

Abstract
Whole genome sequencing, also known as deep sequencing, is becoming a more affordable and efficient way to identify SNP mutations, deletions and insertions in DNA sequences across several different strains. Two major obstacles preventing the widespread use of deep sequencers are the costs involved in services used to prepare DNA libraries for sequencing and the overall accuracy of the sequencing data. This Unit describes the preparation of DNA libraries for multiplexed paired-end sequencing using the Illumina GA series sequencer. Self-preparation of DNA libraries can help reduce overall expenses, especially if optimization is required for the different samples, and use of the Illumina GA Sequencer can improve the quality of the data.
Keywords: Multiplexed, Paired-end, Deep sequencing, Illumina GA, DNA Library
Introduction
Proper identification of new strains and isolates, including important clinical isolates, often requires more than just phenotypic characterization. Important base mutations, sequence deletions or repeats, and even sequence insertions, can go unnoticed from routine assays and have broader implications than one may realize. With the sequencing of the first bacteriophage genome in 1977(Sanger et al. 1977a)and the first bacterial genome in 1995 (Fleischmann et al. 1995), whole genome sequencing (deep sequencing) has become an increasingly popular tool (Borman et al. 2008; Brinkhoff et al. 2008; Bansal et al. 2010; Rounsley and Last 2010; Szpara et al. 2010). With the development of automation and high-throughput machines, such as the Illumina GA-II, deep sequencing can not only be used to help identify previously unknown genetic differences (Bentley et al. 2008; Borman et al. 2008; Brinkhoff et al. 2008; Bansal et al. 2010; Rounsley and Last 2010; Szpara et al. 2010)to shed insight into unexplainable phenotypes, but also provide sequence data to help establish evolutionary drift between or across related strains(Sapp 2007; Brinkhoff et al. 2008)with the appropriate analysis software(Blankenberg et al. 2007; Blankenberg et al. 2010; Milne et al. 2010), all at a relatively short turn-around time and significantly reduced costs.
Although there are commercially available services that can provide the deep sequencing data for varying fees, one can avoid these unnecessary costs by preparing your own DNA libraries that only need to be sequenced. This is especially useful for those that are conducting deep sequencing on a small number of samples with a limited budget. Most laboratories already carry all the components necessary for DNA library construction and would only need to purchase a few of the commercially available reagents and oligos.
The following protocol is adapted from the protocol used by the Illumina GA-II deep sequencer and is designed to help generate a bacterial DNA library that is labeled with one indexed tag. Subsequent use of alternate tags (up to 12) in individual reactions will allow the user to perform multiplexed paired-end sequencing to further reduce sequencing costs and increase efficiency. Each section results in a sample that is then purified and carried over as the starting material in the subsequent section. With a starting material of good quality, clean chromosomal DNA, the user will fragment the DNA using a nebulizer, into a broad range of fragment sizes, with the majority of the fragments between 200 to 800 bp. This fragmented DNA will then be blunt-ended in the second step, followed by the addition of a 3′ adenine, which will then be used to ligate adapter fragments. Using previously designed PCR oligos, the adapter-modified fragments will then be enriched using PCR. This end product, DNA library, is then purified and is now ready to be loaded onto the Illumina GA-II sequencer.
Overall Protocol
Introduction
Preparation of good quality, clean chromosomal DNA is required for efficient DNA library construction. It is good practice to prepare a concentrated stock of the chromosomal DNA, as it is always easier to dilute the DNA than to concentrate it. To obtain good quality clean chromosomal DNA, any commercially available chromosomal DNA preparation kit can be used. The DNA should be resuspended in an EDTA-free and alcohol-free resuspension buffer, as both components will reduce the overall efficiency of DNA library construction.
This protocol is adapted from Illumina’s paired-end multiplexing protocol and is designed for sequencing using the Illumina GA-II sequencer. The overall protocol starts with purified chromosomal DNA (A260/280 = 1.8 – 2.0) at a concentration of 200 – 400 ng/μl. This chromosomal DNA can be sheared using a nebulizer (Sambrook and Russell 2001)to obtain fragment sizes ranging from 200 to 800 bp, which are then blunt-ended and modified with an addition of a 3′ adenine(A). The modified ends are then used to ligate an adapter and the subsequent adapter-modified DNA fragments are then enriched for using standard PCR. This enriched PCR product is now called the DNA library and can be purified and aliquoted for loading onto the Illumina GA-II sequencer.
Basic Protocol 1 – Chromosomal DNA Purification and Fragmentation
Chromosomal DNA purification can be performed using any established protocol or commercially available kit. Because the purity and quantity of chromosomal DNA is critical for maximum efficiency during DNA library preparation, quality and yield should be measured following isolation to verify the sample is free of RNA and other contaminants. This protocol describes expected yields of chromosomal DNA following isolation using a commercially available kit and following manufacturer’s recommendations.
