established.
FIGURE 2.

Chemical structures of soluble guanylyl cyclase (sGC) stimulators YC-1 (A), BAY 41-2272 (B), BAY 41-8543 (C), and BAY 63-2521 (D) and sGC activators BAY 58-2667 (E), and HMR-1766 (F).
In summary, BAY 41-2272 helped to elucidate the mechanisms underlying the relaxation induced by sGC activation nevertheless, there are no clinical trials testing the effects of BAY 41-2272 in humans.
Recently described, BAY 63-2521 (so-called riociguat; Fig. 2D) is being proposed to treat pulmonary hypertension. The first published study was performed in healthy volunteers and it was shown that riociguat caused a decrease in the mean arterial pressure and diastolic blood pressure with no effects in the systolic blood pressure.48 Subsequently, the beneficial effects of riociguat were shown in pulmonary hypertension in both animal models49 or human patients.50 Although riociguat is being investigated in Phase III clinical trials50 and it is expected as a promising drug for the treatment of pulmonary hypertension, only five studies report its effects in the pulmonary vascular bed.
HEME-INDEPENDENT SOLUBLE GUANYLYL CYCLASE ACTIVATORS: AND HMR-1766
BAY 58-2667 (Cinaciguat)
BAY 58-2667 is an amino dicarboxylic acid (Fig. 2E) described to have similar hemodynamics, vascular, platelet, and anticoagulant pharmacologic effects as BAY 41-2272. However, BAY 58-2667 binds to the amino acids 371 on the α1 subunit and 231–310 on the β1 subunit of sGC with no changes in the prosthetic heme group indicating that, unlike BAY 41-2272, it activates sGC in a NO- and heme-independent manner.25 In addition, BAY 58-2667 was shown to be 160-fold more potent in inducing vascular relaxation than BAY 41-2272 and more potent than glyceryltrinitrate to decrease coronary perfusion pressure.51 Furthermore, BAY 58-2667-induced sGC activation is potentiated in the absence of the heme moiety or when it is oxidized.25 This makes the molecule an interesting tool for the treatment of pathologies in which free radicals oxidize sCG such as in cardiovascular diseases. In the rat and dog, BAY 58-2667 protected cardiac damage induced by myocardial infarction.52 BAY 58-2667 is being called cinaciguat, and the first evaluation of its pharmacodynamic and pharmacokinetic actions showed that cinaciguat reduces cardiac preload and afterload in healthy volunteers.53 The pharmacodynamic and pharmacokinetic of cinaciguat were also evaluated in patients with acute decompensated heart failure54 and in these patients, cinaciguat improved cardiopulmonary hemodynamics.51 As a result, BAY 58-2667 became a potential drug for the treatment of cardiovascular diseases, being proposed to treat acute myocardial infarct,55 heart failure,4 and diseased blood vessels.56
HMR-1766 (Ataciguat)
HMR-1766 was described as an anthranilic acid derivative (Fig. 2F) that preferentially activates oxidized heme-containing sGC.57,58 HMR-1766 is being proposed for the treatment of cardiovascular diseases associated with augmented oxidative stress58 and pulmonary hypertension.59 However, a recent study showed that HMR-1766 is not able to prevent sGC degradation induced by oxidation.60 Since the first publication in 2004, only nine studies were published investigating the effects of HMR-1766 and to date, no clinical trials investigated the effects of this compound in either cardiovascular or pulmonary disease.
Perspectives
In this last decade, non-NO-based molecules have been described as a novel pharmacologic approach for the treatment of cardiopulmonary diseases, including arterial and pulmonary hypertension. Two drug classes (classified as heme-dependent sGC stimulators and heme-independent sGC activators) have been extensively studied. Although the effects of heme-dependent molecules are effective in producing vasorelaxation, it seems that their effects are diminished under pathologic conditions associated with increased oxidative stress, which causes sGC oxidation and subsequent degradation. Therefore, heme-independent molecules, which were shown to have higher affinity for the oxidized enzyme, became the promising drugs for the treatment of arterial and pulmonary hypertension. However, such molecules are still undergoing clinical trials. Cinaciguat and riociguat seem to be in the most advanced stage of study for the treatment of heart failure and pulmonary hypertension, respectively. Although these molecules are promising drugs, so far they are not available for clinical use.
