

Abstract
N-acyl-γ-glutamyl-diaminopimelic acid is a prototype ligand for Nod1. We report a detailed SAR of C12-γ-D-Glu-DAP. Analogues with glutaric or γ-aminobutyric acid replacing the glutamic acid show greatly attenuated Nod1-agonistic activity. Substitution of the meso-diaminopimelic (DAP) acid component with monoaminopimelic acid, L- or D-lysine, or cadaverine also results in reduced activity. The free amine on DAP is crucial. However, the N-acyl group on the D-glutamyl residue can be substituted with N-alkyl groups with full preservation of activity. The free carboxylates on the DAP and Glu components can also be esterified, resulting in more lipophilic, but active analogues. Transcriptomal profiling showed a dominant upregulation of IL-19, IL-20, IL-22, and IL-24, which may explain the pronounced Th2-polarizing activity of these compounds, and also implicate cell signaling mediated by TREM-1. These results may explain the hitherto unknown mechanism of synergy between Nod1- and TLR-agonists, and are likely to be useful in designing vaccine adjuvants.
Keywords: Nod1, iE-DAP, Diaminopimelic acid, Vaccine adjuvants, Innate immunity
Introduction
Cellular pattern recognition receptors (PRRs) are ubiquitous innate immune sensors that recognize specific molecular patterns present in molecules that are broadly shared by pathogens, but are structurally distinct from host molecules1–4 PRRs include not only membrane-bound Toll-like receptors (TLRs), members of which are expressed on the cell surface, as well as the endocytoplasmic reticulum, but also cytosolic receptors encompassing the Nod and RIG-I families of proteins.4;5 Nod1 and Nod2 are prototype members of the nucleotide oligomerization domain (NOD) and ligand-recognizing leucine-rich repeat (NLR)- containing proteins which serve to signal the presence of intracytoplasmic peptidoglycan fragments by sensing diaminopimelic acid peptides and muramyl dipeptides, respectively.6–9 Our interest in understanding the structural determinants of the biological properties of ligands of PRRs stems from the potential value in harnessing innate immune stimulatory properties in marshalling, and specifically directing subsequent adaptive immune responses with desired immunophenotypes and profiles.10;11 We had previously reported SAR studies on TLR2 and TLR7,12;13 and whilst studies on several other TLR ligands continue, our attention is focused also on the intracytoplasmic NOD family of receptors, especially Nod1. Whereas the muramyldipeptide chemotype has been extensively studied since the early 1970s14–19 (preceding by several decades the discovery of Nod2 as its primary receptor9;20), structure-activity relationships and signaling pathways involving Nod1-agonistic γ-glutamyl-meso-diaminopimelic acid peptides21;22 have remained sparse. Investigators at the Fujisawa Pharmaceutical Company reported in the early 1980s that D-lactoyl-L-alanyl- γ-D-glutamyl-(L)-meso-diaminopimelyl-(L)-glycine and heptanoyl-γ-D-glutamyl-(L)-meso-diaminopimelyl- D-alanine enhanced host defense in murine models of fatal E. coli sepsis.23–27 Subsequent to the discovery that the γ-glutamyl-meso-diaminopimelic acid (iE-DAP) fragment is the crucial pharmacophore mediating recognition of peptidoglycan fragments by Nod1,21;22 synthetic, lipophilic, N-acyl glutamyl derivatives have been shown to be potent Nod1 agonists.28;29 N-dodecanoyl-γ-D-glutamyl-diaminopimelic acid (C12-iE-DAP) is now available commercially as a reference Nod1 agonist. Also noteworthy is work by Boons and colleagues who have synthesized and evaluated DAP-containing muramyl peptides.30;31
We were keen to explore the structural space around iE-DAP scaffold for the reason that Nod1 agonists are associated with the mobilization of adaptive immune responses with a dominant Th2 bias, which is undesirable when a strong CD8+ cytotoxic T lymphocytic response is required.32 We hypothesized that covalent coupling to iE-DAP of the TLR7/8-agonistic imidazoquinolines12;33 that we have found to be extraordinarily Th1-polarizing10;11 could lead to vaccine adjuvants with balanced Th1/Th2 immunostimulatory phenotypes. In order to accomplish this goal, it was necessary to first determine optimal positions on the iE-DAP backbone that would permit the generation of dual-active ‘hybrid’ molecules, combining the TLR7-agonistic imidazoquinoline and NOD1-active iE-DAP chemotypes in a single covalently coupled construct. We report in this paper an SAR study of Nod1-agonistic iE-DAP derivatives. Secondary screens including transcriptomal profiling in ex vivo models using whole human blood have revealed a possible basis for the pronounced Th2 bias of this chemotype.
Results and Discussion
The construction of orthogonally protected γ-D-glutamyl-diaminopimelic acid synthons (Scheme 1) allowed convenient access to a number of analogues wherein the carboxyl and amino substituents on both amino acids could be varied (Schemes 2–4). For the sake of consistency, we designate the α-amino group on the D-Glu as N, and the γ-amino group on the diaminopimelic acid (DAP) residue as N′. We used the commercially available diaminopimelic acid (6; mixture of 4 isomers), since derivatives of all four isomers have been shown to be active.28;29 Esters of DAP proved surprisingly difficult to obtain by a number of conventional methods, necessitating sequential N-Boc protection of the amines, esterification (as benzyl or ethyl) of the carboxylic acids, deprotection of the N-Boc groups, followed by mono-N-Boc protection using stoichiometric equivalents of Boc2O. N-Fmoc-D-Glu-α-benzyl/allyl esters (4, 5 respectively) were coupled to the mono-N-Boc-diaminopimelic acid diesters (9 or 10) using standard protocols to afford the orthogonally protected synthons (11–13; Scheme 1). N-Fmoc (D-Glu) deprotection, followed by acylation with lauroyl chloride and subsequent deprotection of the N-Boc and O-Bn groups yielded C12-iE-DAP (16; Scheme 2), the activity of which was identical to commercially available reference compound with EC50 values of ~30 pM (Fig. 1A).
Scheme 1.

Reagents: i. ROH, HBTU, TEA, DMAP, DMF; ii. (a) CF3COOH (b) FmocCl, Na2CO3, Dioxane, H2O; iii. (a) (Boc)2O, TEA, H2O (b) R′OH, HBTU, DMAP, TEA, DMF; iv. (a) HCl-dioxane (b) (Boc)2O (1eq.), TEA, MeOH; v. PS-Carbodiimide, PS-DMAP, CH2Cl2.
Scheme 2.

Reagents: i. 30% Piperidine, CH2Cl2; ii. C11H23COCl, pyridine, DMAP; iii. (a) H2, Pd(OH)2/C, MeOH, 60 psi (b) CF3COOH; iv. (a) CF3COOH (b) C11H23COCl, TEA, CH2Cl2; v. H2, Pd(OH)2/C, AcOH, MeOH, 60 psi; vi. CF3COOH; vii. (a) C11H23COCl, pyridine, CH2Cl2 (b) H2, Pd(OH)2/C, MeOH, 60 psi; viii. C11H23CHO (1eq.), MP-CNBH3, CH2Cl2, MeOH, AcOH; ix. RCHO(excess), MP-CNBH3, CH2Cl2, MeOH, AcOH; x. 1H-Pyrazole-1-carboxamidine.HCl, pyridine, MW irradiation, 60°C; xi. H2, Pd(OH)2/C, MeOH, 60 psi; xii. N,N′-Di-Boc-1HPyrazole- 1-carboxamidine•HCl, pyridine, THF, 50°C.
Scheme 4.

Reagents: i. 30% Piperidine, CH2Cl2; ii. (a) (Boc)2O, TEA, MeOH (b) H2, Pd(OH)2/C, MeOH, 60 psi (c) C11H23SH, HBTU, DMAP, TEA, DMF; iii. CF3COOH; iv. C11H23COCl, pyridine; v. (a) H2, Pd(OH)2/C, MeOH, 60 psi (b) CF3COOH.
Figure 1.

A. NF-κB induction activity in stable transfectants of HEK-293 cells expressing human Nod1. B. Rank-order plot of potencies (EC50 values) of iE-DAP analogues.
The N′-acyl (DAP) compound (18), the N,N′-diacyl (D-Glu, and DAP, respectively) analogue (20), as well as N-alkyl (22) and N,N-dialkyl analogues (26–28) were also synthesized from synthon 11 (Scheme 2). Our initial attempts at converting the N′ amine to a guanidine group resulted in an undesired cyclization-elimination reaction to form the aminoimidazolone 30; this was obviated by the use of N,N′-di- Boc-1H-pyrazole-1-carboxamidine as the guanidination reagent, affording the desired product 32 (Scheme 2).
Next, analogues were synthesized from 12 with the acyl group on the α-amine of D-Glu transposed to the α-carboxylic acid group as either the undecanoyl ester (35), the 1-aminoundecane-derived amide (37), as well as the N-acyl long-chain ester 41 (Scheme 3). The N-guanidino ester analogue 43, as well as the α-D-Glu ethyl ester derivative of C12-iE-DAP (45) were also obtained from 12 (Scheme 3). The α-D-Glu thioester DAP ethyl ester analogue (48) and DAP ethyl ester derivative of C12-iE-DAP (50) were synthesized from synthon 13 (Scheme 4).
Scheme 3.

Reagents: i. (a) 30% Piperidine, CH2Cl2 (b) (Boc)2O, TEA, MeOH; ii. (a) Chlorotris (triphenylphosphine) rhodium(I), EtOH, H2O, reflux (b) C11H23OH, HBTU, DMAP, TEA, DMF; iii. (a) H2, Pd(OH)2/C, MeOH, 60 psi (b) CF3COOH; iv. (a) Chlorotris(triphenylphosphine) rhodium(I), EtOH, H2O, reflux (b) C11H23NH2, HBTU, TEA, DMAP, CH2Cl2; v. (a) Chlorotris (triphenylphosphine) rhodium(I), EtOH, H2O, reflux (b) ROH, HBTU, DMAP, DMF; vi. (a) 30% Piperidine, CH2Cl2 (b) C11H23COCl, pyridine, DMAP; vii. (a) 30% Piperidine, CH2Cl2 (b) 1H-Pyrazole-1- carboxamidine•HCl, pyridine, MW irradiation, 60°C.
In order to examine the importance of the α-amino and carboxyl groups of D-Glu, γ-aminobutyric acid (des-carboxyl, 53) or glutaric acid (des-amino, 56) were coupled to the DAP derivative 9 as depicted in Scheme 5. Similarly, suitably protected mono-aminopimelic acid 58, D- and L-lysine derivatives (62–65; Scheme 6), and commercially available N-Boc-cadaverine and D, L-α-amino-ε-caprolactam were used to examine the roles of the amine and carboxyl groups of DAP (Scheme 7).
Scheme 5.

Reagents: i. Fmoc-GABA-OH, PS-Carbodiimide, PS-DMAP, CH2Cl2; ii. (a) 30% Piperidine, CH2Cl2 (b) C11H23COCl, pyridine; iii. (a) H2, Pd(OH)2/C, MeOH, 60 psi (b) CF3COOH; iv. Glutaric anhydride, CH2Cl2; v. C11H23OH, HBTU, DMAP, TEA, DMF.
Scheme 6.

Reagents: i. BnOH, p-TSA, reflux; ii. (a) BnOH, HBTU, DMAP, DMF (b) 30% Piperidine, CH2Cl2; iii. (a) PhCH2OCOCl, Na2CO3, Dioxane, H2O (b) CF3COOH; iv. (a) (Boc)2O, TEA, MeOH (b) H2, Pd(OH)2/C, MeOH, 60 psi.
Scheme 7.

Reagents: i. PS-Carbodiimide, PS-DMAP, TEA, CH2Cl2; ii. 30% Piperidine, CH2Cl2; iii. C11H23COCl, pyridine; iv. H2, Pd(OH)2/C, MeOH, 60 psi; v. CF3COOH.
The N-acyl on the D-Glu residue appears to be optimal for Nod1-agonistic activity since the transposition of the acyl group to the N′ position on DAP (18), or diacylation at N and N′ (20) resulted in substantial decrease in activity (Table 1). The substitution of the N-acyl (C12) group with N-alkyl (22) led to virtually identical activity (27 pM versus 23 pM, Table 1). Compounds with C8 or C16 N,N-dialkyl groups (26, 28) displayed attenuated potency while the C12 N,N-dialkyl compound 27 displayed a rather dramatic increase in potency, with residual partial activity evident even at very high dilutions (Table 1, 27Fig. 1). Compound was the most potent analogue identified in this study. The primary amines on both the DAP and D-Glu segments were found to be crucial; the guanidino analogues 32 and 43, as well as the aminoimidazolone analogue 30 exhibited diminished activity.
Table 1.
EC50 values of hNod1 agonistic activities of analogues
| Cmpd | Structure | EC50 (nM) |
|---|---|---|
| 16 | 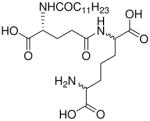 |
0.027 |
| 18 | 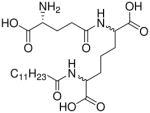 |
0.55 |
| 20 | 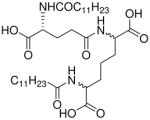 |
0.58 |
| 22 | 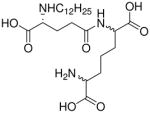 |
0.023 |
| 26 | 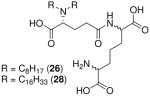 |
26: 0.20 |
| 28 | 28: 0.12 | |
| 27 | 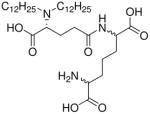 |
0.0015 |
| 30 | 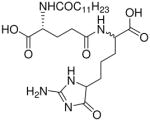 |
1.31 |
| 32 | 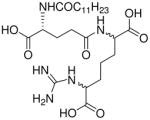 |
0.622 |
| 35 | 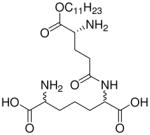 |
0.066 |
| 37 | 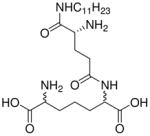 |
0.826 |
| 41 | 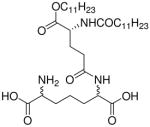 |
0.134 |
| 43 | 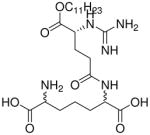 |
0.238 |
| 45 | 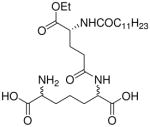 |
0.069 |
| 48 | 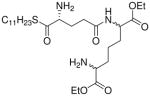 |
0.181 |
| 50 | 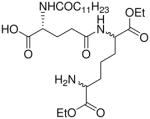 |
0.145 |
| 53 | 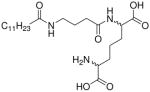 |
0.504 |
| 56 | 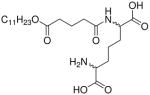 |
0.39 |
| 67 | 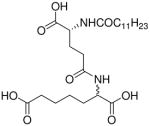 |
>100 |
| 69 | 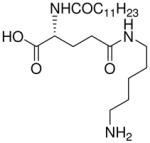 |
>100 |
| 71 | 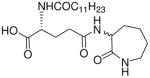 |
1.73 |
| 73 | 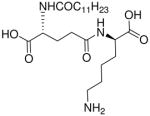 |
>100 |
| 75 | 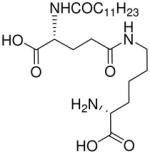 |
0.37 |
| 77 | 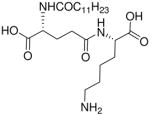 |
0.57 |
| 79 | 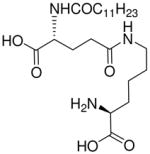 |
0.96 |
We next examined the importance of the free α-carboxyl group on D-Glu. The potencies of the undecanoyl ester of iE-DAP (35), as well as the ethyl ester of C12-iE-DAP (45) were comparable to that of the reference compound, C12-iE-DAP (16). The undecanoyl ester of C12-iE-DAP (41), and the thioester analogue 48 also retained modest activity. The 1-aminoundecane-derived amide analogue (37), however, was substantially less active. Augmented activity in 35 and 45 appears to be related to neither net charge nor bulk hydrophobicity, both of which could be reasonably expected to modulate transmembrane permeation, and consequent presentation of the ligand to the intracellular Nod1 receptor. The net charge at physiological pH of 35 and 37 is zero, whilst that of 41 and 45 is −1. We surmised that the higher potency of 45 relative to 41 could be due to the more facile hydrolytic cleavage by intracellular esterases of the shorter, ethyl ester of 45, but our observation that the rate and kinetics of NF-κB induction for both compounds were very similar (data not shown) is not in agreement with our conjecture, and we do not yet understand the basis for the difference in potencies of these analogues. Substitution of the N-acyl D-Glu fragment with either N-acyl γ-aminobutyric acid (53, des-α-carboxyl analogue), or the undecanoyl ester of glutaric acid (56, des-α-amino analogue) resulted only in considerable loss in Nod1-agonistic activity. Taken together, the above SAR data suggest that considerable ‘plasticity’ exists in the recognition of the D-Glu fragment. Indeed, a plot of the potencies of these analogues displays an unexpectedly diffuse, near-uniform distribution of EC50 values (Fig. 1B), unlike the well-demarcated and discrete SAR that we had previously observed for TLR2-13 and TLR7-active12 compounds.
In contradistinction to the broad and diffuse SAR for D-Glu fragment, the DAP portion of the molecule is far more stringent in determining Nod1 agonism. Whereas esterification of carboxyl groups of DAP (50) is tolerated, substitution with mono-aminopimelic acid (67, des-amino) or cadaverine (69, des-dicarboxyl) resulted in abrogation of activity. The meso-diaminopimelic acid-containing peptidoglycan is specific for Gram-negative bacteria, and the corresponding homologue in Gram-positive organisms is L-lysine coupled via its α-amine to γ-D-glutamyl residue (77), the activity of which is ~10-fold less than that of 16.34;35 The D-lysine analogue 73 is bereft of any activity. The unnatural ε-amine-coupled lysine analogues were also evaluated. The D-lysine analogue 75 was found to be four-fold more active than 79, its L-lysine congener.