The first step in DNA library preparation for deep sequencing is chromosomal DNA fragmentation followed by subsequent purification of those double stranded fragments. Fragmentation of the DNA to the appropriate size range is required not only to ensure maximal adapter ligation efficiency, but more importantly because of the current limitations Illumina deep sequencers have in the length of accurate reads that can be generated. This protocol describes the steps involved in fragmenting and isolating the chromosomal DNA. Although the use of a nebulizer (or any other procedure) to fragment DNA to the proper size range requires some empirical determination, especially in terms of the time required for fragmentation, some approximate times are given as a rough guide. A small amount of the fragmented sample can be visualized on a gel to serve as a checkpoint prior to proceeding (see Critical Parameters and Troubleshooting).
Materials
Overnight cultures
QIAprep Spin Miniprep Kit (Qiagen Cat. #27104)
NanoDrop spectrophotometer(NanoDrop Model #ND100)
200 – 400 ng/μl Chromosomal DNA (A260/280= 1.8 – 2.0)
TE buffer (see recipe)
Nebulization buffer (see recipe)
Low-melt agarose (BRL Cat. #5517UB)
1X TBE (see recipe)
Ice and ice bucket
2.0 ml sterile microfuge tubes (USA Scientific Cat. #1620-2700)
Nebulizer (Invitrogen Cat. #K7025-05)
Vacuum tubing (Tygon Model #R3603)
Worm drive hose clamp that fits the vacuum tubing
Agarose gel electrophoresis set-up
QIAquick PCR Purification kit (Qiagen Cat. #28104)
-
Grow up cultures and isolate chromosomal DNA using established methods/kits.
Resuspend the DNA in the resuspension buffer provided in the kit.
Ensure high quality, clean DNA with A260/280 between 1.8 and 2.0.
-
Quantitate the amount of DNA using the NanoDrop ND100 spectrophotometer, using the resuspension buffer as a blank.
From a 2 ml overnight culture, the DNA resuspended in 150 μl of buffer can be expected to yield a concentration of 200 – 400 ng/μl, with an A260/280 around 1.87.
Aliquot 5 μg of resuspended and purified DNA into a 2 ml microfuge tube and bring the final volume to 50 μl with TE buffer.
Add 700 μl of Nebulization Buffer and mix well, minimizing air bubbles, and transfer the entire mixture to a nebulizer.
Chill the sample in the nebulizer on ice for 5 min.
Place the ice bucket and nebulizer in a fume hood, turn on the fume hood, and connect the nebulizer and the air valve with the vacuum tubing.
Secure the tubing with a worm drive hose clamp.
-
Nebulize the sample until a majority of the fragments are ≤800 bp.
The nebulization time must be determined empirically with the same sample to be prepared for sequencing. This must be done ahead of time by nebulizing purified chromosomal DNA for varying amounts of time (e.g. 1 min, 2 min, 5 min, 10 min, 15 min, 20 min) and running the different nebulized samples on an agarose gel until the majority of the smear is in the desired fragment size range.
Extreme heat potentially generated by nebulization may result in denaturation of smaller fragments and/or de-purination of DNA, and thus, processing on ice is recommended.
At this point, to increase efficiency of subsequent reactions, the fragmented sample can be run on a 2% low-melt agarose gel, and the 200 – 800 bp smear cut out and purified using the QIAquick PCR Purification Kit, and eluted in 30 μl of QIAGEN EB Buffer, thus eliminating unwanted fragments from being further processed and wasting reagents.
Proceed to End Repair of Fragmented DNA.
Basic Protocol 2 – End Repair of Fragmented DNA
The fragmentation of the DNA is a random process and therefore will generate DNA pieces with varying 5′ and 3′ overhangs and blunt-ends in all possible combinations (see Figure 1). However, for the eventual ligation of adapters to the fragments requires the generation of 3′ adenine (A) base overhangs. This protocol describes the end repair of all the DNA fragments to make them blunt-ended using an enzyme mix containing T4 DNA polymerase, Klenow enzyme, and T4 polynucleotide kinase, in preparation for 3′ modification with the addition of an A base.
Figure 1.

Overview of DNA library preparation for deep sequencing. The purified genomic DNA is processed and modified as described in the basic protocols. The adapter-modified DNA fragments (library) are then enriched through PCR. Note the relative positions and characteristics (Inset) of the InPE1.0, InPE2.0 and the Index primers used during PCR. For multiplexed paired-end PCR, the same InPE1.0 and InPE2.0 primers are used, however, up to 12 different Index primers can be added to the enrichment reactions individually. Sequencing is performed in one direction through sequencing by synthesis, and then in the alternate direction as described in the background information. The complementary sequences are shaded darker.