Acknowledgments
We thank Dr. Samuel Barillas for editing the manuscript.
FBMP is supported by a postdoctoral fellowship from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). RCW is funded by grants from the National Institutes of Health (HL-74167; HL-71138).
Footnotes
The authors state that they hold no financial interest in the products/drugs mentioned within this article.
References
- 1.Rees DD, Palmer RM, Hodson HF, et al. A specific inhibitor of nitric oxide formation from L-arginine attenuates endothelium-dependent relaxation. Br J Pharmacol. 1989;96:418–424. doi: 10.1111/j.1476-5381.1989.tb11833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- 3.Chirkov YY, Horowitz JD. Impaired tissue responsiveness to organic nitrates and nitric oxide: a new therapeutic frontier? Pharmacol Ther. 2007;116:287–305. doi: 10.1016/j.pharmthera.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 4.Boerrigter G, Costello-Boerrigter LC, Cataliotti A, et al. Targeting heme-oxidized soluble guanylate cyclase in experimental heart failure. Hypertension. 2007;49:1128–1133. doi: 10.1161/HYPERTENSIONAHA.106.083832. [DOI] [PubMed] [Google Scholar]
- 5.Lucas KA, Pitari GM, Kazerounian S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- 6.Waldman SA, Murad F. Cyclic GMP synthesis and function. Pharmacol Rev. 1987;39:163–196. [PubMed] [Google Scholar]
- 7.Maurice DH, Palmer D, Tilley DG, et al. Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol. 2003;64:533–546. doi: 10.1124/mol.64.3.533. [DOI] [PubMed] [Google Scholar]
- 8.Rybalkin SD, Yan C, Bornfeldt KE, et al. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93:280–291. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- 9.Hwang TL, Wu CC, Teng CM. Comparison of two soluble guanylyl cyclase inhibitors, methylene blue and ODQ, on sodium nitroprusside-induced relaxation in guinea-pig trachea. Br J Pharmacol. 1998;125:1158–1163. doi: 10.1038/sj.bjp.0702181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ignarro LJ, Wood KS, Wolin MS. Activation of purified soluble guanylate cyclase by protoporphyrin IX. Proc Natl Acad Sci U S A. 1982;79:2870–2873. doi: 10.1073/pnas.79.9.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerassimou C, Kotanidou A, Zhou Z, et al. Regulation of the expression of soluble guanylyl cyclase by reactive oxygen species. Br J Pharmacol. 2007;150:1084–1091. doi: 10.1038/sj.bjp.0707179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morita T, Perrella MA, Lee ME, et al. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc Natl Acad Sci U S A. 1995;92:1475–1479. doi: 10.1073/pnas.92.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brüne B, Ullrich V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol Pharmacol. 1987;32:497–504. [PubMed] [Google Scholar]
- 14.Ko FN, Wu CC, Kuo SC, et al. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226–4233. [PubMed] [Google Scholar]
- 15.Wu CC, Ko FN, Kuo SC, et al. YC-1 inhibited human platelet aggregation through NO-independent activation of soluble guanylate cyclase. Br J Pharmacol. 1995;116:1973–1978. doi: 10.1111/j.1476-5381.1995.tb16400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stone JR, Marletta MA. Synergistic activation of soluble guanylate cyclase by YC-1 and carbon monoxide: implications for the role of cleavage of the iron–histidine bond during activation by nitric oxide. Chem Biol. 1998;5:255–261. doi: 10.1016/s1074-5521(98)90618-4. [DOI] [PubMed] [Google Scholar]