Primary assay readouts using cell-culture systems may not always accurately reflect in vivo behavior owing to a variety of reasons, including differential plasma protein binding (that we ourselves have observed and characterized),36 and we therefore wished to confirm that the 27 was indeed more potent than 16, the reference Nod1 agonist, in ex vivo assays employing whole human blood. Although Nod1 agonists have previously been shown to enhance neutrophil recruitment in mice,34 a recent report employing synthetic, non-acylated, γ-glutamyl-meso-diaminopimelic acid dipeptide indicated that Nod1-specific responses could not be elicited in isolated human neutrophils.37 Using an ex vivo system using fresh, whole human blood that we have established for immunoprofiling innate immune stimuli,10;13;38 both p38 MAPK39 and CD11b40 were upregulated in a dose-dependent manner, with 27 being more potent than 16 (Fig. 2). Indeed, the non-acylated, γ-glutamyl-meso-diaminopimelic acid dipeptide was inactive in our primary and secondary screens (data not shown), emphasizing the requirement of an N-acyl functionality on glutamic acid.
Figure 2.

Dose-responses of p38 MAP kinase (A) and CD11b (B) induction in the granulocytic population in whole human blood by 16 and 27 determined by flow cytometry.
Recent findings using murine models of immunization indicate that Nod1 engagement drives B and T cell immunity with a predominant Th2 polarization, and additional Th17 priming.32 However, mechanisms underlying the induction of strong Th2-biased immune responses have remained obscure. Given the significant differences between species-dependent recognition of innate immune ligands,11 we desired to evaluate whether surrogate markers of Th2-skewed responses could be observed in human ex vivo model systems, in the hope that these studies may also help uncover signaling events determining Th2 polarization. Transcriptomal profiling experiments using human blood revealed a very prominent upregulation of Class II helical cytokines by 27, especially members of the IL-10 superfamily41 including IL-19, IL-20, IL-22, and IL-24 (Table 2); these cytokines, especially IL-19,42–45 have been shown to drive Th2-polarized responses. We have never before observed this particular constellation of cytokines in our earlier10 (and ongoing) immunoprofiling studies, and we are excited in no small measure that quantifying the induction of IL-19 and related cytokines may have predictive value as biomarkers and surrogate signatures for Th2 induction in immunoprofiling vaccine adjuvants, and may perhaps supplant the necessity of long and laborious immunization experiments which typically involve large numbers of animals. Also observed, as could be expected in the context of Th17-induction,32 was the upregulation of IL-17. Similar responses were observed with 16, but of lower magnitude, consistent with the lower potency of 16 (data not shown).
Table 2.
Interleukin Transcriptomal Responses in Human PBMCs exposed to 20 μg/mL of 27
| Probe Set ID | Fold change | Regulation | Gene Symbol | Gene Title |
|---|---|---|---|---|
| 220745_at | 30.39 | up | IL19 | interleukin 19 |
| 205207_at | 29.54 | up | IL6 | interleukin 6 (interferon, beta 2) |
| 210118_s_at | 14.34 | up | IL1A | interleukin 1, alpha |
| 220322_at | 11.10 | up | IL1F9 | interleukin 1 family, member 9 |
| 216876_s_at | 10.56 | up | IL17A | interleukin 17A |
| 216244_at | 8.27 | up | IL1RN | interleukin 1 receptor antagonist |
| 212659_s_at | 8.19 | up | IL1RN | interleukin 1 receptor antagonist |
| 216243_s_at | 7.54 | up | IL1RN | interleukin 1 receptor antagonist |
| 207901_at | 6.13 | up | IL12B | interleukin 12B (natural killer cell stimulatory factor 2, cytotoxic lymphocyte maturation factor 2, p40) |
| 207906_at | 5.99 | up | IL3 | interleukin 3 (colony-stimulating factor, multiple) |
| 212657_s_at | 4.94 | up | IL1RN | interleukin 1 receptor antagonist |
| 39402_at | 4.45 | up | IL1B | interleukin 1, beta |
| 205067_at | 4.14 | up | IL1B | interleukin 1, beta |
| 224071_at | 4.00 | up | IL20 | interleukin 20 |
| 221165_s_at | 3.87 | up | IL22 | interleukin 22 |
| 207433_at | 3.62 | up | IL10 | interleukin 10 |
| 211269_s_at | 3.62 | up | IL2RA | interleukin 2 receptor, alpha |
| 206341_at | 3.57 | up | IL2RA | interleukin 2 receptor, alpha |
| 206569_at | 3.41 | up | IL24 | interleukin 24 |
| 227997_at | 3.17 | up | IL17RD | interleukin 17 receptor D |
| 222223_s_at | 3.15 | up | IL1F5 | interleukin 1 family, member 5 (delta) |
| 211506_s_at | 2.29 | up | IL8 | interleukin 8 |
| 224262_at | 2.05 | up | IL1F10 | interleukin 1 family, member 10 (theta) |
| 206295_at | 2.03 | up | IL18 | interleukin 18 (interferon-gammainducing factor) |
| 1555431_a_at | 4.05 | down | IL31RA | interleukin 31 receptor A |
| 220056_at | 3.69 | down | IL22RA1 | interleukin 22 receptor, alpha 1 |
| 207008_at | 2.75 | down | IL8RB | interleukin 8 receptor, beta |
| 220663_at | 2.06 | down | IL1RAPL1 | interleukin 1 receptor accessory proteinlike 1 |
| 205227_at | 2.02 | down | IL1RAP | interleukin 1 receptor accessory protein |
Further analyses of gene transcripts induced by 27 yielded the unexpected finding that exposure of human blood to 27 results in the activation of pathways involving signaling mediated by Triggering Receptor Expressed on Myeloid Cells 1 (TREM-1; Fig. 3). TREM-1 is a recently-discovered receptor expressed on the cell surface of monocytes and neutrophils, and plays an important role in amplifying myeloid cell-activated inflammatory responses.46–48 Our observation of the upregulation of TREM-1 signaling components may explain the hitherto unknown mechanism of synergy between Nod1- and TLRagonists. 32;34;49;50
Figure 3.

Pathway analysis of transcriptomal activation patterns in human PBMCs exposed to 20 μg/mL of 16 and 27. The dashed line corresponds to the threshold of −log (base 10) value for P = 0.05 (after Benjamini-Hochberg multiple testing correction).
The overarching objective that led to our work on Nod1 ligands was not so much to synthesize higher potency compounds, but rather to understand SAR that would permit us to covalently couple other innate immune ligands12;13;33 to the iE-DAP scaffold in order to test the hypothesis that such hybrids may imbue a Th1-Th2 balanced, yet effective vaccine adjuvants. The studies reported here indicate that various substitutions and modifications on the γ-D-glutamyl moiety are tolerated whereas the diaminopimelic acid fragment is far more stringent. The α-carboxylic acid of D-Glu appears to be optimal for coupling with other innate immune ligands, and work in this area is in progress. Secondary and tertiary screens, including transcriptomal profiling have shed light on the possible mechanisms underlying the strong Th2 bias of Nod1 ligands, and of synergy with other innate immune stimuli.
Experimental Section
Chemistry
All of the solvents and reagents used were obtained commercially and used as such unless noted otherwise. Moisture- or air-sensitive reactions were conducted under nitrogen atmosphere in oven-dried (120 °C) glass apparatus. The solvents were removed under reduced pressure using standard rotary evaporators. Flash column chromatography was carried out using RediSep Rf ‘Gold’ high performance silica columns on CombiFlash Rf (Teledyne-Isco, Lincoln, NE) instrument unless otherwise mentioned, while thin-layer chromatography was carried out on silica gel CCM pre-coated aluminum sheets. Purity for all final compounds was confirmed to be at least 97% by LC-MS using a Zorbax Eclipse Plus 4.6 mm x 150 mm, 5 μm analytical reverse phase C18 column with H2O-isopropanol or H2O-CH3CN gradients (with 0.1% CF3COOH in both mobile phases) and an Agilent ESI-TOF mass spectrometer (mass accuracy of 5 ppm) operating in the positive ion (or negative ion, as appropriate) acquisition mode.
Synthesis of Compound 2: (R)-1-Benzyl 5-tert-butyl 2-((tert-butoxycarbonyl)amino) pentanedioate
To a solution of 1 (1 g, 3.29 mmol) in anhydrous DMF, were added HBTU (1.38 g, 3.62 mmol), triethylamine (0.5 mL, 3.62 mmol), benzyl alcohol (682 μL, 6.59 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 4 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in ethyl acetate and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (10% EtOAc/hexanes) to obtain the compound 2 (1.14 g, 88 %). 1H NMR (500 MHz, CDCl3) δ 7.39 – 7.31 (m, 5H), 5.16 (dt, J = 23.2, 11.5 Hz, 3H), 4.35 (dd, J = 13.0, 8.2 Hz, 1H), 2.35 – 2.22 (m, 2H), 2.13 (td, J = 13.3, 7.0 Hz, 1H), 1.92 (tt, J = 14.7, 7.5 Hz, 1H), 1.43 (s, 18H). 13C NMR (126 MHz, CDCl3) δ 172.09, 172.06, 155.39, 131.57, 118.82, 80.74, 79.93, 65.95, 53.11, 31.60, 28.30, 28.06, 27.89, 27.68. MS (ESI) calculated for C21H31NO6, m/z 393.22, found 416.22 (M + Na)+.
Synthesis of Compound 3: (R)-1-Allyl 5-tert-butyl 2-((tert-butoxycarbonyl)amino) pentanedioate
To a solution of 1 (1 g, 3.29 mmol) in anhydrous DMF, were added HBTU (1.38 g, 3.62 mmol), triethylamine (0.5 mL, 3.62 mmol), allyl alcohol (450 μL, 6.59 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 4 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in ethyl acetate and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (10% EtOAc/hexanes) to obtain the compound 3 (1.0 g, 90 %). 1H NMR (500 MHz, CDCl3) δ 5.91 (ddt, J = 16.3, 10.6, 5.8 Hz, 1H), 5.34 (dd, J = 17.2, 1.3 Hz, 1H), 5.26 (dd, J = 10.4, 0.7 Hz, 1H), 5.11 (d, J = 8.0 Hz, 1H), 4.64 - 4.63 (m, 2H), 4.33 (dd, J = 13.2, 8.3 Hz, 1H), 2.38 - 2.26 (m, 2H), 2.17-2.11 (m, 1H), 1.96-1.88 (m, 1H), 1.45 (s, 9H), 1.44 (s, 9 H). 13C NMR (126 MHz, CDCl3) δ 172.05, 172.02, 155.35, 131.53, 118.78, 80.70, 79.89, 65.91, 53.07, 31.56, 28.26, 28.02, 27.85, 27.64. MS (ESI) calculated for C17H29NO6, m/z 343.20, found 366.19 (M + Na)+.
Synthesis of Compound 4: (R)-4-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-5-(benzyloxy)- 5-oxopentanoic acid
The solution of 2 (1 g, 2.54 mmol) in trifluoroacetic acid was stirred for 1 hour, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt. To the solution of trifluoroacetate salt in dioxane, the aqueous solution of sodium carbonate (810 mg, 7.64 mmol) was added at 10 °C. The reaction mixture was stirred at room temperature for 10 min, followed by the addition of FmocCl (722 mg, 2.79 mmol) solution in dioxane. The reaction mixture was stirred at room temperature for 16 hrs, followed by removal of the solvent under reduced pressure. The residue was dissolved in ethyl acetate and water, and the solution was acidified using 10% HCl until pH was ~1. The aqueous layer was washed with ethyl acetate. The ethyl acetate was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (40% EtOAc/hexanes) to obtain the compound 4 (1.0 g, 91 %). 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 7.5 Hz, 2H), 7.56 (d, J = 7.4 Hz, 2H), 7.33 (qd, J = 15.0, 7.2 Hz, 9H), 5.47 (d, J = 8.2 Hz, 1H), 5.19 – 5.06 (m, 2H), 4.53 – 4.30 (m, 3H), 4.16 (dd, J = 15.6, 8.8 Hz, 1H), 2.48 – 2.12 (m, 3H), 1.94 (dt, J = 14.5, 7.7 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 177.59, 171.91, 156.27, 143.99, 143.84, 141.50, 141.49, 135.19, 128.88, 128.81, 128.56, 127.95, 127.30, 125.29, 125.26, 120.21, 120.20, 67.73, 67.34, 53.42, 47.32, 29.91, 27.64. MS (ESI) calculated for C27H25NO6, m/z 459.16, found 482.17 (M + Na)+.
Synthesis of Compound 5: (R)-4-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-5-(allyloxy)-5- oxopentanoic acid
The solution of compound 3 (1 g, 2.91 mmol) in trifluoroacetic acid was stirred for 1 hour, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt. To the solution of trifluoroacetate salt in dioxane, the aqueous solution of sodium carbonate (926 mg, 8.74 mmol) was added at 10 °C. The reaction mixture was stirred at room temperature for 10 min, followed by the addition of FmocCl (830 mg, 3.2 mmol) solution in dioxane. The reaction mixture was stirred at room temperature for 16 hours, followed by removal of the solvent under reduced pressure. The residue was dissolved in ethyl acetate and water, and the solution was acidified using 10% HCl until pH was ~1. The aqueous layer was washed with ethyl acetate. The ethyl acetate layer was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (40% EtOAc/hexanes) to obtain the compound 5 (1.1 g, 92 %). 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H), 7.59 (d, J = 7.1 Hz, 2H), 7.40 (t, J = 7.4 Hz, 2H), 7.31 (t, J = 7.4 Hz, 2H), 5.90 (ddd, J = 22.5, 11.0, 5.7 Hz, 1H), 5.44 (d, J = 8.1 Hz, 1H), 5.30 (dd, J = 27.7, 13.7 Hz, 2H), 4.65 (d, J = 5.5 Hz, 2H), 4.58 – 4.35 (m, 3H), 4.21 (t, J = 6.8 Hz, 1H), 2.58 – 2.17 (m, 3H), 2.07 – 1.92 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 172.70, 172.54, 155.72, 155.57, 135.49, 128.74, 128.57, 128.43, 80.07, 67.19, 67.14, 53.27, 32.24, 28.47, 28.45, 21.48, 21.19, 21.08. MS (ESI) calculated for C23H23NO6, m/z 409.15, found 432.14 (M + Na)+.
Synthesis of Compound 7: Dibenzyl 2,6-bis((tert-butoxycarbonyl)amino)heptanedioate
To a solution of compound 6 (1 g, 5.25 mmol) in water, were added di-tert-butyl dicarbonate (4.58 g, 21.0 mmol) and triethylamine (2.92 mL, 21.0 mmol). The reaction mixture was stirred for 6 hours, followed by removal of the solvent under reduced pressure to obtain the residue. To the solution of residue in anhydrous DMF, were added HBTU (4.38 g, 11.55 mmol), triethylamine (1.6 mL, 11.55 mmol), benzyl alcohol (1.63 mL, 15.75 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 8 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in ethyl acetate and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (8% EtOAc/hexanes) to obtain the compound 7 (2.48 g, 83%). 1H NMR (500 MHz, CDCl3) δ 7.44 – 7.29 (m, 10H), 5.13 (dt, J = 12.4, 8.3 Hz, 6H), 4.29 (s, 2H), 1.85 – 1.71 (m, 2H), 1.68 (s, 2H), 1.49 – 1.31 (m, 20H). 13C NMR (126 MHz, CDCl3) δ 172.70, 172.54, 155.72, 155.57, 135.49, 128.74, 128.57, 128.43, 80.07, 67.19, 67.14, 53.27, 32.24, 28.47, 28.45, 21.48, 21.19, 21.08. MS (ESI) calculated for C31H42N2O8, m/z 570.29, found 593.27 (M + Na)+.
Synthesis of Compound 8: Diethyl 2,6-bis((tert-butoxycarbonyl)amino)heptanedioate
To a solution of compound 6 (1 g, 5.25 mmol) in water, were added di-tert-butyl dicarbonate (4.58 g, 21.0 mmol) and triethylamine (2.92 mL, 21.0 mmol). The reaction mixture was stirred for 6 hours, followed by removal of the solvent under reduced pressure to obtain the residue. To the solution of residue in anhydrous DMF, were added HBTU (4.38 g, 11.55 mmol), triethylamine (1.6 mL, 11.55 mmol), ethyl alcohol (0.92 mL, 15.75 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 6 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in ethyl acetate and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (8% EtOAc/hexanes) to obtain the compound 8 (2.0 g, 85%). 1H NMR (500 MHz, CDCl3) δ 5.17 – 4.99 (m, 2H), 4.30 – 4.08 (m, 6H), 1.84 – 1.74 (m, 2H), 1.72 – 1.55 (m, 4H), 1.48 – 1.31 (m, 18H), 1.25 (t, J = 7.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 173.24, 173.06, 156.07, 155.92, 80.33, 68.61, 61.80, 53.58, 39.17, 32.74, 32.67, 30.80, 29.38, 28.81, 28.79, 28.71, 24.19, 23.45, 21.82, 21.47, 14.64, 14.52. MS (ESI) calculated for C21H38N2O8, m/z 446.26, found 469.26 (M + Na)+.