Materials
Fragmented DNA sample
TE buffer (see recipe)
Qiagen EB buffer (Qiagen Cat. #19086)
T4 DNA Ligase buffer mix (see recipe)
Enzyme master mix (see recipe)
Sterile double-distilled water (nuclease-free)
200 μl sterile thin-walled PCR tubes (Corning Cat. #6571)
Nitrocellulose filter (Millipore Cat. #VSWP02500)
Petri dish (Fisher Cat. #0875712)
PCR thermal cycler to hold 200 μl PCR tubes with heated lid
Vacuum dryer centrifuge (Jouan Model #RC10.10)
-
Prepare the following reaction mix on ice in a 200 μl thin-walled PCR tube:
30 μl (up to 5 ng) Fragmented DNA sample 15 μl T4 DNA Ligase Buffer Master Mix (T4 DNA Ligase Buffer containing 10 mM ATP, 10 mM dNTP Mix) 1 μl Enzyme Master Mix (T4 DNA Polymerase, Klenow Enzyme, T4 PNK) 4 μl Water 50 μl Total Volume Incubate the reaction mix at 20°C for 30 min in a PCR thermal cycler.
Heat-inactivate the reaction mix at 75°C for 20 min.
-
Spot dialyze the reaction mix for 45 min.
Fill a Petri dish with 25 ml of TE Buffer.
Float a nitrocellulose filter on the buffer.
Using a pipettor, carefully add the reaction mix to the center of the filter and allow to dialyze for 45 min.
Carefully recover as much of the reaction mix off of the filter as possible using a pipettor and dry down the sample in vacuum dryer centrifuge.
Resuspend the pellet in 32 μl of QIAGEN EB Buffer.
Proceed to 3′ End Modification.
Basic Protocol 3 – 3′ End Modification: Addition of ‘A’ Bases to the 3′ End of the DNA Fragments
As mentioned previously, the ligation of the adapters used in both library enrichment and the actual sequencing process requires the presence of a 3′ A base on the double stranded DNA fragments. This procedure is described in the following protocol, using the 3′ end repaired double stranded DNA fragments generated in Basic Protocol 2.
Materials
End repaired DNA sample
TE buffer (see recipe)
Qiagen EB buffer (Qiagen Cat. #19086)
1 mM dATP (NEB Cat. #M0440S)
Klenow buffer (NEB Cat. #M0212S)
Klenow Fragment (3′ to 5′ exo minus) (NEB Cat. #M0212S)
PCR thermal cycler to hold 200 μl PCR tubes with heated lid
Petri dish (Fisher Cat. #0875712)
Nitrocellulose filter (Millipore Cat. #VSWP02500)
Vacuum dryer centrifuge (Jouan Model #RC10.10)
-
Prepare the following reaction mix on ice in the same tube containing the End Repaired DNA sample:
32 μl End Repaired DNA sample 5 μl Klenow Buffer 10 μl 1 mM dATP 3 μl Klenow Fragment (3′ to 5′ exo minus) 50 μl Total Volume Incubate the reaction mix at 37°C for 30 min in a PCR thermal cycler.
Heat-inactivate the reaction mix at 75°C for 20 min.
Spot dialyze the reaction mix for 45 min in TE Buffer as before.
Carefully recover as much of the reaction mix off of the filter as possible and dry down the sample in vacuum dryer centrifuge.
Resuspend the pellet in 10 μl of QIAGEN EB Buffer.
Proceed to Adapter Ligation.
Basic Protocol 4 – Adapter Ligation
The sequencing of the DNA library requires the ligation of special adapters to the modified DNA fragments. These adapters are not only required for the final step in library preparation of enrichment, but for the actual sequencing of the fragments by the sequencers. The actual sequences of the adapters are proprietary (Illumina), however, the basic strategy of how the adapters are incorporated is outlined in Figure 1. These adapters will be used in the proprietary sequencing by synthesis reactions once loaded on the Illumina GA sequencer. The adapter-ligated products can be visualized on a gel to serve as another checkpoint prior to proceeding with enrichment (see Critical Parameters and Troubleshooting).