- 17.Becker EM, Alonso-Alija C, Apeler H, et al. NO-independent regulatory site of direct sGC stimulators like YC-1 and BAY 41-2272. BMC Pharmacol. 2001;1:13. doi: 10.1186/1471-2210-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin E, Lee YC, Murad F. YC-1 activation of human soluble guanylyl cyclase has both heme-dependent and heme-independent components. Proc Natl Acad Sci U S A. 2001;98:12938–12942. doi: 10.1073/pnas.231486198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mülsch A, Bauersachs J, Schäfer A, et al. Effect of YC-1, an NO-independent, superoxide-sensitive stimulator of soluble guanylyl cyclase, on smooth muscle responsiveness to nitrovasodilators. Br J Pharmacol. 1997;120:681–689. doi: 10.1038/sj.bjp.0700982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motterlini R, Sawle P, Hammad J, et al. CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005;19:284–286. doi: 10.1096/fj.04-2169fje. [DOI] [PubMed] [Google Scholar]
- 21.Rothermund L, Friebe A, Paul M, et al. Acute blood pressure effects of YC-1-induced activation of soluble guanylyl cyclase in normotensive and hypertensive rats. Br J Pharmacol. 2000;130:205–208. doi: 10.1038/sj.bjp.0703320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linder AE, Weber DS, Whitesall SE, et al. Altered vascular reactivity in mice made hypertensive by nitric oxide synthase inhibition. J Cardiovasc Pharmacol. 2005;46:438–444. doi: 10.1097/01.fjc.0000175879.14994.63. [DOI] [PubMed] [Google Scholar]
- 23.Straub A, Stasch JP, Alonso-Alija C, et al. NO-independent stimulators of soluble guanylate cyclase. Bioorg Med Chem Lett. 2001;11:781–784. doi: 10.1016/s0960-894x(01)00073-7. [DOI] [PubMed] [Google Scholar]
- 24.Stasch JP, Becker EM, Alonso-Alija C, et al. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410:212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- 25.Stasch JP, Schmidt P, Alonso-Alija C, et al. NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol. 2002;136:773–783. doi: 10.1038/sj.bjp.0704778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stasch JP, Alonso-Alija C, Apeler H, et al. Pharmacological actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41-8543: in vitro studies. Br J Pharmacol. 2002;135:333–343. doi: 10.1038/sj.bjp.0704484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sovershaev MA, Egorina EM, Hansen JB, et al. Soluble guanylate cyclase agonists inhibit expression and procoagulant activity of tissue factor. Arterioscler Thromb Vasc Biol. 2009;29:1578–1586. doi: 10.1161/ATVBAHA.109.192690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hobbs AJ, Moncada S. Antiplatelet properties of a novel, non-NO-based soluble guanylate cyclase activator, BAY 41-2272. Vasc Pharmacol. 2003;40:149–154. doi: 10.1016/s1537-1891(03)00046-6. [DOI] [PubMed] [Google Scholar]
- 29.Ahluwalia A, Foster P, Scotland RS, et al. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. Proc Natl Acad Sci U S A. 2004;101:1386–1391. doi: 10.1073/pnas.0304264101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomazzi SM, Moreira J, De Nucci G, et al. Inhibitory effects on human eosinophil chemotaxis in vitro by BAY 41-2272, an activator of nitric oxide-independent site of soluble guanylate cyclase. Biochem Pharmacol. 2005;69:875–882. doi: 10.1016/j.bcp.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 31.Priviero FB, Baracat JS, Teixeira CE, et al. Mechanisms underlying relaxation of rabbit aorta by BAY 41-2272, a nitric oxide-independent soluble guanylate cyclase activator. Clin Exp Pharmacol Physiol. 2005;32:728–734. doi: 10.1111/j.1440-1681.2005.04262.x. [DOI] [PubMed] [Google Scholar]
- 32.Teixeira CE, Priviero FB, Webb RC. Molecular mechanisms underlying rat mesenteric artery vasorelaxation induced by the nitric oxide-independent soluble guanylyl cyclase stimulators BAY 41-2272 [5-cyclopropyl-2-[1-(2-fluorobenzyl)-1H–pyrazolo[3,4-b]pyridin-3-yl] pyrimidin-4-ylamine] and YC-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzyl Indazole] J Pharmacol Exp Ther. 2006;317:258–266. doi: 10.1124/jpet.105.095752. [DOI] [PubMed] [Google Scholar]