Synthesis of Compound 9: Dibenzyl 2-amino-6-((tert-butoxycarbonyl)amino)heptanedioate
The compound 7 (2 g, 3.5 mmol) was dissolved in 10 mL of HCl-dioxane and stirred for 2 hours. The solvent was then removed under vacuum to obtain the hydrochloride salt, which was then dissolved in methanol. The triethylamine was added to solution until pH ~ 7, followed by gradual addition of di-tert-butyl dicarbonate (764 mg, 3.5 mmol). The reaction mixture was stirred for 30 minutes, followed by evaporation of the solvent under reduced pressure to obtain the crude product, which was then purified using column chromatography (5% MeOH/CH2Cl2) to obtain the compound 9 (640 mg, 39%). 1H NMR (500 MHz, CDCl3) δ 7.45 – 7.28 (m, 10H), 5.23 – 5.09 (m, 4H), 5.03 (t, J = 7.3 Hz, 1H), 4.41 – 4.23 (m, 1H), 3.41 (td, J = 8.1, 5.2 Hz, 1H), 1.85 – 1.50 (m, 6H), 1.49 – 1.34 (m, 11H). 13C NMR (126 MHz, CDCl3) δ 176.17, 176.14, 173.01, 155.83, 136.10, 135.84, 129.25, 129.09, 129.06, 128.88, 128.78, 80.36, 67.46, 67.16, 67.14, 54.70, 54.68, 53.80, 53.74, 34.73, 34.66, 32.77, 32.74, 28.77, 21.93. MS (ESI) calculated for C26H34N2O6, m/z 470.24, found 471.24 (M + H)+.
Synthesis of Compound 10: Diethyl 2-amino-6-((tert-butoxycarbonyl)amino)heptanedioate
The compound 8 (1 g, 2.24 mmol) was dissolved in 5 mL of HCl-dioxane and stirred for 2.5 hours. The solvent was then removed under vacuum to obtain the hydrochloride salt, which was then dissolved in methanol. The triethylamine was added to solution until pH ~ 7, followed by gradual addition of di-tert-butyl dicarbonate (490 mg, 2.24 mmol). The reaction mixture was stirred for 30 minutes, followed by evaporation of the solvent under reduced pressure to obtain the crude product, which was then purified using column chromatography (5% MeOH/CH2Cl2) to obtain the compound 10 (271 mg, 35%). 1H NMR (500 MHz, CDCl3) δ 5.31 (dd, J = 31.4, 7.9 Hz, 1H), 4.28 – 4.16 (m, 5H), 3.96 (bs, 1H), 2.04 – 1.88 (m, 2H), 1.88 – 1.76 (m, 1H), 1.73 – 1.62 (m, 1H), 1.62 – 1.43 (m, 2H), 1.42 (s, 9H), 1.32 – 1.24 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 176.13, 176.08, 172.94, 155.59, 80.00, 61.50, 61.08, 54.40, 53.50, 53.44, 34.56, 34.53, 32.65, 32.62, 28.50, 21.73, 21.67, 14.42, 14.37. MS (ESI) calculated for C16H30N2O6, m/z 346.21, found 347.21 (M + H)+.
Synthesis of Compound 11: (5R)-Tribenzyl 1-(9H-fluoren-9-yl)-18,18-dimethyl-3,8,16-trioxo- 2,17-dioxa-4,9,15-triazanonadecane-5,10,14-tricarboxylate
To a solution of 4 (500 mg, 1.08 mmol) in anhydrous dichloromethane, were added 9 (522 mg, 1.11 mmol), polystyrene bound carbodiimide (967 mg, 1.19 mmol) and a catalytic amount of polystyrene bound DMAP. The reaction mixture was stirred at room temperature for 4 hours, followed by filtration to remove the solid resin. The filtrate was evaporated under vacuum to obtain the residue which was then purified using column chromatography (35% EtOAc/hexanes) to obtain the compound 11 (748 mg, 76%). 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H), 7.60 (d, J = 7.3 Hz, 2H), 7.39 – 7.30 (m, 19H), 6.59 – 6.49 (m, 0.50H), 6.28 (s, 0.50H), 5.71 (dd, J = 25.1, 7.7 Hz, 1H), 5.24 – 5.04 (m, 7H), 4.56 (d, J = 6.6 Hz, 1H), 4.50 – 4.33 (m, 3H), 4.33 – 4.15 (m, 2H), 2.31 – 2.16 (m, 2H), 2.03 – 1.90 (m, 2H), 1.85 – 1.75 (m, 2H), 1.70 – 1.51 (m, 2H), 1.40 – 1.37 (m, 11H). 13C NMR (126 MHz, CDCl3) δ 172.65, 172.57, 172.16, 172.09, 171.97, 171.82, 156.53, 155.78, 144.10, 143.85, 141.51, 141.48, 135.55, 135.36, 128.85, 128.83, 128.81, 128.73, 128.64, 128.60, 128.53, 128.45, 127.92, 127.30, 125.36, 120.18, 80.12, 67.59, 67.41, 67.33, 67.24, 53.63, 53.53, 53.33, 52.21, 47.37, 47.34, 32.32, 32.22, 31.73, 28.93, 28.51, 21.54, 21.43, 21.28, 21.13. MS (ESI) calculated for C53H57N3O11, m/z 911.40, found 934.39 (M + Na)+.
Synthesis of Compound 12: (5R)-5-Allyl 10,14-dibenzyl 1-(9H-fluoren-9-yl)-18,18-dimethyl- 3,8,16-trioxo-2,17-dioxa-4,9,15-triazanonadecane-5,10,14-tricarboxylate
To a solution of 5 (500 mg, 1.22 mmol) in anhydrous dichloromethane, were added 9 (585 mg, 1.24 mmol), polystyrene bound carbodiimide (1.09 g, 1.34 mmol) and a catalytic amount of polystyrene bound DMAP. The reaction mixture was stirred at room temperature for 4 hours, followed by filtration to remove the solid resin. The filtrate was evaporated under reduced pressure to obtain the residue which was then purified using column chromatography (35% EtOAc/hexanes) to obtain the compound 12 (830 mg, 79%). 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 7.5 Hz, 2H), 7.63 (d, J = 6.6 Hz, 2H), 7.46 – 7.30 (m, 14H), 6.69 – 6.30 (m, 1H), 5.91 (dd, J = 19.3, 12.8 Hz, 1H), 5.72 (dd, J = 20.3, 7.6 Hz, 1H), 5.35 (d, J = 17.2 Hz, 1H), 5.27 (d, J = 10.4 Hz, 1H), 5.22 – 5.04 (m, 5H), 4.70 – 4.56 (m, 3H), 4.52 – 4.36 (m, 3H), 4.35 – 4.19 (m, 2H), 2.38 – 2.20 (m, 3H), 1.98 (dd, J = 19.2, 8.3 Hz, 1H), 1.91 – 1.56 (m, 4H), 1.44 (s, 11H). 13C NMR (126 MHz, CDCl3) δ 172.66, 172.56, 172.20, 172.12, 171.99, 171.85, 156.56, 155.83, 144.09, 143.86, 141.51, 135.54, 135.45, 131.59, 128.83, 128.71, 128.64, 128.51, 127.94, 127.31, 125.35, 120.19, 119.40, 119.36, 119.32, 80.13, 67.43, 67.33, 67.23, 66.42, 53.61, 53.53, 53.33, 52.24, 47.37, 32.41, 32.34, 31.84, 31.75, 31.61, 29.00, 28.51, 21.46, 21.27, 21.14. MS (ESI) calculated for C49H55N3O11, m/z 861.38, found 884.33 (M + Na)+.
Synthesis of Compound 13: (5R)-5-Benzyl 10,14-diethyl 1-(9H-fluoren-9-yl)-18,18-dimethyl- 3,8,16-trioxo-2,17-dioxa-4,9,15-triazanonadecane-5,10,14-tricarboxylate
To a solution of 4 (195 mg, 0.425 mmol) in anhydrous dichloromethane, were added 10 (150 mg, 0.43 mmol), polystyrene bound carbodiimide (367 mg, 0.467 mmol) and a catalytic amount of polystyrene bound DMAP. The reaction mixture was stirred at room temperature for 4 hours, followed by filtration to remove the solid resin. The filtrate was evaporated under reduced pressure to obtain the residue which was then purified using column chromatography (30% EtOAc/hexanes) to obtain the compound 13 (232 mg, 70%). 1H NMR (500 MHz, CDCl3) δ 7.79 – 7.69 (m, 2H), 7.59 (dd, J = 21.4, 11.5 Hz, 2H), 7.55 – 7.26 (m, 9H), 6.55 (dd, J = 27.8, 7.3 Hz, 1H), 6.31 (dd, J = 18.6, 7.1 Hz, 1H), 5.75 (dd, J = 27.6, 7.7 Hz, 1H), 5.27 – 5.05 (m, 3H), 4.61 – 4.33 (m, 4H), 4.28 – 4.04 (m, 6H), 2.31 – 2.20 (m, 2H), 2.04 – 1.93 (m, 1H), 1.89 – 1.52 (m, 5H), 1.46 – 1.36 (m, 9H), 1.34 – 1.21 (m, 7H). 13C NMR (126 MHz, CDCl3) δ 156.50, 144.11, 143.88, 141.52, 135.38, 131.11, 129.02, 128.87, 128.76, 128.66, 128.59, 127.93, 127.31, 125.37, 120.19, 80.07, 68.37, 67.61, 67.29, 61.75, 61.57, 53.29, 52.13, 47.36, 38.92, 32.48, 32.37, 31.74, 30.56, 29.13, 28.53, 23.94, 23.20, 21.44, 14.37, 14.28, 11.12. MS (ESI) calculated for C43H53N3O11, m/z 787.37, found 810.37 (M + Na)+.
Synthesis of Compound 14: Dibenzyl 2-((R)-4-amino-5-(benzyloxy)-5-oxopentanamido)-6- ((tert-butoxycarbonyl)amino)heptanedioate
The compound 11 (500 mg, 0.55 mmol) was dissolved in 10 mL of dichloromethane, followed by the addition of 3 mL of piperidine. The reaction mixture was stirred for 15 minutes, followed by removal of the solvent under reduced pressure. The crude was purified using column chromatography (5% MeOH/CH2Cl2) to obtain the compound 14 (347 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.31 (m, 15H), 6.57 (dt, J = 36.1, 8.8 Hz, 1H), 5.25 – 5.05 (m, 7H), 4.59 (d, J = 5.6 Hz, 1H), 4.28 (s, 1H), 3.52 (dd, J = 8.2, 5.1 Hz, 1H), 2.36 (qt, J = 21.1, 6.8 Hz, 2H), 2.13 (dd, J = 12.5, 6.0 Hz, 1H), 1.93 – 1.50 (m, 7H), 1.49 – 1.32 (m, 11H). 13C NMR (101 MHz, CDCl3) δ 176.03, 172.79, 172.67, 136.10, 135.85, 129.15, 129.12, 129.01, 128.94, 128.85, 128.76, 67.53, 67.32, 54.38, 54.01, 53.70, 52.40, 33.12, 33.00, 32.17, 30.48, 30.31, 28.82, 21.78, 21.59. MS (ESI) calculated for C38H47N3O9, m/z 689.33, found 690.33 (M + H)+.
Synthesis of Compound 15: (15R)-Tribenzyl 2,2-dimethyl-4,12,17-trioxo-3-oxa-5,11,16- triazaoctacosane-6,10,15-tricarboxylate
To a solution of 14 (200 mg, 0.29 mmol) in pyridine, were added lauroyl chloride (82.6 μL, 0.34 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 6 hours, followed by evaporation of the solvent under reduced pressure to obtain the residue, which was then dissolved in dichloromethane and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (50% EtOAc/hexanes) to obtain the compound 15 (209 mg, 83%). 1H NMR (500 MHz, CDCl3) δ 7.42 – 7.27 (m, 15H), 6.98 (dd, J = 16.0, 7.8 Hz, 0.50H), 6.57 – 6.46 (m, 1.50H), 5.22 – 5.07 (m, 7H), 4.76 – 4.70 (m, 0.50H), 4.61 – 4.51 (m, 1.50H), 4.31 – 4.20 (m, 1H), 2.28 – 2.16 (m, 5H), 1.98 – 1.57 (m, 11H), 1.50 – 1.36 (m, 6H), 1.32 – 1.20 (m, 17H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 172.68, 172.61, 172.46, 172.38, 172.33, 172.24, 172.20, 172.12, 172.04, 135.57, 135.45, 135.40, 128.85, 128.84, 128.82, 128.71, 128.65, 128.62, 128.58, 128.53, 128.48, 128.47, 80.10, 67.53, 67.23, 53.39, 52.29, 51.86, 36.83, 36.77, 32.53, 32.40, 32.12, 31.58, 29.84, 29.82, 29.70, 29.56, 29.51, 29.18, 28.54, 25.86, 25.79, 22.90, 14.34. MS (ESI) calculated for C50H69N3O10, m/z 871.50, found 894.50 (M + Na)+.
Synthesis of Compound 16: 2-Amino-6-((R)-4-carboxy-4-dodecanamidobutanamido) heptanedioic acid
G3The compound 15 (50 mg, 57.3 μmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 16 (35 mg, quantitative yield). 1H NMR (500 MHz, MeOD) δ 4.46 – 4.30 (m, 2H), 3.78 – 3.68 (m, 1H), 2.35 (t, J = 5.6 Hz, 2H), 2.27 – 2.10 (m, 3H), 1.88 (ddd, J = 24.9, 20.5, 16.8 Hz, 4H), 1.75 – 1.66 (m, 1H), 1.65 – 1.47 (m, 4H), 1.36 – 1.21 (m, 16H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 176.64, 175.50, 175.02, 173.29, 55.13, 54.97, 53.42, 53.16, 37.09, 36.99, 33.25, 33.04, 32.52, 32.45, 32.38, 31.64, 31.53, 31.47, 30.92, 30.82, 30.65, 30.50, 28.82, 28.58, 28.30, 27.14, 27.11, 23.89, 23.06, 22.71, 22.63, 14.59. MS (ESI) calculated for C24H43N3O8, m/z 501.31, found 502.33 (M + H)+.
Synthesis of Compound 17: (5R)-Tribenzyl 1-(9H-fluoren-9-yl)-3,8,16-trioxo-2-oxa-4,9,15- triazaheptacosane-5,10,14-tricarboxylate
The compound 11 (100 mg, 0.10 mmol) was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt. To the solution of trifluoroacetate salt in anhydrous dichloromethane, were added lauroyl chloride (39 μL, 0.16 mmol), triethylamine (30 μL, 0.22 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 2 hours, followed by complete removal of the solvent under reduced pressure. The residue was then dissolved in dichloromethane and washed with water. The organic solvent was dried over sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (40% EtOAc/hexanes) to obtain the compound 17 (103 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 7.83 – 7.70 (m, 2H), 7.68 – 7.52 (m, 2H), 7.46 – 7.27 (m, 19H), 6.27 – 6.06 (m, 1H), 5.93 – 5.74 (m, 1H), 5.30 – 5.04 (m, 6H), 4.67 – 4.28 (m, 5H), 4.28 – 4.18 (m, 1H), 2.33 – 2.26 (m, 2H), 2.22 – 2.13 (m, 2H), 2.01 (dd, J = 15.1, 7.4 Hz, 1H), 1.92 – 1.79 (m, 2H), 1.79 – 1.66 (m, 3H), 1.64 – 1.56 (m, 2H), 1.48 – 1.17 (m, 19H), 0.90 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.48, 172.36, 172.06, 172.02, 156.61, 143.74, 141.50, 135.39, 128.84, 128.70, 128.47, 127.92, 127.30, 125.35, 120.17, 67.55, 67.41, 67.31, 53.83, 51.87, 51.64, 47.35, 36.69, 32.19, 32.11, 31.62, 29.83, 29.69, 29.54, 29.46, 25.81, 22.88, 21.67, 14.32. MS (ESI) calculated for C60H71N3O10, m/z 993.51, found 1016.53 (M + Na)+.
Synthesis of Compound 18: 2-((R)-4-Amino-4-carboxybutanamido)-6-dodecanamido) heptanedioic acid
The compound 17 (70 mg, 0.07 mmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the crude product which was purified using column chromatography (50% MeOH/CH2Cl2) to obtain the compound 18 (32 mg, 92%). 1H NMR (500 MHz, MeOD) δ 4.34 (s, 2H), 3.78 – 3.69 (m, 2H), 2.50 (t, J = 17.3 Hz, 2H), 2.23 (t, J = 7.4 Hz, 2H), 2.17 – 2.06 (m, 2H), 1.86 – 1.66 (m, 5H), 1.64 – 1.55 (m, 2H), 1.50 – 1.41 (d, J = 15.2 Hz, 2H), 1.39 – 1.19 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 176.56, 53.81, 36.98, 33.24, 32.21, 30.92, 30.82, 30.64, 30.47, 27.13, 25.50, 23.90, 23.51, 14.59. MS (ESI) calculated for C24H43N3O8, m/z 501.31, found 502.32 (M + H)+.
Synthesis of Compound 19: Dibenzyl 2-amino-6-((R)-5-(benzyloxy)-4-dodecanamido-5- oxopentanamido)heptanedioate
The compound 15 (200 mg, 0.23 mmol) was dissolved in 10 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of compound 19 (203 mg, quantitative yield). MS (ESI) calculated for C45H61N3O8, m/z 771.44, found 772.48 (M + H)+.
Synthesis of Compound 20: 2-((R)-4-Carboxy-4-dodecanamidobutanamido)-6-dodecanamidoheptanedioic acid
To a solution of 19 (50 mg, 56.4 μmol) in pyridine, were added lauroyl chloride (16.1 μL, 67.7 μmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 6 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in dichloromethane and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product. The crude was purified using column chromatography (35% EtOAc/hexanes) to obtain the pure product, which was dissolved in methanol and subjected to catalytic hydrogenolysis using Pd(OH)2/C at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the compound 20 (34.3 mg, 89%). 1H NMR (500 MHz, MeOD) δ 4.45 – 4.25 (m, 3H), 2.34 (t, J = 6.7 Hz, 2H), 2.28 – 2.07 (m, 5H), 1.98 – 1.79 (m, 3H), 1.70 (dd, J = 19.9, 14.8 Hz, 2H), 1.64 – 1.55 (m, 4H), 1.49 – 1.39 (m, 3H), 1.37 – 1.15 (m, 31H), 0.87 (t, J = 6.8 Hz, 6H). 13C NMR (126 MHz, MeOD) δ 176.64, 175.98, 175.72, 175.21, 53.63, 36.99, 33.35, 33.25, 32.27, 30.92, 30.83, 30.65, 30.49, 28.88, 28.73, 27.10, 23.89, 23.54, 14.59. MS (ESI) calculated for C36H65N3O9, m/z 683.47, found 682.49 (M – H)−.
