

Abstract
Doxorubicin (DOX) is a broad spectrum antineoplastic drug widely used in the treatment of several hematogenous and solid human malignancies. Despite its excellent clinical efficacy as a chemotherapeutic agent, its therapeutic usage has been restricted due to its cardiotoxicity. Phosphodiesterase-5 (PDE-5) inhibitors or erectile dysfunction drugs including sildenafil, have been shown to have powerful cardioprotective effect against injuries under a variety of experimental situations including ischemia/reperfusion injury, myocardial infarction and DOX-induced cardiomyopathy. We studied the effect of – tadalafil, a long acting PDE-5 inhibitor in preventing damage in the heart with DOX treatment. Our results showed that tadalafil improved left ventricular function and survival by attenuating DOX-induced apoptosis and cardiac oxidative stress without interfering with the anti-tumor efficacy of DOX in both in vitro and in vivo tumor models. Herein, we present an overview of our study, and consider the potential mechanisms by which tadalafil, at therapeutically relevant concentrations mediate beneficial cardioprotective effects in DOX cardiotoxicity. Based on our current and previously published studies, we propose that the class of PDE-5 inhibitors can represent a novel approach which can be exploited for achieving therapeutic benefit in the treatment of DOX-induced cardiotoxicity in patients.
Keywords: Doxorubicin, Cardiomyopathy, Phosphodiesterase inhibitors, PKG, cGMP, ROS
Introduction
Doxorubicin (DOX) is an effective antineoplastic drug widely used in the treatment of several hematogenous and solid human malignancies (1, 2). However, despite its therapeutic efficacy, its clinical usage is limited by the development of cumulative dose-dependent cardiomyopathy (3) which may occur many years after the cessation of DOX treatment (4, 5). The acute DOX-induced cardiotoxicity is characterized by hypotension, arrhythmia, tachycardia while the chronic effects are manifested as cardiac dysfunction eventually leading to congestive heart failure (3, 6). The deterioration of cardiac functions is often accompanied with high mortality rates although many patients with ventricular dysfunction may lack overt symptoms until late in their disease.
The exact pathogenesis of DOX-induced cardiotoxicity is still not entirely clear although a diverse set of mechanisms have been proposed, including oxidative stress, mitochondrial DNA damage, intracellular calcium overload, inhibition of protein synthesis, disturbance of myocardial adrenergic function, cytokine release, myofibrillar degeneration and cardiomyocyte apoptosis (7–12). Among the multiple mechanisms, it is widely accepted that DOX-induced cardiomyocyte apoptosis is primarily due to the generation of reactive oxygen species (ROS) in the myocardium which triggers intrinsic mitochondria-dependent apoptotic pathway in cardiomyocytes (13). Numerous earlier studies indicate that free radical scavengers and antioxidants may combat DOX-induced cardiotoxicity (14–16), however, despite various therapeutic interventions adapted to protect the heart against DOX-induced cardiotoxicity, most of the antioxidant therapies remained particularly ineffective due to pronounced clinical side effects and due to their inability to specifically target the oxidative damage to cardiac mitochondria (17).
Previous studies from our laboratory have shown that PDE-5 inhibitors like sildenafil (Viagra) and tadalafil (Cialis) which were developed for the treatment of erectile dysfunction in men induced powerful cardioprotective effect during ischemia/reperfusion injury (18–20). Recently, we demonstrated that both sildenafil and tadalafil attenuated cardiac dysfunction in DOX-induced cardiomyopathy (21, 22). The pharmacokinetic properties of tadalafil like quick onset of action, longer half-life, relatively slow metabolism, and greater selectivity for PDE-5 enzyme over other PDE isoenzymes and fewer side effects makes tadalafil the preferred choice in the treatment for long-term management of patients receiving DOX for malignant tumors (23). Moreover, the rate and extent of tadalafil absorption is unaffected by food and therefore it could be used at a relatively lower daily dosage during chronic treatment (24). In Koka et al 2010 (22), we have shown that tadalafil has significantly improved the cardiac contractile function which was impaired by DOX with assessment of the left ventricular function by transthoracic echocardiograpghy and millar conductance catheter. In addition, we have also demonstrated, that tadalafil induced the in vivo cardioprotective effects and improved survival rates in mice without interfering with the anti-tumor efficacy of DOX by the following mechanisms: 1) attenuating DOX-induced apoptosis, 2) restoring the depleted prosurvival proteins including Bcl-2 and GATA-4, in the myocardium 3) reducing the oxidative stress via the up-regulation of mitochondrial superoxide dismutase (MnSOD) 4) up-regulating of cardiac cGMP and protein kinase G (PKG) activity. In this report, we present an overview on the role of PDE-5 inhibition in protection against DOX-induced cardiotoxicity and further continue to elaborate on the mechanisms by which tadalafil attenuates DOX-induced cardiotoxicity.