Materials
3′ Modified DNA sample
T4 DNA ligase buffer (NEB Cat. #M0202S)
Index PE Adapter oligo mix (Illumina Cat. #PE400-1001)
T4 DNA ligase (NEB Cat. #M0202S)
Low-melt agarose (BRL Cat. #5517UB)
1X TBE (see recipe)
2.0 ml sterile microfuge tubes (USA Scientific Cat. #1620-2700)
Agarose gel electrophoresis set-up
Transilluminator (Clare Chemical Research Model #DR-45M)
QIAquick Gel Extraction kit (Qiagen Cat. #28704)
NanoDrop spectrophotometer(NanoDrop Model #ND100)
-
Prepare the following reaction mix on ice:
10 μl 3′ End Modified DNA sample 3.1 μl T4 DNA Ligase Buffer 10X 10 μl Index PE Adapter Oligo Mix 1 μl T4 DNA Ligase 6.9 μl Sterile double-distilled water 31 μl Total Volume Incubate the reaction mix at 20°C for 30 min in a PCR thermal cycler.
-
Add 5 μl of the gel loading dye to the reaction sample and load the entire mix onto a 2% low-melt agarose gel.
Leave an empty lane between the marker and the first sample, and separate all samples by one lane to prevent carry-over contamination.
-
Quickly place the gel on a transilluminator and carefully excise the smear between 300 – 500 bp.
Note: The size range of the input DNA fragments was 200 to 800 bp, but a 300 to 500 bp range is excised here. The lower end of the range is increased by 100 bp because of the ligation of the adapters. The higher end of the range is reduced to 500 bp because of the length of the sequencing reads. Sequencing is conducted from both ends in paired-end sequencing and the length of the reads is currently limited to 150 bp using Illumina GA deep sequencers. Limiting the size of the fragments to 500 bp with adapters allows almost complete sequencing of the fragment from both 5′ and 3′ ends. If the sizes of the fragments are too large (e.g. > 600 bp) then after sequencing from both ends, there will still be a large section of the fragment that is not sequenced.
Place the gel slice in a sterile 2.0 ml microfuge tube.
Purify the DNA from the 2% agarose gel using QIAquick Gel Extraction Kit and elute sample in 30 μl of QIAGEN EB Buffer.
Quantitate amount of DNA as before using the NanoDrop ND100.
Proceed to Enrichment of Adapter-Modified DNA Fragments by PCR.
Basic Protocol 5 – Enrichment of Adapter-Modified DNA Fragments by PCR
The final step in DNA library preparation is the enrichment of the adapter-modified fragments. Efficient enrichment ensures a higher probability for greater fold coverage simply because there are more fragments to sequence. This protocol details the PCR reaction and the primers used to enrich the fragments prior to loading onto the sequencers. Note that there are three primers, InPE1.0, InPE2.0 and an Index primer, added to this PCR enrichment reaction; the sense of each primer (sense or anti-sense) should also be noted (see Figure 1). Using the twelve (12) Index primers individually, up to 12 enrichment reactions can be performed simultaneously and loaded together for multiplexed paired-end deep sequencing of up to twelve (12) different genomes. Gel analysis a small aliquot of the enriched product can be used as a final checkpoint prior to sequencing as described in the Critical Parameters and Troubleshooting section.
Materials
Adapter-ligated DNA sample
PCR Primer InPE1.0 (Illumina Cat. #PE400-1001)
PCR Primer InPE2.0 (Illumina Cat. #PE400-1001)
PCR Index primer (Illumina Cat. #PE400-1001)
5x HF Phusion buffer (NEB Cat. #F-540S)
10 mM dNTP (NEB Cat. #N0447S)
Phusion DNA polymerase (NEB Cat. #F-540S)
6X gel loading dye with Gel Green (see recipe)
100 bp Marker (NEB Cat. #N3231S)
1X TBE (see recipe)
5% TBE DNA acrylamide gel (BioRad Cat. #161-1109)
Sterile double-distilled water (nuclease-free)
PCR thermal cycler to hold 200 μl PCR tubes with heated lid
DNA acrylamide gel electrophoresis set-up
QIAquick PCR Purification kit (Qiagen Cat. #28104)
NanoDrop spectrophotometer (NanoDrop Model #ND100)
-
Prepare the following reaction mix on ice in a new 200 μl PCR tube, making sure that the Phusion DNA polymerase is the last component to be added to the mix:
-
The PCR Index Primer used should be different for each strain DNA library constructed (e.g. strain 1 – Index Primer 1; strain 2 – Index Primer 2; strain 3 – Index Primer 3; etc)
175 ng Adapter-Ligated DNA sample 1 μl PCR Primer InPE1.0 for fragment amplification 1 μl PCR Primer InPE2.0 for fragment amplification 1 μl PCR Index Primer 10 μl 5X HF Phusion Buffer 1 μl 10 mM dNTP 0.5 μl Phusion DNA Polymerase up to 50 μl Sterile Double-Distilled Water 50 μl Total Volume
-
-
Amplify using the following PCR protocol:
30 sec at 98°C
-
18 cycles of:
10 sec at 98 °C
30 sec at 65 °C
30 sec at 72 °C
5 min at 72 °C
Hold at 4 °C until ready to proceed with next step.