Synthesis of Compound 21: (15R)-Tribenzyl 2,2-dimethyl-4,12-dioxo-3-oxa-5,11,16- triazaoctacosane-6,10,15-tricarboxylate
To a solution of 14 (190 mg, 0.28 mmol) in anhydrous dichloromethane (10 mL) and methanol (2 mL), were added dodecyl aldehyde (51.5 mg, 0.28 mmol), 3 – 4 drops of acetic acid, and macroporous resin bound sodium cyanoborohydride (150 mg, 0.33 mmol). The solution was stirred for 8 hours, followed by filtration to remove the resin. The solvent was removed under vacuum to obtain the residue, which was purified using column chromatography (15% EtOAc/hexanes) to obtain the compound 21 (124 mg, 51%). 1H NMR (500 MHz, CDCl3) δ 7.43 – 7.27 (m, 15H), 5.25 – 5.09 (m, 6H), 4.50 (dd, J = 12.7, 7.7 Hz, 1H), 4.35 – 4.16 (m, 1H), 2.73 – 2.64 (m, 1H), 2.62 – 2.13 (m, 4H), 2.02 (t, J = 20.0 Hz, 1H), 1.91 – 1.72 (m, 3H), 1.71 – 1.50 (m, 4H), 1.41 (d, J = 12.6 Hz, 7H), 1.38 – 1.17 (m, 25H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 172.53, 172.11, 155.94, 155.65, 155.52, 135.36, 134.56, 130.91, 128.62, 128.43, 128.29, 79.94, 68.16, 67.97, 67.06, 60.40, 59.91, 53.22, 52.10, 47.94, 32.38, 32.20, 32.04, 31.95, 31.43, 30.99, 30.37, 29.64, 29.58, 29.36, 29.09, 28.92, 28.66, 28.34, 28.12, 27.43, 27.00, 23.76, 23.00, 22.72, 21.45, 21.24, 14.16. MS (ESI) calculated for C50H71N3O9, m/z 857.52, found 858.53 (M + H)+.
Synthesis of Compound 22: 2-Amino-6-((R)-4-carboxy-4-(dodecylamino)butanamido) heptanedioic acid
The compound 21 (50 mg, 58.3 μmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 22 (35 mg, quantitative yield in 2 steps). 1H NMR (500 MHz, MeOD) δ 4.40 (d, J = 3.5 Hz, 1H), 3.88 – 3.56 (m, 3H), 3.21 – 3.07 (m, 1H), 3.06 – 2.94 (m, 2H), 2.53 (t, J = 6.7 Hz, 2H), 2.14 (d, J = 6.0 Hz, 2H), 1.91 (s, 3H), 1.79 – 1.43 (m, 6H), 1.43 – 1.20 (m, 15H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 175.33, 62.44, 62.15, 54.58, 53.37, 46.97, 46.32, 45.72, 45.40, 44.90, 33.10, 32.83, 32.34, 31.97, 31.13, 30.78, 30.68, 30.52, 30.24, 27.61, 27.46, 26.59, 26.28, 23.75, 23.09, 22.55, 22.34, 14.44. MS (ESI) calculated for C24H45N3O7, m/z 487.33, found 488.34 (M + H)+.
Synthesis of Compound 23: (15R)-Tribenzyl 2,2-dimethyl-16-octyl-4,12-dioxo-3-oxa-5,11,16- triazatetracosane-6,10,15-tricarboxylate
To a solution of 14 (100 mg, 0.15 mmol) in anhydrous dichloromethane (10 mL) and methanol (5 mL), were added octyl aldehyde (56.5 μL, 0.36 mmol), 3 – 4 drops of acetic acid, and macroporous resin bound sodium cyanoborohydride (163.5 mg, 0.36 mmol). The reaction mixture was then stirred for 10 hours, followed by filtration to remove the resin. The solvent was removed under vacuum to obtain the compound 23 as crude. MS (ESI) calculated for C54H79N3O9, m/z 913.58, found 914.60 (M + H)+.
Synthesis of Compound 24: (15R)-Tribenzyl 16-dodecyl-2,2-dimethyl-4,12-dioxo-3-oxa-5,11,16- triazaoctacosane-6,10,15-tricarboxylate
To the solution of 14 (100 mg, 0.15 mmol) in anhydrous dichloromethane (10 mL) and methanol (5 mL), were added dodecyl aldehyde (66.8 mg, 0.36 mmol), 3 – 4 drops of acetic acid, and macroporous resin bound sodium cyanoborohydride (163.5 mg, 0.36 mmol). The solution was then stirred for 14 hours, followed by filtration to remove the resin. The solvent was removed under vacuum to obtain the residue, which was purified using column chromatography (15% EtOAc/hexanes) to obtain the compound 24 (105 mg, 71%). 1H NMR (500 MHz, CDCl3) δ 7.45 – 7.28 (m, 15H), 5.28 (dd, J = 12.0, 3.7 Hz, 1H), 5.23 – 5.01 (m, 6H), 4.44 (bs, 1H), 4.34 – 4.11 (m, 2H), 3.22 – 2.85 (m, 4H), 2.58 – 2.24 (m, 4H), 1.85 – 1.74 (m, 2H), 1.72 – 1.53 (m, 6H), 1.48 – 1.34 (m, 11H), 1.34 – 1.12 (m, 37H), 0.97 – 0.82 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 172.69, 172.23, 171.94, 171.40, 162.45, 135.64, 135.61, 134.64, 131.10, 129.21, 129.16, 129.03, 129.01, 128.92, 128.89, 128.82, 128.78, 128.62, 128.45, 128.38, 128.35, 80.10, 68.36, 68.11, 67.20, 63.20, 62.48, 53.51, 53.40, 52.72, 52.55, 38.92, 32.71, 32.12, 30.55, 29.82, 29.69, 29.63, 29.61, 29.54, 29.22, 29.20, 29.13, 28.51, 27.11, 24.94, 23.93, 23.20, 22.90, 21.88, 21.71, 14.34. MS (ESI) calculated for C62H95N3O9, m/z 1025.71, found 1026.73 (M + H)+.
Synthesis of Compound 25: (15R)-Tribenzyl 16-hexadecyl-2,2-dimethyl-4,12-dioxo-3-oxa- 5,11,16-triazadotriacontane-6,10,15-tricarboxylate
To a solution of 14 (100 mg, 0.15 mmol) in anhydrous dichloromethane (10 mL) and methanol (5 mL), were added hexadecanal (86.4 mg, 0.36 mmol), 3 – 4 drops of acetic acid, and macroporous resin bound sodium cyanoborohydride (163.5 mg, 0.36 mmol). The solution was then stirred for 14 hours, followed by filtration to remove the resin. The solvent was removed under vacuum to obtain the residue, which was purified using column chromatography (12% EtOAc/hexanes) to obtain the compound 25 (112 mg, 68%). 1H NMR (500 MHz, CDCl3) δ 7.45 – 7.28 (m, 15H), 5.31 – 4.96 (m, 7H), 4.48 (d, J = 5.2 Hz, 1H), 4.26 – 4.18 (m, 1H), 3.24 – 2.06 (m, 9H), 1.77 – 1.55 (m, 8H), 1.46 – 1.33 (m, 11H), 1.33 – 1.07 (m, 53H), 0.88 (t, J = 6.8 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 172.62, 172.24, 172.11, 171.63, 155.80, 155.69, 135.57, 131.10, 128.93, 128.83, 128.64, 128.57, 128.47, 128.41, 80.10, 68.36, 67.24, 67.18, 63.06, 62.51, 53.47, 53.37, 52.40, 52.14, 38.92, 32.82, 32.32, 32.14, 30.55, 29.92, 29.87, 29.86, 29.76, 29.74, 29.70, 29.58, 29.42, 29.36, 29.12, 28.51, 27.25, 25.07, 23.93, 23.19, 22.90, 21.78, 21.65, 14.34, 14.27. MS (ESI) calculated for C70H111N3O9, m/z 1137.83, found 1138.85 (M + H)+.
Synthesis of Compound 26: 2-Amino-6-((R)-4-carboxy-4-(dioctylamino)butanamido) heptanedioic acid
The crude of compound 23 was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the residue which was further purified using column chromatography to obtain the trifluoroacetate salt of compound 26 (60 mg, 64%). 1H NMR (500 MHz, 10% CDCl3 in MeOD) δ 4.34 (dd, J = 9.2, 4.6 Hz, 1H), 3.91 – 3.79 (m, 2H), 3.21 – 3.00 (m, 4H), 2.51 (ddd, J = 29.4, 15.8, 6.3 Hz, 2H), 2.20 – 2.09 (m, 1H), 2.08 – 1.95 (m, 1H), 1.95 – 1.78 (m, 3H), 1.78 – 1.39 (m, 8H), 1.34 – 1.16 (m, 19H), 0.82 (t, J = 6.8 Hz, 6H). 13C NMR (126 MHz, 10% CDCl3 in MeOD) δ 175.14, 174.73, 174.41, 171.93, 53.92, 53.35, 33.09, 32.93, 32.48, 31.97, 31.09, 30.27, 30.21, 30.19, 27.71, 25.66, 23.75, 23.42, 22.53, 14.55. MS (ESI) calculated for C28H53N3O7, m/z 543.39, found 544.41 (M + H)+.
Synthesis of Compound 27: 2-Amino-6-((R)-4-carboxy-4-(didodecylamino)butanamido) heptanedioic acid
The compound 24 (50 mg, 0.048 mmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 27 (37 mg, quantitative yield). 1H NMR (500 MHz, MeOD) δ 4.43 – 4.37 (m, 1H), 3.87 (dd, J = 29.3, 7.5 Hz, 2H), 3.20 (dd, J = 19.5, 10.2 Hz, 2H), 3.15 – 3.05 (m, 2H), 2.73 – 2.46 (m, 3H), 2.27 – 1.99 (m, 3H), 1.98 – 1.83 (m, 3H), 1.72 (s, 5H), 1.63 – 1.43 (m, 3H), 1.31 (d, J = 38.3 Hz, 33H), 0.88 (t, J = 6.8 Hz, 6H). 13C NMR (126 MHz, MeOD) δ 33.24, 30.92, 30.80, 30.69, 30.64, 30.34, 27.78, 23.90, 14.60. MS (ESI) calculated for C36H69N3O7, m/z 655.51, found 656.51 (M + H)+.
Synthesis of Compound 28: 2-Amino-6-((R)-4-carboxy-4-(dihexadecylamino)butanamido) heptanedioic acid
The compound 25 (50 mg, 0.043 mmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 28 (38 mg, quantitative yield). 1H NMR (500 MHz, 20% CDCl3 in MeOD) δ 4.48 – 4.20 (m, 2H), 3.86 – 3.73 (m, 3H), 3.29 – 3.04 (m, 4H), 2.72 – 2.44 (m, 3H), 2.31 – 1.80 (m, 7H), 1.80 – 1.48 (m, 8H), 1.46 – 1.12 (m, 46H), 0.89 (t, J = 6.9 Hz, 6H). 13C NMR (126 MHz, 20% CDCl3 in MeOD) δ 31.40, 29.14, 29.11, 29.09, 29.04, 28.97, 28.94, 28.88, 28.82, 28.53, 26.03, 22.10, 13.07. MS (ESI) calculated for C44H85N3O7, m/z 767.64, found 768.65 (M + H)+.
Synthesis of Compound 30: (2R)-5-((4-(2-Amino-4-oxo-4,5-dihydro-1H-imidazol-5-yl)-1- carboxybutyl)amino)-2-dodecanamido-5-oxopentanoic acid
To a solution of 19 (100 mg, 0.13 mmol) in pyridine, was added 1H-pyrazole-1-carboxamidine.HCl (55.2 mg, 0.37 mmol). The reaction mixture was heated in microwave at 65 °C for 1 hour. The solvent was removed under vacuum and the residue was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the crude product which was purified using column chromatography (30% MeOH/CH2Cl2) to obtain the compound 30 (38 mg, 58%). 1H NMR (500 MHz, MeOD) δ 4.40 – 4.27 (m, 2H), 4.21 – 4.18 (m, 1H), 2.34 (t, J = 5.9 Hz, 2H), 2.24 (dd, J = 13.3, 6.9 Hz, 2H), 2.20 – 1.95 (m, 2H), 1.95 – 1.65 (m, 5H), 1.64 – 1.54 (m, 2H), 1.54 – 1.43 (m, 2H), 1.37 – 1.21 (m, 15H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 176.67, 176.56, 176.20, 175.20, 61.16, 54.19, 54.01, 53.67, 37.11, 37.03, 33.35, 33.25, 32.54, 32.46, 31.92, 31.80, 30.92, 30.81, 30.65, 30.51, 28.87, 27.09, 23.90, 22.56, 22.33, 22.19, 14.60. MS (ESI) calculated for C25H43N5O7, m/z 525.32, found 524.32 (M – H)−.
Synthesis of Compound 32: 2-((R)-4-Carboxy-4-dodecanamidobutanamido)-6-guanidinoheptanedioic acid
To a solution of 19 (50 mg, 0.06 mmol) in THF, was added N,N′-di-Boc-1H-pyrazole-1- carboxamidine.HCl (57.4 mg, 0.19 mmol) and pyridine (1 mL). The reaction mixture was then stirred at 50 °C for 3 hours. The solvent was removed under reduced pressure and the residue was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 1 hour, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the crude, which was purified using column chromatography (8% MeOH/CH2Cl2). The product was dissolved in 5 mL of trifluoroacetic acid and the reaction mixture was stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 32 (30 mg, 63%). 1H NMR (500 MHz, MeOD) δ 4.34 (s, 2H), 4.00 (s, 1H), 2.38 – 2.28 (m, 2H), 2.24 (dd, J = 14.4, 7.0 Hz, 2H), 2.18 – 2.08 (m, 1H), 2.07 – 1.80 (m, 4H), 1.79 – 1.65 (m, 2H), 1.61 – 1.55 (m, 2H), 1.55 – 1.36 (m, 3H), 1.36 – 1.19 (m, 14H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, 5% CDCl3 in MeOD) δ 176.46, 175.71, 175.05, 158.34, 57.03, 54.25, 37.11, 33.35, 33.20, 33.11, 32.83, 32.64, 30.88, 30.78, 30.62, 30.51, 29.28, 27.06, 23.86, 22.90, 22.58, 14.59. MS (ESI) calculated for C25H45N5O8, m/z 543.33, found 544.35 (M + H)+.
Synthesis of Compound 33: (6R)-6-Allyl 11,15-dibenzyl 2,2,19,19-tetramethyl-4,9,17-trioxo- 3,18-dioxa-5,10,16-triazaicosane-6,11,15-tricarboxylate
The compound 12 (250 mg, 0.29 mmol) was dissolved in 10 mL of dichloromethane, followed by the addition of 3 mL of piperidine. The reaction mixture was stirred for 15 minutes, followed by removal of the solvent under reduced pressure. The residue was dissolved in dichloromethane, followed by the addition of di-tert-butyl dicarbonate (95 mg, 0.43 mmol) and triethylamine (80 μL, 0.58 mmol). The reaction mixture was stirred at room temperature for 1 hour, followed by removal of the solvent under reduced pressure. The crude was purified using column chromatography (30% EtOAc/Hexanes) to obtain the compound 33 (147 mg, 69%). 1H NMR (500 MHz, CDCl3) δ 7.41 – 7.29 (m, 10H), 5.97 – 5.82 (m, 1H), 5.30 (ddd, J = 37.2, 23.6, 5.8 Hz, 3H), 5.21 – 5.06 (m, 5H), 4.68 – 4.51 (m, 3H), 4.38 – 4.20 (m, 2H), 2.31 (dd, J = 14.5, 7.3 Hz, 2H), 2.25 – 2.13 (m, 1H), 1.99 – 1.54 (m, 6H), 1.43 (s, J = 2.9 Hz, 20H). 13C NMR (126 MHz, CDCl3) δ 172.67, 172.58, 172.32, 172.18, 171.85, 156.06, 155.83, 135.55, 135.53, 135.47, 132.32, 132.24, 131.68, 128.83, 128.81, 128.69, 128.64, 128.61, 128.51, 128.46, 119.23, 119.13, 80.34, 80.17, 80.08, 77.48, 77.23, 76.98, 67.37, 67.27, 67.22, 67.18, 66.26, 53.34, 53.16, 53.04, 52.25, 52.18, 32.47, 32.24, 31.85, 31.76, 31.64, 29.88, 29.26, 29.01, 28.86, 28.50. MS (ESI) calculated for C39H53N3O11, m/z 739.37, found 762.37 (M + Na)+.
Synthesis of Compound 34: (6R)-11,15-Dibenzyl 6-undecyl 2,2,19,19-tetramethyl-4,9,17-trioxo- 3,18-dioxa-5,10,16-triazaicosane-6,11,15-tricarboxylate
To a solution of compound 33 (80 mg, 0.10 mmol) in ethanol:water (9:1), was added Wilkinson’s catalyst (10 mg, 0.01 mmol). The reaction mixture was refluxed for 3 hours at 90 °C, followed by the removal of the solvent under reduced pressure. The crude was purified using column chromatography (5% MeOH/CH2Cl2) and dried under vacuum. To the solution of afforded product (52 mg, 0.07 mmol) in anhydrous DMF, were added 1-undecanol (30 μL, 0.15 mmol), HBTU (33.7 mg, 0.09 mmol), triethylamine (20 μl, 0.15 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred at room temperature for 14 hours, followed by removal of the solvent under reduced pressure. The residue was dissolved in ethyl acetate and washed with water. The organic fraction was dried over anhydrous sodium sulfate, filtered, concentrated and purified using column chromatography (35% EtOAc/hexanes) to obtain the compound 34 (44.4 mg, 70%). 1H NMR (500 MHz, CDCl3) δ 7.40 – 7.30 (m, 10H), 5.27 (dd, J = 22.5, 8.4 Hz, 1H), 5.20 – 4.96 (m, 5H), 4.65 – 4.49 (m, 1H), 4.41 – 4.20 (m, 2H), 4.16 – 4.07 (m, 2H), 2.30 (t, J = 6.8 Hz, 2H), 2.24 – 2.13 (m, 1H), 1.93 – 1.72 (m, 3H), 1.67- 1.59 (m, 3H), 1.43 (d, J = 2.7 Hz, 20H), 1.35 – 1.18 (m, 18H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.93, 170.59, 170.19, 154.13, 133.94, 127.20, 127.03, 126.85, 115.67, 78.69, 78.48, 65.63, 64.37, 51.77, 51.70, 51.47, 51.42, 50.64, 50.59, 30.87, 30.48, 30.17, 28.18, 28.07, 27.91, 27.82, 27.09, 26.90, 24.36, 21.27, 12.95. MS (ESI) calculated for C47H71N3O11, m/z 853.51, found 876.47 (M + Na)+.