- 33.Baracat JS, Teixeira CE, Okuyama CE, et al. Relaxing effects induced by the soluble guanylyl cyclase stimulator BAY 41-2272 in human and rabbit corpus cavernosum. Eur J Pharmacol. 2003;477:163–169. doi: 10.1016/j.ejphar.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 34.Teixeira CE, Priviero FB, Webb RC. Effects of 5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridine-3-yl]pyrimidin-4-ylamine (BAY 41-2272) on smooth muscle tone, soluble guanylyl cyclase activity, and NADPH oxidase activity/expression in corpus cavernosum from wild-type, neuronal, and endothelial nitric-oxide synthase null mice. J Pharmacol Exp Ther. 2007;322:1093–1102. doi: 10.1124/jpet.107.124594. [DOI] [PubMed] [Google Scholar]
- 35.Pyriochou A, Beis D, Koika V, et al. Soluble guanylyl cyclase activation promotes angiogenesis. J Pharmacol Exp Ther. 2006;319:663–671. doi: 10.1124/jpet.106.108878. [DOI] [PubMed] [Google Scholar]
- 36.Toque HA, Antunes E, Teixeira CE, et al. Increased cyclic guanosine monophosphate synthesis and calcium entry blockade account for the relaxant activity of the nitric oxide-independent soluble guanylyl cyclase stimulator BAY 41–2272 in the rabbit penile urethra. Urology. 2008;72:711–715. doi: 10.1016/j.urology.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 37.Teixeira CE, Priviero FB, Claudino MA, et al. Stimulation of soluble guanylyl cyclase by BAY 41-2272 relaxes anococcygeus muscle: interaction with nitric oxide. Eur J Pharmacol. 2006;530:157–165. doi: 10.1016/j.ejphar.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 38.Mullershausen F, Russwurm M, Friebe A, et al. Inhibition of phosphodiesterase type 5 by the activator of nitric oxide-sensitive guanylyl cyclase BAY 41-2272. Circulation. 2004;109:1711–1713. doi: 10.1161/01.CIR.0000126286.47618.BD. [DOI] [PubMed] [Google Scholar]
- 39.Teixeira CE, Priviero FB, Todd J, Jr, et al. Vasorelaxing effect of BAY 41–2272 in rat basilar artery: involvement of cGMP-dependent and independent mechanisms. Hypertension. 2006;47:596–602. doi: 10.1161/01.HYP.0000199914.36936.1b. [DOI] [PubMed] [Google Scholar]
- 40.Bau FR, Monica FZ, Priviero FB, et al. Evaluation of the relaxant effect of the nitric oxide-independent soluble guanylyl cyclase stimulator BAY 41-2272 in isolated detrusor smooth muscle. Eur J Pharmacol. 2010;637:171–177. doi: 10.1016/j.ejphar.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 41.Buys ES, Sips P, Vermeersch P, et al. Gender-specific hypertension and responsiveness to nitric oxide in sGCalpha1 knockout mice. Cardiovasc Res. 2008;79:179–186. doi: 10.1093/cvr/cvn068. [DOI] [PubMed] [Google Scholar]
- 42.Evgenov OV, Ichinose F, Evgenov NV, et al. Soluble guanylate cyclase activator reverses acute pulmonary hypertension and augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs. Circulation. 2004;110:2253–2259. doi: 10.1161/01.CIR.0000144469.01521.8A. [DOI] [PubMed] [Google Scholar]
- 43.Freitas CF, Morganti RP, Annichino-Bizzacchi JM, et al. Effect of BAY 41-2272 in the pulmonary hypertension induced by heparin–protamine complex in anaesthetized dogs. Clin Exp Pharmacol Physiol. 2007;34:10–14. doi: 10.1111/j.1440-1681.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- 44.Dumitrascu R, Weissmann N, Ghofrani HA, et al. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation. 2006;113:286–295. doi: 10.1161/CIRCULATIONAHA.105.581405. [DOI] [PubMed] [Google Scholar]
- 45.Priviero FB, Zemse SM, Teixeira CE, et al. Oxidative stress impairs vasorelaxation induced by the soluble guanylyl cyclase activator BAY 41-2272 in spontaneously hypertensive rats. Am J Hypertens. 2009;22:493–499. doi: 10.1038/ajh.2009.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zanfolin M, Faro R, Araujo EG, et al. Protective effects of BAY 41-2272 (sGC stimulator) on hypertension, heart, and cardiomyocyte hypertrophy induced by chronic L-NAME treatment in rats. J Cardiovasc Pharmacol. 2006;47:391–395. [PubMed] [Google Scholar]