To verify that the PCR worked, load 5 μl of the uncleaned PCR reaction and 5 μl of the adapter-ligation mix from the previous section on a 5% TBE DNA acrylamide gel.
-
Run the DNA acrylamide gel.
There should be an increase in the intensity of the PCR band/smear as compared to the adapter-ligation mix, as well as a shift in the size range of the smear because of the adapters and PCR primers(see Figure 2).
Purify the PCR product using the QIAquick PCR Purification Kit and elute the DNA library sample in 50 μl of QIAGEN EB Buffer.
-
Quantitate the amount of DNA as before using the NanoDrop ND100.
Yield typically ranges around 1200 ng.
Aliquot ~150 ng of each prepared DNA library sample for sequencing on the Illumina GA-II deep sequencer.
Store the rest of the library stocks at −20°C until needed.
Figure 2.
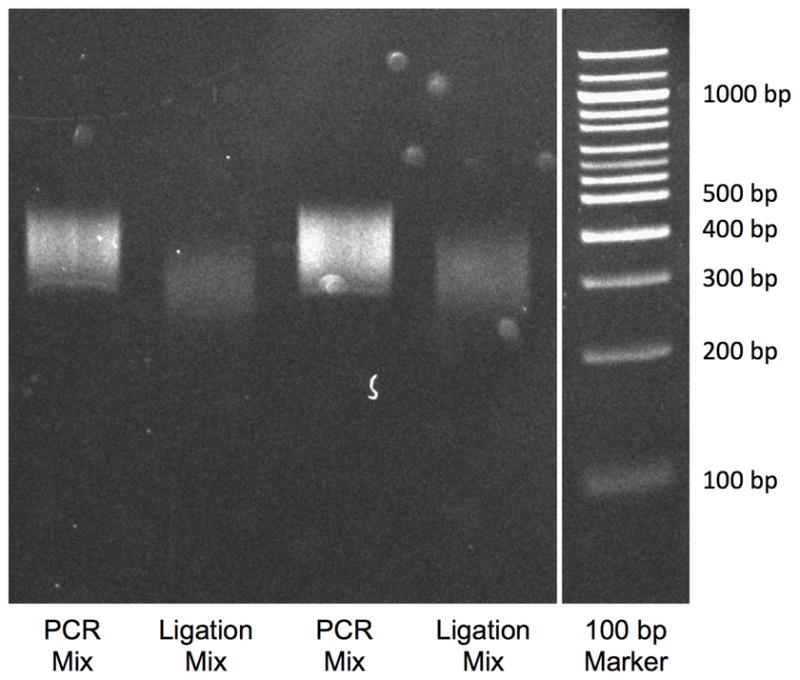
Fragmented DNA smearing patterns after ligation of adapters (Ligation Mix) and after PCR (PCR Mix). Note increase in smear intensity as well as a shift up in the size range of the smear.
Reagents and Solutions
-
TE Buffer (1000 ml)
1 M Tris-HCl (pH 8.0) – 10.0 ml(Final Concentration 10 mM)
0.5 M EDTA (pH 8.0) – 0.2 ml(Final Concentration 0.1 mM)
Sterile double-distilled water – up to 1000 ml
-
1X TBE (Tris-borate-EDTA) Buffer (1000 ml)
Tris base – 5.4 g(Final Concentration 44.58 mM)
Boric acid – 2.75 g(Final Concentration 44.48 mM)
0.5 M EDTA (pH 8.0) – 2.0 ml(Final Concentration 0.1 mM)
Sterile double-distilled water – up to 1000 ml
-
Nebulization Buffer (50 ml)
100% Glycerol – 2.475 ml(Final Concentration 4.95% v/v)
1 M Tris-HCl (pH 8.0) – 0.5 ml(Final Concentration 10 mM)
0.5 M EDTA (pH 8.0) – 0.1 ml(Final Concentration 1 mM)
Sterile double-distilled water – up to 50.0 ml
-
Enzyme Master Mix(11 μl) – add 1 μl per End Repair reaction
400,000 units/ml T4 DNA ligase – 5.0 μl (2,000 units) (NEB Cat. #M0202S)
5,000 units/ml Klenow Fragment – 1.0 μl(5 units) (NEB Cat. #M0210S)
10,000 units/ml T4 PNK – 5.0 μl(50 units) (NEB Cat. #M0201S)
-
T4 DNA Ligase Buffer Mix (15 μl)
10X T4 DNA ligase buffer containing 10 mM ATP – 10.0 μl (Final Concentration 6.67X) (NEB Cat. #M0202S)
10 mM dNTP – 4.0 μl(Final Concentration 2.67 mM) (NEB Cat. #N0447S)
Sterile double-distilled water – up to 15 μl
-
6X Loading Dye (20 ml)
Bromophenol Blue – 0.05 g(Final Concentration 0.075 mM)
100% Glycerol – 6.0 ml(Final Concentration 30% v/v)
0.5 M EDTA (pH 8.0) – 40.0 μl(Final Concentration 1 mM)
1 M Tris-HCl (pH 8.0) – 1.0 ml(Final Concentration 50 mM)
90X Gel Green – 2.0 ml (Final Concentration 9X) (Biotium Cat. #41005-0.5 ml)
Sterile double-distilled water – up to 20.0 ml
Commentary
Background Information
First developed by Maxam and Gilbert (Maxam and Gilbert 1977) and Sanger (Sanger et al. 1977a; Sanger et al. 1977b)independently in 1977, DNA sequencing has come a long way. Most scientists are familiar with sequencing at a specific target gene level with a specifically designed set of primers. Although this technique is well established (Sambrook and Russell 2001) and has allowed for the advancement of genetic manipulation and the discovery of various polymorphisms (Borman et al. 2008; Brinkhoff et al. 2008; Bansal et al. 2010; Rounsley and Last 2010; Szpara et al. 2010), it had generated a demand for high-throughput protocols and techniques. Recent advancements in technology in conjunction with the basic ideas behind sequencing and shotgun sequencing (Rounsley and Last 2010), has resulted in the introduction of automated high-throughput sequencing. The first of its kind was the 454 Sequencer (Roche), which miniaturized and created a high-throughput version of Sanger sequencing and laid the groundwork for the development of other systems, such as the Illumina GA Sequencer series.