Synthesis of Compound 35: 2-Amino-6-((R)-4-amino-5-oxo-5-(undecyloxy)pentanamido) heptanedioic acid
The compound 34 (40 mg, 46.9 μmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 35 (32.8 mg, quantitative yield). 1H NMR (500 MHz, CDCl3) δ 4.40 (dd, J = 9.2, 4.8 Hz, 1H), 4.32 – 4.22 (m, 2H), 4.11 (dd, J = 11.8, 6.0 Hz, 1H), 3.82 – 3.74 (m, 1H), 2.53 (t, J = 6.9 Hz, 2H), 2.23 (dt, J = 13.3, 6.7 Hz, 1H), 2.18 – 2.09 (m, 1H), 1.90 (ddd, J = 21.0, 13.9, 5.3 Hz, 3H), 1.80 – 1.67 (m, 3H), 1.64 – 1.46 (m, 2H), 1.43 – 1.22 (m, 16H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 176.90, 172.93, 70.30, 57.13, 56.15, 56.04, 55.92, 55.86, 35.59, 34.66, 34.59, 33.84, 33.76, 33.26, 33.17, 32.99, 32.88, 32.08, 29.78, 29.71, 29.39, 26.26, 25.15, 24.95, 16.95. MS (ESI) calculated for C23H43N3O7, m/z 473.31, found 474.31 (M + H)+.
Synthesis of Compound 36: Dibenzyl 2-((tert-butoxycarbonyl)amino)-6-((R)-4-((tert-butoxycarbonyl) amino)-5-oxo-5-(undecylamino)pentanamido)heptanedioate
To a solution of 33 (240 mg, 0.32 mmol) in ethanol:water (9:1), was added Wilkinson’s catalyst (30 mg, 0.03 mmol). The reaction mixture was refluxed for 3 hours at 90 °C, followed by removal of the solvent under reduced pressure. The crude was purified using column chromatography (5% MeOH/CH2Cl2). To the solution of afforded product (193 mg, 0.27 mmol) in anhydrous dichloromethane, were added undecylamine (118 μL, 0.55 mmol), HBTU (157 mg, 0.41 mmol), triethylamine (77 μl, 0.55 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred at room temperature for 14 hours, followed by removal of the solvent under reduced pressure. The residue was dissolved in ethyl acetate and washed with water. The organic fraction was dried over anhydrous sodium sulfate, filtered, concentrated and purified using column chromatography (25% EtOAc/hexanes) to obtain the compound 36 (188 mg, 81%). 1H NMR (500 MHz, CDCl3) δ 7.44 – 7.27 (m, 10H), 6.65 – 6.49 (m, 1H), 5.63 (d, J = 8.1 Hz, 1H), 5.62 – 5.52 (m, 1H), 5.23 – 4.92 (m, 5H), 4.60 – 4.45 (m, 2H), 4.27 (d, J = 8.2 Hz, 1H), 4.09 (d, J = 6.7 Hz, 1H), 3.31 – 3.14 (m, 2H), 2.38 – 2.22 (m, 1H), 2.00 – 1.53 (m, 5H), 1.49 – 1.32 (m, 23H), 1.31 – 1.21 (m, 16H), 0.90 – 0.84 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 174.07, 173.52, 172.99, 172.68, 171.76, 171.40, 157.18, 156.56, 135.51, 135.42, 128.88, 128.81, 128.74, 128.66, 128.59, 128.50, 128.38, 80.44, 80.17, 67.43, 67.28, 67.19, 53.47, 53.21, 52.65, 52.28, 39.89, 39.73, 32.98, 32.75, 32.47, 32.11, 31.96, 31.51, 30.67, 29.82, 29.76, 29.54, 28.53, 27.12, 22.89, 22.03, 21.87, 21.48, 21.35, 14.34. MS (ESI) calculated for C47H72N4O10, m/z 852.52, found 875.53 (M + Na)+.
Synthesis of Compound 37: 2-Amino-6-((R)-4-amino-5-oxo-5-(undecylamino)pentanamido) heptanedioic acid
The compound 36 (40 mg, 46.9 μmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 37 (32.8 mg, quantitative yield). 1H NMR (500 MHz, MeOD) δ 4.47 – 4.32 (m, 1H), 3.92 – 3.79 (m, 1H), 3.64 – 3.54 (m, 1H), 3.28 – 3.13 (m, 2H), 2.56 – 2.34 (m, 2H), 2.19 – 2.01 (m, 2H), 2.00 – 1.60 (m, 4H), 1.58 – 1.41 (m, 4H), 1.30 (d, J = 21.4 Hz, 16H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 175.94, 175.78, 174.26, 174.03, 169.72, 55.49, 53.98, 53.77, 53.47, 40.75, 33.08, 32.29, 31.95, 31.82, 31.51, 31.31, 30.75, 30.46, 30.32, 28.74, 28.49, 28.04, 23.74, 22.62, 22.07, 14.44. MS (ESI) calculated for C23H44N4O6, m/z 472.33, found 473.33 (M + H)+.
Synthesis of Compound 38: (5R)-10,14-Dibenzyl 5-undecyl 1-(9H-fluoren-9-yl)-18,18-dimethyl- 3,8,16-trioxo-2,17-dioxa-4,9,15-triazanonadecane-5,10,14-tricarboxylate
To a solution of 12 (100 mg, 0.11 mmol) in ethanol:water (9:1), was added Wilkinson’s catalyst (10 mg, 0.01 mmol). The reaction mixture was refluxed for 3 hours at 90 °C, followed by removal of the solvent under reduced pressure. The crude was purified using column chromatography (5% MeOH/CH2Cl2) and dried under vacuum. To the solution of afforded product (70 mg, 85.2 μmol) in anhydrous DMF, were added 1-undecanol (35 μL, 170 μmol), HBTU (38.8 mg, 102.2 μmol) and a catalytic amount of DMAP. The reaction mixture was stirred at room temperature for 14 hours, followed by removal of the solvent under reduced pressure. The crude was dissolved in ethyl acetate and washed with water. The organic fraction was dried over anhydrous sodium sulfate, filtered, concentrated and purified using column chromatography (15% EtOAc/hexanes) to obtain the compound 38 (63 mg, 75%). 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H), 7.61 (d, J = 6.4 Hz, 2H), 7.50 – 7.26 (m, 14H), 6.64 (s, 0.50H), 6.37 (s, 0.50H), 5.67 (dd, J = 32.4, 7.9 Hz, 1H), 5.21 – 4.99 (m, 5H), 4.62 – 4.53 (m, 1H), 4.47 – 4.31 (m, 3H), 4.30 – 4.19 (m, 2H), 4.13 (td, J = 6.5, 2.6 Hz, 2H), 2.34 – 2.16 (m, 3H), 1.99 – 1.89 (m, 1H), 1.87 – 1.74 (m, 2H), 1.72 – 1.56 (m, 6H), 1.39 (d, J = 20.0 Hz, 10H), 1.33 – 1.20 (m, 15H), 0.90 (dt, J = 13.9, 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 172.77, 172.70, 172.64, 144.54, 144.29, 141.96, 135.98, 129.25, 129.14, 129.06, 128.95, 128.88, 128.36, 127.73, 125.79, 120.62, 80.57, 67.85, 67.75, 67.67, 66.58, 53.76, 52.66, 47.80, 32.91, 32.54, 32.19, 30.23, 30.13, 29.96, 29.85, 29.59, 29.13, 28.94, 26.41, 23.32, 21.58, 14.77. MS (ESI) calculated for C57H73N3O11, m/z 975.52, found 998.46 (M + Na)+.
Synthesis of Compound 39: (5R)-10,14-Dibenzyl 5-ethyl 1-(9H-fluoren-9-yl)-18,18-dimethyl- 3,8,16-trioxo-2,17-dioxa-4,9,15-triazanonadecane-5,10,14-tricarboxylate
To a solution of 12 (100 mg, 0.11 mmol) in ethanol:water (9:1), was added Wilkinson’s catalyst (10 mg, 0.01 mmol). The reaction mixture was refluxed for 3 hours at 90 °C, followed by removal of the solvent under reduced pressure. The crude was purified using column chromatography (5% MeOH/CH2Cl2) and dried under vacuum. To the solution of afforded product (75 mg, 91.3 μmol) in anhydrous DMF, were added ethyl alcohol (11 μL, 182 μmol), HBTU (41.6 mg, 109.5 μmol) and a catalytic amount of DMAP. The reaction mixture was stirred at room temperature for 14 hours, followed by removal of the solvent under reduced pressure. The residue was dissolved in ethyl acetate and washed with water. The organic fraction was dried over anhydrous sodium sulfate, filtered, concentrated and purified using column chromatography (15% EtOAc/hexanes) to obtain the compound 39 (58.9 mg, 76%). 1H NMR (500 MHz, CDCl3) δ 7.79 – 7.69 (m, 2H), 7.57 – 7.52 (m, 2H), 7.57 – 7.26 (m, 14H), 6.61 (d, J = 6.9 Hz, 0.50H), 6.37 (t, J = 6.9 Hz, 0.50H), 5.65 (dd, J = 29.3, 7.3 Hz, 1H), 5.23 – 5.00 (m, 5H), 4.64 – 4.54 (m, 1H), 4.45 – 4.37 (m, 2H), 4.29 – 4.15 (m, 4H), 2.36 – 2.17 (m, 3H), 1.98 – 1.88 (m, 1H), 1.87 – 1.74 (m, 2H), 1.73 – 1.62 (m, 2H), 1.48 – 1.33 (m, 10H), 1.33 – 1.23 (m, 5H). 13C NMR (126 MHz, CDCl3) δ 172.67, 172.18, 171.99, 156.58, 155.85, 144.11, 143.88, 141.52, 135.55, 135.46, 131.10, 129.00, 128.83, 128.71, 128.64, 128.53, 127.95, 127.31, 125.36, 120.20, 80.12, 68.37, 67.43, 67.24, 61.98, 53.55, 53.35, 52.22, 47.39, 38.92, 32.45, 31.79, 30.56, 29.12, 28.82, 28.52, 23.94, 23.20, 21.47, 21.29, 21.14, 14.36, 14.29, 11.17. MS (ESI) calculated for C48H55N3O11, m/z 849.38, found 872.37 (M + Na)+.
Synthesis of Compound 40: (15R)-10,6-Dibenzyl 15-undecyl 2,2-dimethyl-4,12,17-trioxo-3-oxa- 5,11,16-triazaoctacosane-6,10,15-tricarboxylate
The compound 38 (50 mg, 0.05 mmol) was dissolved in 10 mL of dichloromethane, followed by the addition of 3 mL of piperidine. The reaction mixture was stirred for 15 minutes, followed by removal of the solvent under reduced pressure. The residue (0.05 mmol) was dissolved in pyridine, followed by the addition of lauroyl chloride (14.6 μL, 0.06 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 4 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in dichloromethane and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (18% EtOAc/hexanes) to obtain the compound 40 (36.4 mg, 76%). 1H NMR (500 MHz, CDCl3) δ 7.74 – 7.29 (m, 10H), 7.18 – 7.06 (m, 1H), 6.59 – 6.41 (m, 2H), 5.31 – 5.08 (m, 5H), 4.73 – 4.63 (m, 1H), 4.54 (ddd, J = 12.1, 10.0, 5.6 Hz, 2H), 4.30 – 4.20 (m, 1H), 4.15 – 4.07 (m, 2H), 2.38 – 2.14 (m, 5H), 1.97 – 1.54 (m, 17H), 1.41 (d, J = 19.6 Hz, 7H), 1.34 – 1.19 (m, 26H), 0.98 – 0.81 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 172.71, 172.50, 172.32, 172.18, 135.62, 128.86, 128.82, 128.65, 128.52, 128.46, 80.11, 67.31, 67.24, 66.15, 53.36, 52.27, 51.86, 36.87, 32.12, 29.82, 29.72, 29.56, 29.52, 29.44, 28.70, 28.55, 25.99, 25.90, 22.90, 14.34. MS (ESI) calculated for C54H85N3O10, m/z 935.62, found 958.59 (M + Na)+.
Synthesis of Compound 41: 2-Amino-6-((R)-4-dodecanamido-5-oxo-5-(undecyloxy) pentanamido)heptanedioic acid
The compound 40 (30 mg, 32.0 μmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 41 (24.6 mg, quantitative yield). 1H NMR (500 MHz, CDCl3) δ 4.51 – 4.26 (m, 2H), 4.21 (ddd, J = 19.6, 10.9, 5.9 Hz, 1H), 4.15 – 3.95 (m, 2H), 2.51 – 2.13 (m, 4H), 2.12 – 1.48 (m, 21H), 1.48 – 1.38 (m, 2H), 1.37 – 1.05 (m, 21H), 0.96 – 0.80 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 168.30, 68.63, 50.65, 50.48, 50.31, 50.14, 49.97, 39.16, 32.38, 30.79, 30.15, 29.86, 29.83, 29.37, 26.21, 24.17, 23.44, 23.14, 14.53, 14.51, 11.41. MS (ESI) calculated for C35H65N3O8, m/z 655.48, found 654.51 (M – H+)−.
Synthesis of Compound 43: 2-Amino-6-((R)-4-guanidino-5-oxo-5-(undecyloxy)pentanamido) heptanedioic acid
The compound 38 (50 mg, 0.05 mmol) was dissolved in 10 mL of dichloromethane, followed by the addition of 3 mL of piperidine. The reaction mixture was stirred for 15 minutes, followed by removal of the solvent under reduced pressure. The crude mixture was purified using column chromatography (5% MeOH/CH2Cl2) to obtain the free primary amine derivative, which was dissolved in pyridine, followed by the addition of 1H-pyrazole-1-carboxamidine.HCl (22.2 mg, 0.15 mmol). The sealed reaction mixture was then heated in microwave at 65 °C for 1 hour. The solvent was removed under vacuum and the residue was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 4 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the crude product which was purified using column chromatography (30% MeOH/CH2Cl2) to obtain the free carboxylic acid derivative, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 43 (20.3 mg, 47% in 4 steps). 1H NMR (500 MHz, MeOD) δ 4.43 – 4.08 (m, 5H), 3.58 (bs, 1H), 2.46 – 2.27 (m, 3H), 2.12 – 1.90 (m, 2H), 1.84 (bs, 3H), 1.66 (dd, J = 13.7, 6.9 Hz, 2H), 1.53 – 1.41 (m, 2H), 1.40 – 1.19 (m, 15H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 67.16, 33.09, 30.76, 30.68, 30.49, 30.39, 29.66, 26.97, 23.75, 14.46. MS (ESI) calculated for C24H45N5O7, m/z 515.33, found 516.31 (M + H)+.
Synthesis of Compound 44: (15R)-10,6-Dibenzyl 15-ethyl 2,2-dimethyl-4,12,17-trioxo-3-oxa- 5,11,16-triazaoctacosane-6,10,15-tricarboxylate
The compound 39 (100 mg, 0.11 mmol) was dissolved in 10 mL of dichloromethane, followed by the addition of 3 mL of piperidine. The reaction mixture was stirred for 15 minutes, followed by removal of the solvent under reduced pressure to obtain the residue which was dissolved in pyridine, followed by the addition of lauroyl chloride (41.9 μL, 0.18 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 4 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in dichloromethane and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (35% EtOAc/hexanes) to obtain the compound 44 (65.4 mg, 69%). 1H NMR (500 MHz, CDCl3) δ 7.73 – 7.27 (m, 10H), 7.06 (dd, J = 17.7, 8.8 Hz, 1H), 6.65 – 6.53 (m, 1H), 6.47 – 6.36 (m, 1H), 5.20 – 5.05 (m, 4H), 4.53 – 4.50 (m, 3H), 4.33 – 4.09 (m, 4H), 2.35 – 2.11 (m, 5H), 1.93 – 1.69 (m, 4H), 1.68 – 1.65 (m, 3H), 1.47 – 1.33 (m, 9H), 1.32 – 1.17 (m, 18H), 0.92 – 0.83 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 172.42, 172.26, 135.58, 131.11, 129.02, 128.82, 128.65, 128.52, 128.47, 80.10, 68.37, 67.24, 61.95, 53.43, 52.32, 51.83, 38.93, 36.85, 32.12, 31.64, 30.56, 29.83, 29.71, 29.56, 29.51, 29.13, 28.55, 25.89, 23.94, 23.20, 22.91, 14.34, 14.28, 11.18. MS (ESI) calculated for C45H67N3O10, m/z 809.48, found 832.51 (M + Na)+.