- 47.Masuyama H, Tsuruda T, Kato J, et al. Soluble guanylate cyclase stimulation on cardiovascular remodeling in angiotensin II-induced hypertensive rats. Hypertension. 2006;48:972–978. doi: 10.1161/01.HYP.0000241087.12492.47. [DOI] [PubMed] [Google Scholar]
- 48.Frey R, Mück W, Unger S, et al. Single-dose pharmacokinetics, pharma-codynamics, tolerability, and safety of the soluble guanylate cyclase stimulator BAY 63-2521: an ascending-dose study in healthy male volunteers. J Clin Pharmacol. 2008;48:926–934. doi: 10.1177/0091270008319793. [DOI] [PubMed] [Google Scholar]
- 49.Schermuly RT, Stasch JP, Pullamsetti SS, et al. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur Respir J. 2008;32:881–891. doi: 10.1183/09031936.00114407. [DOI] [PubMed] [Google Scholar]
- 50.Mittendorf J, Weigand S, Alonso-Alija C, et al. Discovery of riociguat (BAY 63-2521): a potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertension. Chem Med Chem. 2009;4:853–865. doi: 10.1002/cmdc.200900014. [DOI] [PubMed] [Google Scholar]
- 51.Lapp H, Mitrovic V, Franz N, et al. Cinaciguat (BAY 58-2667) improves cardiopulmonary hemodynamics in patients with acute decompensated heart failure. Circulation. 2009;119:2781–2788. doi: 10.1161/CIRCULATIONAHA.108.800292. [DOI] [PubMed] [Google Scholar]
- 52.Korkmaz S, Radovits T, Barnucz E, et al. Pharmacological activation of soluble guanylate cyclase protects the heart against ischemic injury. Circulation. 2009;120:677–686. doi: 10.1161/CIRCULATIONAHA.109.870774. [DOI] [PubMed] [Google Scholar]
- 53.Frey R, Mück W, Unger S, et al. Pharmacokinetics, pharmacodynamics, tolerability, and safety of the soluble guanylate cyclase activator cinaciguat (BAY 58-2667) in healthy male volunteers. J Clin Pharmacol. 2008;48:1400–1410. doi: 10.1177/0091270008322906. [DOI] [PubMed] [Google Scholar]
- 54.Mueck W, Frey R. Population pharmacokinetics and pharmacodynamics of cinaciguat, a soluble guanylate cyclase activator, in patients with acute decompensated heart failure. Clin Pharmacokinet. 2010;49:119–129. doi: 10.2165/11317590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 55.Krieg T, Liu Y, Rütz T, et al. BAY 58-2667, a nitric oxide-independent guanylyl cyclase activator, pharmacologically post-conditions rabbit and rat hearts. Eur Heart J. 2009;30:1607–1613. doi: 10.1093/eurheartj/ehp143. [DOI] [PubMed] [Google Scholar]
- 56.Gladwin MT. Deconstructing endothelial dysfunction: soluble guanylyl cyclase oxidation and the NO resistance syndrome. J Clin Invest. 2006;116:2330–2332. doi: 10.1172/JCI29807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schindler U, Strobel H, Schönafinger K, et al. Biochemistry and pharmacology of novel anthranilic acid derivatives activating heme-oxidized soluble guanylyl cyclase. Mol Pharmacol. 2006;69:1260–1268. doi: 10.1124/mol.105.018747. [DOI] [PubMed] [Google Scholar]
- 58.Zhou Z, Pyriochou A, Kotanidou A, et al. Soluble guanylyl cyclase activation by HMR-1766 (ataciguat) in cells exposed to oxidative stress. Am J Physiol. 2008;295:H1763–H1771. doi: 10.1152/ajpheart.51.2008. [DOI] [PubMed] [Google Scholar]
- 59.Weissmann N, Hackemack S, Dahal BK, et al. The soluble guanylate cyclase activator HMR1766 reverses hypoxia-induced experimental pulmonary hypertension in mice. Am J Physiol. 2009;297:L658–L665. doi: 10.1152/ajplung.00189.2009. [DOI] [PubMed] [Google Scholar]
- 60.Hoffmann LS, Schmidt PM, Keim Y, et al. Distinct molecular requirements for activation or stabilization of soluble guanylyl cyclase upon haem oxidation-induced degradation. Br J Pharmacol. 2009;157:781–795. doi: 10.1111/j.1476-5381.2009.00263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]