Two major considerations in deep sequencing are costs and coverage, including the total number of reads. Costs of deep sequencing can include DNA library construction, which includes all the reagents and consumables, plus optimizations involving repeated trials of library construction, as well as costs of loading the libraries on the flow cell and running the actual sequencing reactions. Coverage, which includes the total number of reads generated by the sequencer, is a major consideration in determining the quality or reliability of the data, which can ultimately determine which sequencing machine you will choose. The reads are the short sequences of DNA that are generated by the sequencer. The longer the reads are, the easier and more reliably the entire DNA sequence can be pieced together, similar to shotgun sequencing. The coverage is the redundancy of the reads, or in other words, the number of times the same sequence is generated by another piece of the DNA library. So if a particular stretch of sequence is said to have 50-fold coverage, it would mean that that stretch of sequence has been generated by 50 different reads. Intuitively, if a sequence has been verified by more reads, and all the reads show the exact same sequence, then it is more likely that the sequence is correct and any single miscalled base in a single read can be safely assumed to be due to PCR amplification error and not a true base mutation in the sequence. Alternatively, if a sequence has 2-fold coverage, and the two reads have differing bases at any given position, one would not be able to confidently determine if it is a PCR artifact or a true base mutation.
The efficiency and quality of data from deep sequencing is constantly being improved, especially with the Illumina GA Sequencer series. Initially, the GA-I made 2 major advancements over the existing and popularly used 454 Sequencer (Roche): 1) a significant drop in costs of upwards of 30X, and 2) an increase in the depth of sequence coverage and up to 150X more reads. In addition to these two improvements, the new GA-II series has also increased the length of the reads, reaching upwards of 150 bp, and the costs have been additionally reduced with the ability to multiplex with paired-end sequencing. Paired-end sequencing has also improved the quality of the reads. Paired-end sequencing is the process of using a pair of primers or oligos to label both the 3′ and 5′ ends of each fragment in the DNA library. The sequencer generates a read from the 3′ end, and then another read from the 5′ end, effectively double-checking the sequence for each fragment. Multiplexing sequencing reactions is very much akin to multiplexing real-time PCR. Up to 12 different Index Primers are used to label the 12 different individually prepared DNA libraries. These 12 different Index Primers can then be used to separate all the reads generated from the sequencer. Multiplexing allows the user to run up to 12 samples to be sequenced in a single lane of the 7 available lanes of the 8-lane flow cell (one lane is left for a control). This brings the total number of sequences generated from a single flow cell to 84 different sequencing reactions, cutting costs and time. The only major disadvantage to this system is the amount of data generated by the multiplexed paired-end sequencing reactions, especially if the flow cell is run at capacity. One will require a computer system almost solely dedicated to analyzing the sequencing data, a system with a large hard-drive memory (minimum suggestion of 1 TB) and significant processor speeds (minimum suggestion of dual quad-core processors).