Synthesis of Compound 45: 2-Amino-6-((R)-4-dodecanamido-5-ethoxy-5-oxopentanamido) heptanedioic acid
The compound 44 (30 mg, 37.0 μmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 45 (23.4 mg, quantitative yield). 1H NMR (500 MHz, MeOD) δ 4.35 – 4.26 (m, 2H), 4.08 (qd, J = 7.1, 3.1 Hz, 2H), 3.73 (d, J = 11.0 Hz, 1H), 2.32 – 2.24 (m, 2H), 2.20 – 2.01 (m, 3H), 1.92 – 1.74 (m, 4H), 1.74 – 1.31 (m, 6H), 1.30 – 1.07 (m, 18H), 0.81 (dt, J = 13.9, 4.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 173.46, 62.57, 53.58, 53.23, 37.02, 36.92, 33.23, 33.13, 32.83, 31.46, 30.91, 30.82, 30.63, 30.46, 30.44, 28.53, 28.23, 27.18, 27.11, 23.89, 14.65, 14.59. MS (ESI) calculated for C26H47N3O8, m/z 529.33, found 528.36 (M – H)−.
Synthesis of Compound 46: Diethyl 2-((R)-4-amino-5-(benzyloxy)-5-oxopentanamido)-6-((tert-butoxycarbonyl) amino)heptanedioate
The compound 13 (100 mg, 0.12 mmol) was dissolved in 10 mL of dichloromethane, followed by the addition of 3 mL of piperidine. The reaction mixture was stirred for 15 minutes, followed by removal of the solvent under reduced pressure to obtain the crude product which was purified using column chromatography (5% MeOH/CH2Cl2) to obtain the compound 46 (66 mg, 92%). MS (ESI) calculated for C28H43N3O9, m/z 565.30, found 588.30 (M + Na)+.
Synthesis of Compound 48: Diethyl 2-amino-6-((R)-4-amino-5-oxo-5-(undecylthio) pentanamido)heptanedioate
To a solution of compound 46 (50 mg, 0.08 mmol) in dichloromethane, were added di-tert-butyl dicarbonate (28.9 mg, 0.13 mmol) and triethylamine (24.6 μl, 0.17 mmol). The reaction mixture was stirred for 30 minutes, followed by complete removal of the solvent under reduced pressure. The residue was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was then subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in anhydrous DMF, followed by the addition of 1-undecanethiol (40 μL, 0.17 mmol), HBTU (40.2 mg, 0.10 mmol), triethylamine (24.7 μL, 0.17 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred at room temperature for 14 hours, followed by removal of the solvent under reduced pressure. The residue was dissolved in ethyl acetate and washed with water. The organic fraction was dried over anhydrous sodium sulfate, filtered, concentrated and purified using column chromatography (30% EtOAc/hexanes) to obtain the compound 47 (41 mg, 63%). The compound 47 (40 mg, 0.05 mmol) was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 48 (41 mg, quantitative yield). 1H NMR (500 MHz, 20% CDCl3 in MeOD) δ 4.32 – 4.26 (m, 1H), 4.26 – 4.13 (m, 3H), 4.09 (q, J = 7.1 Hz, 2H), 3.91 (dt, , J = 2.0, 6.5 Hz, 1H), 2.95 (t, J = 7.3 Hz, 2H), 2.50 – 2.35 (m, 2H), 2.28 – 1.69 (m, 6H), 1.69 – 1.59 (m, 1H), 1.57 – 1.35 (m, 4H), 1.34 – 1.11 (m, 21H), 0.80 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, 20% CDCl3 in MeOD) δ 197.50, 174.15, 63.80, 62.71, 62.66, 59.98, 53.86, 53.83, 53.78, 53.74, 33.18, 31.98, 31.79, 31.65, 31.19, 31.16, 30.84, 30.71, 30.58, 30.30, 29.94, 28.52, 28.44, 23.85, 22.77, 22.63, 14.64, 14.59, 14.56. MS (ESI) calculated for C27H51N3O6S, m/z 545.35, found 568.36 (M + Na)+.
Synthesis of Compound 50: (2R)-5-((6-Amino-1,7-diethoxy-1,7-dioxoheptan-2-yl)amino)-2- dodecanamido-5-oxopentanoic acid
To a solution of 46 (36 mg, 0.06 mmol) in pyridine, were added lauroyl chloride (22.6 μL, 0.09 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 4 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in dichloromethane and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (18% EtOAc/hexanes) to obtain the compound 49 (39.0 mg, 82%). The compound 49 (30 mg, 0.04 mmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was then dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 50 (26.4 mg, quantitative yield in two steps). 1H NMR (500 MHz, MeOD) δ 4.45 – 4.37 (m, 2H), 4.29 (q, J = 7.1 Hz, 2H), 4.16 (q, J = 6.6 Hz, 2H), 4.05 – 3.97 (m, 1H), 2.34 (d, J = 2.6 Hz, 2H), 2.28 – 2.11 (m, 3H), 2.01 – 1.80 (m, 4H), 1.78 – 1.40 (m, 6H), 1.40 – 1.19 (m, 21H), 0.90 (dt, J = 13.6, 4.7 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 176.49, 175.06, 174.90, 173.29, 170.44, 63.70, 62.49, 53.88, 53.80, 53.33, 53.13, 52.83, 49.52, 49.35, 49.18, 49.01, 48.84, 48.67, 48.50, 36.96, 36.87, 33.10, 32.79, 32.09, 32.04, 31.95, 31.02, 30.89, 30.78, 30.68, 30.51, 30.38, 28.84, 28.47, 27.01, 26.97, 23.76, 22.65, 22.42, 22.32, 14.44. MS (ESI) calculated for C28H51N3O8, m/z 557.37, found 558.40 (M + H)+.
Synthesis of Compound 51: Dibenzyl 2-(4-((((9H-fluoren-9-yl)methoxy)carbonyl)amino) butanamido)-6-((tert-butoxycarbonyl)amino)heptanedioate
To a solution of 9 (221 mg, 0.47 mmol) in anhydrous dichloromethane, were added 4-Fmoc-amino-butyric acid (150 mg, 0.46 mmol), polystyrene bound carbodiimide (382.11 mg, 0.47 mmol) and a catalytic amount of polystyrene bound DMAP. The reaction mixture was stirred at room temperature for 8 hours, followed by filtration to remove the solid resin. The filtrate was evaporated under vacuum to obtain the residue which was then purified using column chromatography (50% EtOAc/hexanes) to obtain the compound 51 (230 mg, 63%). 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 7.5 Hz, 2H), 7.57 (d, J = 7.4 Hz, 2H), 7.44 – 7.25 (m, 14H), 6.47 (d, J = 7.2 Hz, 1H), 5.21 – 5.00 (m, 6H), 4.61 – 4.50 (m, 1H), 4.46 – 4.33 (m, 2H), 4.31 – 4.15 (m, 2H), 3.29 – 3.11 (m, 2H), 2.26 – 2.12 (m, 2H), 1.76 (dd, J = 19.9, 6.1 Hz, 4H), 1.70 – 1.52 (m, 2H), 1.38 (d, J = 13.3 Hz, 11H). 13C NMR (126 MHz, CDCl3) δ 172.70, 172.59, 172.45, 172.29, 157.10, 155.76, 144.15, 141.53, 135.54, 128.85, 128.82, 128.72, 128.64, 128.57, 128.45, 127.88, 127.25, 125.28, 120.16, 80.19, 67.42, 67.34, 67.24, 66.76, 53.34, 52.18, 47.49, 40.21, 33.33, 32.45, 32.30, 31.65, 28.53, 26.21, 21.59, 21.28. MS (ESI) calculated for C45H51N3O9, m/z 777.36, found 800.33 (M + Na)+.
Synthesis of Compound 52: Dibenzyl 2-((tert-butoxycarbonyl)amino)-6-(4-dodecanamidobutanamido) heptanedioate
The compound 51 (140 mg, 0.18 mmol) was dissolved in 10 mL of dichloromethane, followed by the addition of 3 mL of piperidine. The reaction mixture was stirred for 15 minutes, followed by removal of the solvent under reduced pressure to obtain the residue which was dissolved in pyridine, followed by the addition of lauroyl chloride (64.0 μL, 0.27 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 4 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in dichloromethane and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (30% EtOAc/hexanes) to obtain the compound 52 (100 mg, 75%). 1H NMR (500 MHz, CDCl3) δ 7.41 – 7.29 (m, 10H), 6.84 – 6.76 (m, 1H), 5.97 – 5.88 (m, 1H), 5.26 – 5.09 (m, 5H), 4.59 – 4.50 (m, 1H), 4.26 (d, J = 8.5 Hz, 1H), 3.40 – 3.30 (m, 1H), 3.25 – 3.17 (m, 1H), 2.29 – 2.22 (m, 2H), 2.17 – 2.14 (m, 2H), 1.87 – 1.76 (m, 4H), 1.66 (s, 4H), 1.63 – 1.58 (m, 2H), 1.42 (d, J = 14.4 Hz, 9H), 1.33 – 1.20 (m, 16H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 174.14, 173.13, 172.73, 172.62, 172.43, 172.27, 155.92, 155.80, 135.57, 128.84, 128.69, 128.65, 128.56, 128.46, 80.12, 67.27, 53.39, 52.34, 38.80, 37.10, 33.62, 32.30, 32.12, 31.57, 29.84, 29.82, 29.72, 29.57, 29.55, 28.55, 26.06, 25.93, 22.90, 21.66, 21.34, 14.34. MS (ESI) calculated for C42H63N3O8, m/z 737.46, found 760.41 (M + Na)+.
Synthesis of Compound 53: 2-Amino-6-(4-dodecanamidobutanamido)heptanedioic acid
The compound 52 (50 mg, 0.06 mmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 53 (38 mg, quantitative yield). 1H NMR (500 MHz, MeOD) δ 4.39 (dd, J = 9.3, 4.7 Hz, 1H), 3.80 – 3.73 (m, 1H), 3.20 (t, J = 7.0 Hz, 2H), 2.32 – 2.24 (m, 2H), 2.21 – 2.14 (m, 2H), 2.00 – 1.68 (m, 7H), 1.66 – 1.42 (m, 5H), 1.36 – 1.24 (m, 14H), 0.90 (dd, J = 13.1, 6.2 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 176.66, 175.79, 175.46, 173.04, 54.83, 53.43, 39.81, 37.36, 34.18, 33.23, 32.45, 32.39, 31.61, 31.48, 30.90, 30.80, 30.63, 30.52, 27.24, 26.85, 23.89, 23.07, 22.75, 14.58. MS (ESI) calculated for C23H43N3O6, m/z 457.32, found 458.39 (M + H)+.
Synthesis of Compound 54: 5-((1,7-Bis(benzyloxy)-6-((tert-butoxycarbonyl)amino)-1,7- dioxoheptan-2-yl)amino)-5-oxopentanoic acid
To a solution of 9 (100 mg, 0.21 mmol) in anhydrous dichloromethane, was added glutaric anhydride (25.5 mg, 0.22 mmol). The reaction mixture was stirred at room temperature for 1 hour, followed by removal of the solvent under reduced pressure. The residue was then purified using column chromatography (5% MeOH/CH2Cl2) to obtain the compound 54 (118 mg, 95%). 1H NMR (500 MHz, CDCl3) δ 7.73 – 7.29 (m, 10H), 6.87 – 6.34 (m, 1H), 5.22 – 5.08 (m, 4H), 4.60 (bs, 1H), 4.32 – 3.92 (m, 1H), 2.54 – 2.17 (m, 4H), 2.13 – 1.54 (m, 6H), 1.52 – 1.20 (m, 13H). 13C NMR (126 MHz, CDCl3) δ 176.46, 172.80, 172.69, 172.41, 135.41, 131.11, 129.01, 128.86, 128.84, 128.74, 128.69, 128.54, 80.46, 68.37, 67.51, 67.43, 67.15, 53.35, 52.10, 51.82, 38.92, 35.67, 35.08, 33.61, 33.49, 32.99, 32.44, 31.80, 31.66, 31.00, 30.55, 29.91, 29.13, 28.53, 28.31, 23.93, 23.20, 21.55, 21.21, 20.61, 19.92. MS (ESI) calculated for C31H40N2O9, m/z 584.27, found 607.26 (M + Na)+.
Synthesis of Compound 55: Dibenzyl 2-((tert-butoxycarbonyl)amino)-6-(5-oxo-5-(undecyloxy) pentanamido)heptanedioate
To a solution of 54 (90 mg, 0.15 mmol) in anhydrous DMF, were added 1- undecanol (63 μL, 0.30 mmol), HBTU (70 mg, 0.18 mmol), triethylamine (43 μL, 0.30 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred at room temperature for 12 hours, followed by removal of the solvent under reduced pressure. The residue was dissolved in ethylacetate and washed with water. The organic fraction was dried over anhydrous sodium sulfate, filtered, concentrated and purified using column chromatography (10% EtOAc/hexanes) to obtain the compound 55 (86.4 mg, 78%).1H NMR (500 MHz, CDCl3) δ 7.40 – 7.29 (m, 10H), 6.15 (d, J = 6.3 Hz, 1H), 5.20 – 5.02 (m, 5H), 4.62 – 4.54 (m, 1H), 4.32 – 4.19 (m, 1H), 4.05 (t, J = 6.8 Hz, 2H), 2.40 – 2.32 (m, 2H), 2.30 – 2.22 (m, 2H), 2.00 – 1.90 (m, 2H), 1.87 – 1.57 (m, 7H), 1.43 (s, 9H), 1.33 – 1.19 (m, 17H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.49, 173.48, 172.70, 172.57, 172.42, 172.27, 172.26, 155.85, 155.70, 135.55, 135.42, 128.86, 128.82, 128.73, 128.66, 128.55, 128.47, 80.20, 80.09, 67.43, 67.35, 67.25, 64.90, 53.31, 52.03, 51.99, 35.39, 33.47, 32.49, 32.26, 32.11, 31.96, 31.84, 29.80, 29.79, 29.73, 29.54, 29.47, 28.82, 28.52, 26.11, 22.89, 21.48, 21.20, 20.97, 14.33. MS (ESI) calculated for C42H62N2O9, m/z 738.45, found 761.43 (M + Na)+.
Synthesis of Compound 56: 2-Amino-6-(5-oxo-5-(undecyloxy)pentanamido)heptanedioic acid
The compound 55 (35 mg, 0.04 mmol) was dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the residue, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 56 (26 mg, quantitative yield). 1H NMR (500 MHz, MeOD) δ 4.34 (dd, J = 8.7, 5.0 Hz, 1H), 4.02 (t, J = 6.7 Hz, 2H), 3.82 – 3.73 (m, 1H), 2.34 (t, J = 7.5 Hz, 2H), 2.27 (t, J = 7.2 Hz, 2H), 1.97 – 1.80 (m, 5H), 1.74 – 1.63 (m, 1H), 1.62 – 1.42 (m, 4H), 1.29 – 1.25 (m, 16H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 175.63, 175.60, 175.35, 175.32, 175.12, 172.76, 65.83, 54.68, 54.54, 53.40, 53.36, 35.86, 34.37, 33.19, 32.40, 32.32, 31.51, 31.39, 30.85, 30.84, 30.78, 30.59, 30.51, 29.86, 27.16, 23.86, 22.99, 22.73, 22.27, 14.60. MS (ESI) calculated for C23H42N2O7, m/z 458.30, found 459.32 (M + H)+.
General procedure for the syntheses of compounds (66–79)
Synthesis of compound 66: Dibenzyl 2-((R)-4-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-5- (benzyloxy)-5-oxopentanamido)heptanedioate
To a solution of 4 (100 mg, 0.21 mmol) in dichloromethane, were added 58 (78.8 mg, 0.22 mmol), polystyrene bound carbodiimide (170 mg, 0.22 mmol), triethylamine (60 μL, 0.43 mmol) and a catalytic amount of polystyrene bound DMAP. The reaction mixture was stirred at room temperature for 8 hours, followed by filtration to remove the solid resin. The filtrate was evaporated under vacuum to obtain the residue which was then purified using column chromatography (55% EtOAc/hexanes) to obtain the compound 66 (154 mg, 89%). 1H NMR (500 MHz, CDCl3,) δ 7.74 (d, J = 7.5 Hz, 2H), 7.57 (d, J = 7.0 Hz, 2H), 7.41 – 7.24 (m, 19H), 6.47 (d, J = 7.8 Hz, 0.50H), 6.19 (d, J = 7.8 Hz, 0.50H), 5.67 (dd, J = 20.4, 8.0 Hz, 1H), 5.20 – 5.03 (m, 6H), 4.59 (ddd, J = 13.0, 7.5, 5.3 Hz, 1H), 4.49 – 4.33 (m, 3H), 4.18 (t, J = 6.8 Hz, 1H), 2.27 – 2.19 (m, 5H), 2.02 – 1.77 (m, 3H), 1.63 – 1.61 (m, 1H), 1.38 – 1.16 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 173.17, 171.06, 170.13, 170.04, 169.40, 153.91, 141.79, 141.42, 139.22, 133.04, 126.56, 126.55, 126.53, 126.51, 126.46, 126.31, 126.24, 126.21, 126.13, 126.09, 125.63, 125.00, 123.03, 117.90, 65.30, 65.07, 64.95, 64.08, 51.35, 50.01, 45.06, 31.74, 29.88, 26.66, 26.14, 22.55, 22.27. MS (ESI) calculated for C48H48N2O9, m/z 796.34, found 819.31 (M + Na)+.