The basic flow of Illumina sequencing starts with fragmented DNA. These DNA fragments are then modified at both ends with the addition of adapter sequences, and in the case of multiplex paired-end reactions, 1 of 12 additional Index Primers to prepare the library. This library of adapter-modified DNA fragments are denatured, loaded onto the flow cell, and hybridized to the flow cell. The single stranded library molecules hybridize to complementary oligos already covalently linked to the cell surface to form a single-stranded bridge, and the first round of PCR is performed. The double-stranded bridge is then denatured and the single-stranded molecules experience another round of hybridization and PCR amplification. This process is repeated (in essence, PCR on a solid substrate) and eventually clusters of up to 1000 copies of the template are generated on the surface of the flow cell. At this point, the cluster is made up of a mixture of 3′-end-free (sense) and 5′-end-free (anti-sense) single-stranded molecules. In order to sequence the molecules, one form (e.g. the anti-sense) is removed from the flow cell and washed out of the flow cell using proprietary technology, leaving behind a cluster made up of single-stranded molecules all in the same orientation (e.g. sense strands only).
Then by a process called sequencing by synthesis using proprietary reversible terminator technology, four fluorescently labeled terminators are incorporated, using sequencing primers and DNA polymerase. Since all four labeled dNTPs are present for each reaction, unlike traditional Sanger sequencing where one base is added at a time, any bias or non-specific addition of the wrong base is avoided, resulting in more accurate sequencing reactions. After the addition of each base, a laser excites the fluorescent labels and the color of the cluster of identical templates is read. The terminators are removed, and the next cycle of labeled terminator incorporation is repeated for the designated number of cycles, which determines the length of the reads.
For paired-end sequencing, the same fragments are additionally sequenced from the alternate end by flipping the single-stranded templates into the opposite orientation. This is done by hybridization of the single-stranded molecules to the already existing covalently linked oligos and the entire process of bridge amplification and PCR being repeated. However, this round involves the removal of the opposite strand (e.g. sense) and sequencing is performed as before for the designated number of cycles. Most often, the final reads are assembled by aligning the reads against an existing published sequence, and therefore this is referred to as re-sequencing. Re-sequencing is particularly useful when looking for SNPs and other genetic mutations (e.g. deletions or single base insertions) in different isolates of the same strain, or random mutants. Although de novo sequence assembly can be performed, the length of the individual reads will determine the accuracy of the final assembled genome and the efficiency with which the genome is obtained. In the case of the Illumina GA Sequencer series, the length of the reads is being increased while maintaining the accuracy, making de novo synthesis easier.
Critical Parameters and Troubleshooting
As with any protocol that is being attempted for the first time, optimization may be required for individual samples. When using various kits, all manufacturer recommendations should be followed and any adjustments to optimize yield or quality be made after an initial run if it is required. Although this protocol has been successfully used to generate high quality sequence data from DNA libraries for five different strains after multiplex paired-end sequencing, caution should be used when attempting this modified protocol.
The quality of the starting material will ultimately determine the quality of the final output material, and therefore, a good stock of high quality, clean intact chromosomal DNA as a starting material is crucial. Each subsequent step in the protocol should be processed on ice to prevent unwanted enzyme activity, especially in the final PCR enrichment step. Keeping all samples and enzymes on ice will also insure maximal activity of the enzyme for maximum efficiency of the entire process.
Be advised that successful generation of good quality DNA libraries does not guarantee high quality results, as this library must then be processed on a separate machine handled by another user. Thus, it is highly recommended that, as stated in the protocol, only small aliquots of each sample be given at a time for sequencing.
To maximize quality and to insure the subsequent steps are successful, several points exist that allow for the researcher to check the quality of the samples as they are being processed. After fragmentation of the chromosomal DNA, the nebulized sample can be run on an agarose gel to verify that the majority of the fragmented DNA (visible as a smear) falls in the range of 200 to 800 bp. This portion of the smear can then be excised and purified out of the gel and used in subsequent steps to avoid processing of DNA fragments of the wrong size. A picture of the smear before and after excision is a good reference point for comparison of later samples, such as the adapter-modified sample.
The next checkpoint is after adapter ligation, where the product is then run on a gel and a smaller smear (300 to 500 bp) is excised and purified out of the gel. A quick comparison of this smear should show that the smear has shifted slightly higher (~60 bp) than the fragmented smear, as a result of the addition of the adapters to the DNA fragments. The final checkpoint prior to sequencing can be made comparing a small amount of the adapter-modified sample and the PCR enriched DNA library on a DNA acrylamide gel. As seen in Figure 2, the enrichment of the DNA library results in another shift of the entire smear (~68 bp) because of the addition of sequence on the PCR oligos and the PCR primer indices, as compared to the adapter-modified sample. The intensity of the smear after enrichment should also be brighter, indicative of successful PCR.
Anticipated Results
The final DNA library should be a mix of chromosomal DNA fragments that are approximately 130 bp larger than the original fragment sizes that were processed. These fragments will contain adapters and PCR primers and PCR primer indices. These DNA libraries are now ready to be sequenced on the Illumina GA-II sequencer. It is fully anticipated that the final amount of DNA in the library will be significantly less than the original starting amount, but since only picogram amounts of the library are required for the sequencing run, this should not pose a problem.
Time Considerations
The basic protocol begins with purifying chromosomal DNA for fragmentation. The time required to grow cultures and purify the DNA will vary according to the growth rate of the bacterial strains used and the chromosomal DNA isolation kits used. However, an overnight culture is recommended to ensure a high-density of cells to extract DNA from. DNA fragmentation and purification times can also vary, largely due to the time required for fragmentation (to be determined empirically) and the speed at which the sample is run on the gel for purification of the 200 to 800 bp smear. It should be noted that increasing the voltage during electrophoresis can be used to reduce run time, but should be done with caution. Overall this first step should take around 1.5 hrs, and the purified sample can be stored at 4°C, should a stopping point be required. End repair of the fragmented DNA is fairly straightforward and will require approximately 2 hrs, depending on the availability of a vacuum dryer centrifuge and how long it takes for the sample to completely dry. The dried sample can be stored at 4°C, should a stopping point be required.
Adapter ligation steps will vary depending on the voltage at which the sample is exposed to during electrophoresis. Generally, this step can be completed in approximately 2 hrs and again, the sample can be stored at 4°C, should a stopping point be required.
The final step of PCR enrichment of the library will vary depending on the ramping speed of the PCR thermal cycler used, but should be completed within 45 mins. This final step, including the electrophoresis of the sample on the DNA acrylamide gel for verification and purification using the kit, should be completed in approximately 2 hrs and is ready for sequencing.
Although the overall process from DNA fragmentation to final enriched DNA libraries takes approximately 7.5 hrs, the entire protocol can be broken down into individual steps, at the end of which, the samples can be stored at 4°C, should a stopping point be required.
Acknowledgments
This work was supported by NIAID grants AI025096 and AI039654.
Literature Cited
- Bansal V, Harismendy O, Tewhey R, Murray SS, Schork NJ, Topol EJ, Frazer KA. Accurate detection and genotyping of SNPs utilizing population sequencing data. Genome Res. 2010;20:537–545. doi: 10.1101/gr.100040.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Taylor J, Schenck I, He J, Zhang Y, Ghent M, Veeraraghavan N, Albert I, Miller W, Makova KD, et al. A framework for collaborative analysis of ENCODE data: making large-scale analyses biologist-friendly. Genome Res. 2007;17:960–964. doi: 10.1101/gr.5578007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010;19:19.10.11–21. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borman AM, Linton CJ, Miles S-J, Johnson EM. Molecular identification of pathogenic fungi. J Antimicrob Chemother. 2008;61:i7–12. doi: 10.1093/jac/dkm425. [DOI] [PubMed] [Google Scholar]
- Brinkhoff T, Giebel H-A, Simon M. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch Microbiol. 2008;189:531–539. doi: 10.1007/s00203-008-0353-y. [DOI] [PubMed] [Google Scholar]
- Fleischmann R, Adams M, White O, Clayton R, Kirkness E, Kerlavage A, Bult C, Tomb J, Dougherty B, Merrick J. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci USA. 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. Tablet--next generation sequence assembly visualization. Bioinformatics. 2010;26:401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rounsley SD, Last RL. Shotguns and SNPs: how fast and cheap sequencing is revolutionizing plant biology. Plant J. 2010;61:922–927. doi: 10.1111/j.1365-313X.2009.04030.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2001. [Google Scholar]
- Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes JC, Hutchison CA, III, Slocombe PM, Smith M. Nucleotide sequence of bacteriophage phiX174 DNA. Nature. 1977a;265:687–695. doi: 10.1038/265687a0. [DOI] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977b;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp J. The structure of microbial evolutionary theory. Stud Hist Philos Biol Biomed Sci. 2007;38:780–795. doi: 10.1016/j.shpsc.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Szpara ML, Parsons L, Enquist LW. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol. 2010;84:5303–5313. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]