Synthesis of compound 67: 2-((R)-4-carboxy-4-dodecanamidobutanamido)heptanedioic acid
The compound 66 (147 mg, 0.18 mmol) was dissolved in 10 mL of dichloromethane, followed by the addition of 3 mL of piperidine. The reaction mixture was stirred for 15 minutes, followed by removal of the solvent under reduced pressure to obtain the residue, which was dissolved in pyridine, followed by the addition of lauroyl chloride (64.0 μL, 0.27 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred for 4 hours, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in dichloromethane and washed with water. The organic solvent was dried over anhydrous sodium sulfate, filtered, evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (25% EtOAc/hexanes) to obtain the protected intermediate of compound 67 (100 mg, 73%). The protected intermediate (50 mg, 0. 06 mmol) was then dissolved in methanol, followed by the addition of Pd(OH)2/C. The solution was subjected to catalytic hydrogenolysis at 60 psi hydrogen pressure for 2 hours, followed by filtration through celite bed. The solvent was removed under reduced pressure to obtain the compound 67 (29 mg, quantitative yield). 1H NMR (500 MHz, MeOD) δ 4.42 – 4.28 (m, 2H), 2.33 (t, J = 6.8 Hz, 2H), 2.28 (t, J = 7.4 Hz, 2H), 2.24 – 2.18 (m, 2H), 2.18 – 2.10 (m, 1H), 1.93 (dt, J = 13.8, 6.3 Hz, 1H), 1.85 – 1.82 (m, 1H), 1.72 – 1.54 (m, 5H), 1.41 (dd, J = 14.5, 7.4 Hz, 3H), 1.30 – 1.26 (m, 15H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 177.59, 176.63, 176.63, 175.03, 54.04, 53.66, 37.03, 34.85, 33.36, 33.24, 32.50, 32.45, 30.91, 30.81, 30.64, 30.48, 28.97, 28.79, 27.07, 26.66, 25.78, 23.90, 14.60. MS (ESI) calculated for C24H42N2O8, m/z 486.29, found 485.29 (M – H)−.
In the case of N-Boc protected intermediates (compound 68, 72, 76, 78), the compound after hydrogenolysis was dissolved in trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 69, 73, 77, 79.
Synthesis of compound 68: (R)-Benzyl 2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-5-((5- ((tert-butoxycarbonyl)amino)pentyl)amino)-5-oxopentanoate
1H NMR (500 MHz, CDCl3) δ 7.77 (d, J = 7.5 Hz, 2H), 7.71 (dt, J = 7.3, 3.6 Hz, 1H), 7.61 (t, J = 6.8 Hz, 2H), 7.54 (dd, J = 5.7, 3.3 Hz, 1H), 7.40 (t, J = 7.5 Hz, 2H), 7.37 – 7.30 (m, 5H), 5.18 (q, J = 12.1 Hz, 2H), 4.46 – 4.32 (m, 3H), 4.25 – 4.17 (m, 2H), 3.42 (dd, J = 5.5, 3.9 Hz, 1H), 3.18 (ddd, J = 20.5, 13.5, 7.0 Hz, 2H), 3.07 (s, 2H), 2.20 – 2.18 (m, 3H), 1.52 – 1.38 (m, 10H), 1.37 – 1.27 (m, 7H). 13C NMR (126 MHz, CDCl3) δ 168.20, 141.65, 135.35, 131.45, 129.30, 129.14, 129.03, 128.87, 128.24, 127.59, 125.59, 120.48, 79.88, 68.70, 67.86, 50.32, 50.15, 49.98, 47.61, 39.76, 39.20, 30.83, 30.05, 29.41, 28.88, 24.34, 24.21, 23.47. MS (ESI) calculated for C37H45N3O7, m/z 643.33, found 666.34 (M + Na)+.
Synthesis of compound 69: (R)-5-((5-Aminopentyl)amino)-2-dodecanamido-5-oxopentanoic acid
1H NMR (400 MHz, MeOD) δ 4.21 (dd, J = 8.4, 4.9 Hz, 1H), 3.30 – 3.18 (m, 2H), 2.92 (t, J = 6.7 Hz, 2H), 2.27 – 2.20 (m, 4H), 2.08 (td, J = 13.5, 6.5 Hz, 1H), 1.94 (td, J = 13.6, 7.3 Hz, 1H), 1.72 – 1.39 (m, 8H), 1.37 – 1.20 (m, 16H), 0.88 (dd, J = 12.0, 5.4 Hz, 3H). 13C NMR (101 MHz, 10% CDCl3 in MeOD) δ 174.03, 53.83, 38.93, 37.98, 36.02, 32.27, 31.46, 29.16, 29.04, 28.87, 27.91, 26.29, 25.40, 22.64, 22.17, 13.26. MS (ESI) calculated for C22H43N3O4, m/z 413.33, found 414.37(M+H)+.
Synthesis of compound 70: (2R)-Benzyl 2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-5-oxo- 5-((2-oxoazepan-3-yl)amino)pentanoate
1H NMR (500 MHz, CDCl3, Ethyl Acetate) δ 7.76 (d, J = 7.5 Hz, 2H), 7.61 (d, J = 7.4 Hz, 2H), 7.35 (qd, J = 14.7, 7.3 Hz, 9H), 6.98 – 6.90 (m, 1H), 6.06 (bs, 1H), 5.88 – 5.81 (m, 1H), 5.23 – 5.13 (m, 2H), 4.54 – 4.48 (m, 1H), 4.46 – 4.33 (m, 3H), 4.21 (t, J = 7.1 Hz, 1H), 3.30 – 3.18 (m, 2H), 2.37 – 2.18 (m, 3H), 1.99 – 1.97 (m, 1H), 1.84 – 1.76 (dd, J = 14.6, 9.4 Hz, 1H), 1.62 (bs, 3H), 1.49 – 1.35 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 175.57, 172.14, 171.16, 156.34, 144.15, 143.99, 141.49, 135.45, 128.85, 128.71, 128.54, 127.90, 127.30, 125.43, 120.17, 67.51, 67.29, 53.98, 53.91, 52.46, 47.38, 42.38, 32.37, 32.32, 31.76, 31.70, 29.05, 28.10, 27.98, 27.93. MS (ESI) calculated for C33H35N3O6, m/z 569.25, found 592.23 (M + Na)+.
Synthesis of compound 71: (2R)-2-Dodecanamido-5-oxo-5-((2-oxoazepan-3-yl)amino)pentanoic acid
1H NMR (500 MHz, MeOD) δ 4.54 (dd, J = 11.4, 1.5 Hz, 1H), 4.41 – 4.31 (m, 1H), 3.30 – 3.13 (m, 3H), 2.34 (tt, J = 6.4, 5.0 Hz, 2H), 2.27 – 2.11 (m, 3H), 2.02 – 1.72 (m, 5H), 1.65 – 1.49 (m, 3H), 1.43 – 1.22 (m, 16H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 177.24, 176.35, 176.30, 174.12, 174.02, 53.35, 49.46, 49.29, 42.47, 36.94, 33.37, 33.27, 33.10, 32.21, 32.16, 30.78, 30.77, 30.67, 30.51, 30.50, 30.36, 29.94, 29.16, 28.87, 28.80, 26.92, 23.75, 14.45. MS (ESI) calculated for C23H41N3O5, m/z 439.30, found 462.31 (M + Na)+.
Synthesis of compound 72: (R)-Benzyl 2-((R)-4-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)- 5-(benzyloxy)-5-oxopentanamido)-6-((tert-butoxycarbonyl)amino)hexanoate
1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H), 7.60 (d, J = 7.2 Hz, 2H), 7.45 – 7.28 (m, 14H), 6.32 (d, J = 6.9 Hz, 1H), 5.71 (d, J = 8.0 Hz, 1H), 5.22 – 5.08 (m, 4H), 4.59 (t, J = 10.9 Hz, 2H), 4.40 (d, J = 6.9 Hz, 3H), 4.20 (t, J = 6.9 Hz, 1H), 3.02 (bs, 2H), 2.24 (s, 3H), 1.95 (dd, J = 15.4, 7.9 Hz, 1H), 1.83 (ddd, J = 15.7, 10.6, 5.3 Hz, 1H), 1.67 (d, J = 8.6 Hz, 1H), 1.41 (s, 11H), 1.36 – 1.23 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 172.37, 172.01, 171.81, 156.51, 156.28, 144.10, 143.86, 141.53, 135.48, 135.36, 128.87, 128.84, 128.78, 128.73, 128.63, 128.56, 127.94, 127.31, 125.34, 120.20, 120.18, 79.31, 67.61, 67.36, 67.27, 53.63, 52.36, 47.37, 40.17, 32.26, 32.02, 29.71, 28.62, 22.50. MS (ESI) calculated for C45H51N3O9, m/z 777.36, found 800.35 (M + Na)+.
Synthesis of compound 73: (R)-6-Amino-2-((R)-4-carboxy-4-dodecanamidobutanamido) hexanoic acid
1H NMR (500 MHz, MeOD) δ 4.31 (dd, J = 9.1, 4.4 Hz, 1H), 4.26 (dd, J = 9.6, 4.3 Hz, 1H), 2.89 – 2.79 (m, 2H), 2.26 (t, J = 7.2 Hz, 2H), 2.21 – 2.08 (m, 3H), 1.88 – 1.75 (m, 2H), 1.68 – 1.48 (m, 5H), 1.47 – 1.08 (m, 18H), 0.80 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 176.55, 174.81, 53.42, 53.24, 40.56, 36.97, 33.10, 33.04, 32.34, 30.79, 30.77, 30.68, 30.52, 30.50, 30.38, 28.64, 27.95, 27.00, 23.78, 23.75, 14.45. MS (ESI) calculated for C23H43N3O6, m/z 457.32, found 458.33 (M + H)+.
Synthesis of compound 74: (R)-Benzyl 6-((R)-4-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)- 5-(benzyloxy)-5-oxopentanamido)-2-(((benzyloxy)carbonyl)amino)hexanoate
1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 7.5 Hz, 2H), 7.61 – 7.52 (m, 2H), 7.38 – 7.25 (m, 19H), 5.80 (bs, 1H), 5.71 (d, J = 7.8 Hz, 1H), 5.45 – 5.41 (m, 1H), 5.19 – 5.09 (m, 4H), 5.06 (s, 2H), 4.44 – 4.30 (m, 4H), 4.16 (t, J = 6.8 Hz, 1H), 3.14 (dt, J = 13.8, 6.8 Hz, 2H), 2.17 – 2.11 (m, 3H), 1.93 (dd, J = 17.2, 11.2 Hz, 1H), 1.80 (dt, J = 10.2, 7.3 Hz, 1H), 1.70 – 1.62 (m, 1H), 1.49 – 1.38 (m, 2H), 1.33 – 1.24 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 172.41, 172.02, 156.59, 156.24, 144.10, 143.81, 141.53, 141.48, 136.40, 135.46, 135.37, 128.86, 128.84, 128.74, 128.72, 128.62, 128.53, 128.41, 128.30, 127.94, 127.29, 125.35, 125.31, 120.20, 67.58, 67.37, 67.28, 67.20, 53.88, 53.75, 47.34, 39.29, 32.59, 32.34, 29.00, 28.82, 22.53. MS (ESI) calculated for C48H49N3O9, m/z 811.35, found 834.34 (M + Na)+.
Synthesis of compound 75: (R)-2-Amino-6-((R)-4-carboxy-4-dodecanamidobutanamido) hexanoic acid
1H NMR (500 MHz, MeOD) δ 4.34 (dd, J = 9.1, 4.9 Hz, 1H), 3.80 (t, J = 6.1 Hz, 1H), 3.22 (dt, J = 13.3, 6.7 Hz, 1H), 3.13 (dt, J = 13.5, 6.7 Hz, 1H), 2.30 – 2.12 (m, 5H), 1.97 – 1.79 (m, 3H), 1.64 – 1.56 (m, 2H), 1.52 (dd, J = 13.8, 6.7 Hz, 2H), 1.49 – 1.39 (m, 2H), 1.30 – 1.26 (d, J = 17.6 Hz, 16H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 176.53, 174.85, 54.57, 53.08, 39.85, 36.89, 33.27, 33.10, 31.40, 30.78, 30.68, 30.52, 30.50, 30.34, 30.00, 28.69, 26.96, 23.76, 23.40, 14.46. MS (ESI) calculated for C23H43N3O6, m/z 457.32, found 456.33 (M – H)−.
Synthesis of compound 76: (S)-Tert-butyl 2-((R)-4-((((9H-fluoren-9- yl)methoxy)carbonyl)amino)-5-(benzyloxy)-5-oxopentanamido)-6-(((benzyloxy)carbonyl)amino) hexanoate
1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 7.5 Hz, 2H), 7.61 – 7.54 (m, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.35 – 7.25 (m, 12H), 6.30 (dd, J = 84.8, 7.6 Hz, 1H), 5.74 (dd, J = 17.5, 7.9 Hz, 1H), 5.14 (t, J = 12.2 Hz, 2H), 5.08 – 5.01 (m, 2H), 4.88 (bs, 1H), 4.42 – 4.30 (ddt, J = 31.5, 17.9, 7.6 Hz, 4H), 4.17 (t, J = 6.8 Hz, 1H), 3.14 (dd, J = 12.7, 6.4 Hz, 2H), 2.31 – 2.13 (m, 3H), 1.98 (dd, J = 14.5, 7.7 Hz, 1H), 1.76 (ddd, J = 15.5, 10.2, 5.3 Hz, 1H), 1.64 – 1.61 (m, 1H), 1.51 – 1.44 (m, 2H), 1.44 – 1.42 (m, 9H), 1.37 – 1.27 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 172.11, 171.84, 171.77, 156.72, 156.51, 144.12, 143.85, 141.50, 136.79, 135.36, 128.85, 128.73, 128.70, 128.57, 128.28, 127.92, 127.30, 125.35, 120.19, 120.17, 82.33, 67.57, 67.31, 66.76, 53.67, 52.76, 47.34, 40.77, 32.41, 32.16, 29.58, 28.70, 28.19, 22.29. MS (ESI) calculated for C45H51N3O9, m/z 777.36, found 800.36 (M + Na)+.
Synthesis of compound 77: (S)-6-Amino-2-((R)-4-carboxy-4-dodecanamidobutanamido) hexanoic acid
1H NMR (500 MHz, MeOD) δ 4.35 (bs, 2H), 2.91 (t, J = 7.4 Hz, 2H), 2.32 (t, J = 6.7 Hz, 2H), 2.22 (t, J = 7.5 Hz, 2H), 2.17 – 2.08 (m, 1H), 1.96 – 1.87 (m, 2H), 1.77 – 1.55 (m, 5H), 1.48 (dd, J = 15.2, 7.6 Hz, 2H), 1.30 – 1.27 (m, 16H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 176.53, 175.19, 53.81, 40.68, 37.07, 33.41, 33.23, 32.47, 32.36, 30.92, 30.81, 30.65, 30.50, 29.17, 28.86, 28.12, 27.10, 23.95, 23.89, 14.60. MS (ESI) calculated for C23H43N3O6, m/z 457.32, found 458.32 (M + H)+.
Synthesis of compound 78: (S)-Tert-butyl 6-((R)-4-((((9H-fluoren-9- yl)methoxy)carbonyl)amino)-5-(benzyloxy)-5-oxopentanamido)-2-((tert-butoxycarbonyl)amino) hexanoate
1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 7.5 Hz, 2H), 7.61 – 7.55 (m, 2H), 7.41 – 7.25 (m, 9H), 5.89 (bs, 1H), 5.78 (d, J = 7.9 Hz, 1H), 5.16 (q, J = 12.2 Hz, 2H), 5.07 (d, J = 8.1 Hz, 1H), 4.37 (ddd, J = 28.8, 10.6, 7.1 Hz, 3H), 4.19 (t, J = 7.0 Hz, 1H), 3.19 (ddt, J = 25.6, 13.1, 6.5 Hz, 2H), 2.18 (t, J = 10.6 Hz, 2H), 2.01 – 1.92 (m, 1H), 1.78 – 1.62 (m, 3H), 1.58 (ddd, J = 11.4, 9.3, 5.7 Hz, 1H), 1.53 – 1.45 (m, 2H), 1.41 (d, J = 8.0 Hz, 18H), 1.38 – 1.27 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.29, 170.24, 170.19, 154.78, 153.95, 142.30, 142.03, 139.72, 139.67, 133.58, 127.06, 126.95, 126.81, 126.13, 126.12, 125.48, 123.57, 123.54, 118.40, 118.38, 80.27, 78.07, 65.77, 65.53, 52.05, 52.00, 45.54, 37.78, 31.12, 30.81, 27.32, 26.97, 26.73, 26.40, 20.95. MS (ESI) calculated for C42H53N3O9, m/z 743.38, found 766.36 (M + Na)+.
Synthesis of compound 79: (S)-2-Amino-6-((R)-4-carboxy-4-dodecanamidobutanamido) hexanoic acid
1H NMR (500 MHz, MeOD) δ 4.35 (dd, J = 8.9, 4.9 Hz, 1H), 3.74 (t, J = 5.9 Hz, 1H), 3.24 – 3.12 (m, 2H), 2.30 – 2.13 (m, 5H), 1.95 – 1.80 (m, 3H), 1.66 – 1.37 (m, 7H), 1.31 – 1.27 (m, 15H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 176.28, 175.26, 174.74, 173.09, 54.56, 53.07, 39.70, 36.88, 33.15, 32.90, 31.17, 30.61, 30.59, 30.50, 30.35, 30.31, 30.23, 30.16, 29.73, 28.66, 26.78, 23.59, 23.11, 14.54. MS (ESI) calculated for C23H43N3O6, m/z 457.32, found 458.35 (M + H)+.
NF-κB induction
The induction of NF-κB was quantified using HEK-Blue™ cells as previously described by us.10;51 HEK293 cells stably transfected with human Nod1 and alkaline phosphatase (sAP) were obtained from InvivoGen (San Diego, CA), and were maintained in HEK-Blue™ Selection medium containing zeocin and normocin. Stable expression of secreted alkaline phosphatase (sAP) under control of NF-κB/AP-1 promoters is inducible by Nod1 agonists, and extracellular sAP in the supernatant is proportional to Nod1-mediated NF-κB induction. HEK-Blue cells were incubated at a density of ~105 cells/mL in a volume of 80 μL/well, in 384-well, flat-bottomed, cell culture-treated microtiter plates until confluency was achieved, and subsequently graded concentrations of compounds. sAP was assayed spectrophotometrically using an alkaline phosphatase-specific chromogen (present in the HEK-detection medium as supplied by the vendor) at 620 nm.
Experiments involving human blood
Human blood was obtained from healthy adults by antecubital venipuncture in accordance with University of Kansas Human Subjects Experimentation protocols (Protocol # HSCL 12397).
Phosflow™ flow cytometric assay for p38MAPK
Assays were performed as described by us previously.10;38;52 Briefly, 1 mL aliquots of fresh whole blood, anticoagulated with heparin were incubated with 25 μL an equal volume of graded concentrations of compounds diluted in saline for 15 minutes at 37°C. Erythrocytes were lysed and leukocytes were fixed in one step by mixing 200 μL of the samples in 4 mL pre-warmed Whole Blood Lyse/Fix Buffer (Becton-Dickinson Biosciences, San Jose, CA). After washing the cells at 500 g for 8 minutes in buffer, the cells were permeabilized in ice-cold methanol for 30 min, washed twice in phosphate-buffered saline and transferred to a Millipore MultiScreen BV 1.2μ filter plate and stained with either phycoerythrin (PE)-conjugated mouse anti-p38MAPK (pT180/pY182; BD Biosciences) mAb, or a matched PE-labeled mouse IgG1 κ isotype control mAb for 60 minutes. The cells were washed twice in the plate by aspiration as per protocols supplied by the vendor. Cytometry was performed using a BD FACSArray instrument in the single-color mode for PE acquisition on 20,000 gated events. Post-acquisition analyses were performed using FlowJo v 7.0 software (Treestar, Ashland, OR).
CD11b flow cytometric assay
Assays were performed as described by us previously.10;38;52 Briefly, 1 mL aliquots of fresh anticoagulated whole blood were incubated with 25 μL of graded dilutions of the compounds for 1 hour at 37°C. Negative (saline) controls were included in each experiment. Samples were placed on ice for 15 minutes before 20 μL of anti-CD11b/Mac-1 antibody (Becton-Dickinson) were added to each sample tube and allowed to incubate on ice for 30 minutes. This 0°C incubation step prevented internalization of antibody, and ensured staining of only extracellularly expressed CD11b. Erythrocytes were lysed and leukocytes were fixed in one step by mixing 200 μL of the samples in 4 mL pre-warmed Whole Blood Lyse/Fix Buffer (Becton-Dickinson Biosciences, San Jose, CA). After washing the cells twice at 200 g for 5 minutes in CBA buffer, the cells were transferred to a 96-well plate. Flow cytometry was performed using a BD FACSArray instrument in the single-color mode for PE acquisition on 20,000 gated events. Post-acquisition analyses were performed using FlowJo v 7.0 software.
Transcriptomal profiling in whole human blood
Assays were performed as described by us previously.10 Briefly, peripheral blood mononuclear cells (PBMCs) isolated from fresh, heparin-anticoagulated human blood was stimulated with 20 μg/mL of the compounds for two hours, and total RNA was extracted from treated and negative control PBMC samples with QIAamp RNA Blood Mini Kit (Qiagen). 4 μg of each of the RNA samples was used for transcriptomal profiling, employing the Human Genome GeneChip U133 plus 2.0 oligonucleotide array (Affymetrix, Santa Clara, CA). Established standard protocols at the KU Genomics Facility were performed on cRNA target preparation, array hybridization, washing, staining and image scanning. The microarray data were collected using the Affymetrix GeneChip Command Console Software (AGCC) and then subjected to quality assessment before further analyses. QC criteria included low background, low noise, detection of positive controls, and a 5′/3′ ratio of < 3.0. To facilitate direct comparison of gene expression data between different samples, the GeneChip data were first subjected to preprocessing, which included scaling of data from all chips to a target intensity value of 500 (in Affymetrix Expression Console Software), and further normalization in GeneSpring GX (Agilent Technologies, Santa Clara, CA). Prior to identifying target genes, genes that were detected as non-expressed in all samples, i.e., those with absence (A) cells were filtered out. Genes whose expression was changed by our compounds by at least 2-fold (compared to the negative control) were identified to be differentially expressed. Pathway analyses of gene expression were performed using IPA® (Ingenuity Systems, Redwood, CA).
Supplementary Material
Acknowledgments
This work was supported by NIH/NIAID contract HHSN272200900033C.
Abbreviations
- AP-1
Activator protein-1
- CD
Cluster of differentiation
- DAP
Diaminopimelic acid
- DMAP
4- Dimethylaminopyridine
- EC50
Half-maximal effective concentration
- ESI-TOF
Electrospray ionizationtime of flight
- FmocCl
Fluorenylmethyloxycarbonyl chloride
- Glu
Glutamic Acid
- HBTU
O-Benzotriazole- N,N,N,N′-tetramethyl uronium hexafluorophosphate
- HEK
Human embryonic kidney
- iE-DAP
γ-D-Glutamyl-diaminopimelic acid
- IL
Interleukin
- NF-κB
Nuclear factor-κB
- Nod1
Nucleotide oligomerization domain-1
- Nod2
Nucleotide oligomerization domain-2
- NLR
Nod like receptor
- p38MAPK
p38 Mitogen activated protein kinase
- PBMCs
Peripheral blood mononuclear cells
- PE
Phycoerythrin
- PRRs
Pattern recognition receptors
- RIG-I
Retinoic acid inducible gene-I
- RNA
Ribonucleic acid
- sAP
Secreted alkaline phosphatase
- SAR
Structure activity relationship
- Th1
Helper T lymphocyte, type 1
- Th2
Helper T lymphocyte, type 2
- TLR
Toll like receptor
- TREM-1
Triggering Receptor Expressed on Myeloid Cells-1
References
- 1.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Kumagai Y, Takeuchi O, Akira S. Pathogen recognition by innate receptors. J Infect Chemother. 2008;14:86–92. doi: 10.1007/s10156-008-0596-1. [DOI] [PubMed] [Google Scholar]
- 3.Kumagai Y, Akira S. Identification and functions of pattern-recognition receptors. J Allergy Clin Immunol. 2010;125:985–992. doi: 10.1016/j.jaci.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 4.Akira S. Pathogen recognition by innate immunity and its signaling. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:143–156. doi: 10.2183/pjab.85.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Philpott DJ, Girardin SE. Nod-like receptors: sentinels at host membranes. Curr Opin Immunol. 2010;22:428–434. doi: 10.1016/j.coi.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–128. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rietdijk ST, Burwell T, Bertin J, Coyle AJ. Sensing intracellular pathogens-NOD-like receptors. Curr Opin Pharmacol. 2008;8:261–266. doi: 10.1016/j.coph.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 10.Hood JD, Warshakoon HJ, Kimbrell MR, Shukla NM, Malladi S, Wang X, David SA. Immunoprofiling toll-like receptor ligands: Comparison of immunostimulatory and proinflammatory profiles in ex vivo human blood models. Hum Vaccin. 2010;6:1–14. doi: 10.4161/hv.6.4.10866. [DOI] [PubMed] [Google Scholar]
- 11.Warshakoon HJ, Hood JD, Kimbrell MR, Malladi S, Wu WY, Shukla NM, Agnihotri G, Sil D, David SA. Potential adjuvantic properties of innate immune stimuli. Hum Vaccin. 2009;5:381–394. doi: 10.4161/hv.5.6.8175. [DOI] [PubMed] [Google Scholar]
- 12.Shukla NM, Malladi SS, Mutz CA, Balakrishna R, David SA. Structure-activity relationships in human toll-like receptor 7-active imidazoquinoline analogues. J Med Chem. 2010;53:4450–4465. doi: 10.1021/jm100358c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu W, Li R, Malladi SS, Warshakoon HJ, Kimbrell MR, Amolins MW, Ukani R, Datta A, David SA. Structure-activity relationships in toll-like receptor-2 agonistic diacylthioglycerol lipopeptides. J Med Chem. 2010;53:3198–3213. doi: 10.1021/jm901839g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chedid L, Audibert F, Johnson AG. Biological activities of muramyl dipeptide, a synthetic glycopeptide analogous to bacterial immunoregulating agents. Prog Allergy. 1978(25):63–105. [PubMed] [Google Scholar]
- 15.Chedid L, Lederer E. Past, present and future of the synthetic immunoadjuvant MDP and its analogs. Biochem Pharmacol. 1978;27:2183–2186. doi: 10.1016/0006-2952(78)90074-6. [DOI] [PubMed] [Google Scholar]
- 16.Chedid L, Carelli C, Audibert F. Recent developments concerning muramyl dipeptide, a synthetic immunoregulating molecule. J Reticuloendothel Soc. 1979;26:631–641. [PubMed] [Google Scholar]
- 17.Adam A, Petit JF, Lefrancier P, Lederer E. Muramyl peptides. Chemical structure, biological activity and mechanism of action. Mol Cell Biochem. 1981;41:27–47. doi: 10.1007/BF00225295. [DOI] [PubMed] [Google Scholar]
- 18.Warren HS, Vogel FR, Chedid LA. Current status of immunological adjuvants. Annu Rev Immunol. 1986;4:369–388. doi: 10.1146/annurev.iy.04.040186.002101. [DOI] [PubMed] [Google Scholar]
- 19.Lise LD, Audibert F. Immunoadjuvants and analogs of immunomodulatory bacterial structures. Curr Opin Immunol. 1989;2:269–274. doi: 10.1016/0952-7915(89)90199-4. [DOI] [PubMed] [Google Scholar]
- 20.Girardin SE, Philpott DJ. Mini-review: the role of peptidoglycan recognition in innate immunity. Eur J Immunol. 2004;34:1777–1782. doi: 10.1002/eji.200425095. [DOI] [PubMed] [Google Scholar]
- 21.Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 22.Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, Ogura Y, Kawasaki A, Fukase K, Kusumoto S, Valvano MA, Foster SJ, Mak TW, Nunez G, Inohara N. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4:702–707. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- 23.Izumi S, Nakahara K, Gotoh T, Hashimoto S, Kino T, Okuhara M, Aoki H, Imanaka H. Antitumor effects of novel immunoactive peptides, FK-156 and its synthetic derivatives. J Antibiot (Tokyo) 1983;36:566–574. doi: 10.7164/antibiotics.36.566. [DOI] [PubMed] [Google Scholar]
- 24.Mine Y, Yokota Y, Wakai Y, Fukada S, Nishida M, Goto S, Kuwahara S. Immunoactive peptides, FK-156 and FK-565. I. Enhancement of host resistance to microbial infection in mice. J Antibiot (Tokyo) 1983;36:1045–1050. doi: 10.7164/antibiotics.36.1045. [DOI] [PubMed] [Google Scholar]
- 25.Yokota Y, Mine Y, Wakai Y, Watanabe Y, Nishida M, Goto S, Kuwahara S. Immunoactive peptides, FK-156 and FK-565. II. Restoration of host resistance to microbial infection in immunosuppressed mice. J Antibiot (Tokyo) 1983;36:1051–1058. doi: 10.7164/antibiotics.36.1051. [DOI] [PubMed] [Google Scholar]
- 26.Mine Y, Watanabe Y, Tawara S, Yokota Y, Nishida M, Goto S, Kuwahara S. Immunoactive peptides, FK-156 and FK-565. III. Enhancement of host defense mechanisms against infection. J Antibiot (Tokyo) 1983;36:1059–1066. doi: 10.7164/antibiotics.36.1059. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe Y, Tawara S, Mine Y, Kikuchi H. Immunoactive peptides, FK 156 and FK 565. IV. Activation of mouse macrophages. J Antibiot (Tokyo) 1985;38:1781–1787. doi: 10.7164/antibiotics.38.1781. [DOI] [PubMed] [Google Scholar]
- 28.Fujimoto Y, Inamura S, Kawasaki A, Shiokawa Z, Shimoyama A, Hashimoto T, Kusumoto S, Fukase K. Chemical synthesis of peptidoglycan fragments for elucidation of the immunostimulating mechanism. J Endotoxin Res. 2007;13:189–196. doi: 10.1177/0968051907080739. [DOI] [PubMed] [Google Scholar]
- 29.Hasegawa M, Kawasaki A, Yang K, Fujimoto Y, Masumoto J, Breukink E, Nunez G, Fukase K, Inohara N. A role of lipophilic peptidoglycan-related molecules in induction of Nod1-mediated immune responses. J Biol Chem. 2007;282:11757–11764. doi: 10.1074/jbc.M700846200. [DOI] [PubMed] [Google Scholar]
- 30.Wolfert MA, Roychowdhury A, Boons GJ. Modification of the structure of peptidoglycan is a strategy to avoid detection by nucleotide-binding oligomerization domain protein 1. Infect Immun. 2007;75:706–713. doi: 10.1128/IAI.01597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roychowdhury A, Wolfert MA, Boons GJ. Synthesis and proinflammatory properties of muramyl tripeptides containing lysine and diaminopimelic acid moieties. Chembiochem. 2005;6:2088–2097. doi: 10.1002/cbic.200500181. [DOI] [PubMed] [Google Scholar]
- 32.Fritz JH, le Bourhis L, Sellge G, Magalhaes JG, Fsihi H, Kufer TA, Collins C, Viala J, Ferrero RL, Girardin SE, Philpott DJ. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity. 2007;26:445–459. doi: 10.1016/j.immuni.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Shukla NM, Mutz CA, Ukani R, Warshakoon HJ, Moore DS, David SA. Syntheses of fluorescent imidazoquinoline conjugates as probes of Toll-like receptor 7. Bioorg Med Chem Lett. 2010;20:6384–6386. doi: 10.1016/j.bmcl.2010.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masumoto J, Yang K, Varambally S, Hasegawa M, Tomlins SA, Qiu S, Fujimoto Y, Kawasaki A, Foster SJ, Horie Y, Mak TW, Nunez G, Chinnaiyan AM, Fukase K, Inohara N. Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J Exp Med. 2006;203:203–213. doi: 10.1084/jem.20051229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schleifer KH, Kandler O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev. 1972;36:407–477. doi: 10.1128/br.36.4.407-477.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen TB, Kumar EV, Sil D, Wood SJ, Miller KA, Warshakoon HJ, Datta A, David SA. Controlling plasma protein binding: structural correlates of interactions of hydrophobic polyamine endotoxin sequestrants with human serum albumin. Mol Pharm. 2008;5:1131–1137. doi: 10.1021/mp8001123. [DOI] [PubMed] [Google Scholar]
- 37.Ekman AK, Cardell LO. The expression and function of Nod-like receptors in neutrophils. Immunology. 2010;130:55–63. doi: 10.1111/j.1365-2567.2009.03212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimbrell MR, Warshakoon H, Cromer JR, Malladi S, Hood JD, Balakrishna R, Scholdberg TA, David SA. Comparison of the immunostimulatory and proinflammatory activities of candidate Gram-positive endotoxins, lipoteichoic acid, peptidoglycan, and lipopeptides, in murine and human cells. Immunol Lett. 2008;118:132–141. doi: 10.1016/j.imlet.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 40.Davis HM, Carpenter DC, Stahl JM, Zhang W, Hynicka WP, Griswold DE. Human granulocyte CD11b expression as a pharmacodynamic biomarker of inflammation. J Immunol Methods. 2000;240:125–132. doi: 10.1016/s0022-1759(00)00183-6. [DOI] [PubMed] [Google Scholar]
- 41.Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–979. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- 42.Gallagher G, Eskdale J, Jordan W, Peat J, Campbell J, Boniotto M, Lennon GP, Dickensheets H, Donnelly RP. Human interleukin-19 and its receptor: a potential role in the induction of Th2 responses. Int Immunopharmacol. 2004;4:615–626. doi: 10.1016/j.intimp.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Liao SC, Cheng YC, Wang YC, Wang CW, Yang SM, Yu CK, Shieh CC, Cheng KC, Lee MF, Chiang SR, Shieh JM, Chang MS. IL-19 induced Th2 cytokines and was up-regulated in asthma patients. J Immunol. 2004;173:6712–6718. doi: 10.4049/jimmunol.173.11.6712. [DOI] [PubMed] [Google Scholar]
- 44.Oral HB, Kotenko SV, Yilmaz M, Mani O, Zumkehr J, Blaser K, Akdis CA, Akdis M. Regulation of T cells and cytokines by the interleukin-10 (IL-10)-family cytokines IL-19, IL-20, IL-22, IL-24 and IL-26. Eur J Immunol. 2006;36:380–388. doi: 10.1002/eji.200425523. [DOI] [PubMed] [Google Scholar]
- 45.Gallagher G. Interleukin-19: Multiple roles in immune regulation and disease. Cytokine Growth Factor Rev. 2010 doi: 10.1016/j.cytogfr.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 46.Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001;410:1103–1107. doi: 10.1038/35074114. [DOI] [PubMed] [Google Scholar]
- 47.Nathan C, Ding A. TREM-1: a new regulator of innate immunity in sepsis syndrome. Nat Med. 2001;7:530–532. doi: 10.1038/87846. [DOI] [PubMed] [Google Scholar]
- 48.Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol. 2006;7:1266–1273. doi: 10.1038/ni1411. [DOI] [PubMed] [Google Scholar]
- 49.Klesney-Tait J, Colonna M. Uncovering the TREM-1-TLR connection. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1374–L1376. doi: 10.1152/ajplung.00415.2007. [DOI] [PubMed] [Google Scholar]
- 50.Bleharski JR, Kiessler V, Buonsanti C, Sieling PA, Stenger S, Colonna M, Modlin RL. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J Immunol. 2003;170:3812–3818. doi: 10.4049/jimmunol.170.7.3812. [DOI] [PubMed] [Google Scholar]
- 51.Shukla NM, Kimbrell MR, Malladi SS, David SA. Regioisomerism-dependent TLR7 agonism and antagonism in an imidazoquinoline. Bioorg Med Chem Lett. 2009;19:2211–2214. doi: 10.1016/j.bmcl.2009.02.100. [DOI] [PubMed] [Google Scholar]
- 52.Miller KA, Suresh Kumar EVK, Wood SJ, Cromer JR, Datta A, David SA. Lipopolysaccharide Sequestrants: Structural Correlates of Activity and Toxicity in Novel Acylhomospermines. J Med Chem. 2005;48:2589–2599. doi: 10.1021/jm049449j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.