Phosphodiesterase-5 inhibitors attenuate DOX cardiotoxicity
Phosophodiesterase-5 (PDE-5) inhibitors are a class of vasoactive drugs that were developed for treatment of erectile dysfunction (ED) in men. The mechanism of action involves active inhibition of the PDE-5A enzyme and resulting increase in cGMP and smooth muscle relaxation in the penis. Sildenafil and tadalafil have also been approved for pulmonary hypertension, but cardiac indications were still considered unlikely given its modest impact on arterial tone and low expression and activity in resting myocytes. This view began to change with the publication of several preclinical landmark studies from our laboratory showing a powerful protective effect of PDE-5 inhibitors including sildenafil (Viagra), Vardenafil (Levitra) and tadalafil (Cialis) against ischemia/reperfusion injury in the rabbit and mouse hearts (18–20). We demonstrated that sildenafil and other PDE-5 inhibitors increase eNOS/iNOS in the heart which leads to downstream protective mechanisms involving cGMP-dependent activation of protein kinase G (PKG) and opening of mitochondrial KATP channels (25, 26). Opening the mitoKATP channel partially compensates the membrane potential, which enables additional protons to be pumped out to form an H+ electrochemical gradient for both ATP synthesis and Ca2+ transport. The mitochondrial stabilizing effect of sildenafil was further confirmed in our isolated cardiomyocyte study (27), which showed an increase of Bcl-2/Bax ratio and preservation of mitochondrial membrane potential (ΔΨm), in the sildenafil-pretreated myocytes. Because DOX-induced cardiotoxicity involvesthe generation of ROS in the mitochondria, we hypothesized thatcardiomyocyte protection by PDE-5 inhibition via opening of mitoKATP channels, may be extended in demonstrating the prevention of cardiomyocyte apoptosis and subsequent development of cardiomyopathy. Previous studies have shown that the accumulation of ROS results in dissipation of the ΔΨm, direct activation of the mitochondrial permeability transition pore (MPTP), and cytochrome c release followed by caspase-3 activation and DNA fragmentation consistent with apoptosis (28).
In a landmark investigation from our laboratory (21), we demonstrated that in vivo treatment of mice with sildenafil before DOX administration conferred protective effects in the heart. We observed significant attenuation of cardiomyocyte apoptosis, preservation of myofibrillar integrity, prevention of left ventricular dysfunction and ST prolongation consistent with chronic DOX-induced toxicity 8 weeks after the final of 3 treatments. Sildenafil inhibited DOX-induced ΔΨm dissipation, caspase-3 activation and cardiomyocyte apoptosis (21). Moreover, exposure of adult mouse ventricular myocytes to DOX resulted in dissipation of ΔΨm, as illustrated via JC-1 immunofluorescent staining. Moreover, adult cardiomyocytes treated with sildenafil before the treatment with DOX demonstrated preservation of the ΔΨm. This protection with sildenafil in DOX-damaged cardiomyocytes was completely abolished by a nitric oxide synthase (NOS) inhibitor (L-NAME) and 5-hydroxydecanoate 5-HD (21). These findings implied that sildenafil-mediated protection from DOX-induced cardiomyocyte apoptosis was NOS dependant and opening of mitoKATP channel played critical role in attenuation of damage caused by DOX. Additionally, we observed a significant decline in Bcl-2 expression at both 2 weeks and 8 weeks after treatment in the DOX group compared with the sildenafil + DOX and control groups, suggesting a pivotal role of Bcl-2 in altering the pathological process leading to end-stage heart failure.
There is increasing evidence that intermediate filaments such as desmin are involved in cardiomyopathy. Heling et al (29) illustrated the disorganization and accumulation of desmin in explanted human heart specimens from patients with dilated cardiomyopathy. Moreover, desmin-related cardiomyopathy in the knockout (desmin−/−) mice was prevented by Bcl-2 overexpression, as evidenced by prevention of cardiomyocyte apoptosis and preservation of cardiac contractility (30). Consistent with findings by Heling et al (29) and Wang et al (31), we observed disruption of desmin distribution in the DOX group compared with the sildenafil/DOX and control groups. Additionally, because desmin is known to adhere to the mitochondria in the same location where the MPTP is formed, it is conceivable that disruption of desmin either through repeated strain on the contractile apparatus resulting from impaired contractility or through direct cleavage from activated caspases may contribute to MPTP formation, cytochrome c release, and apoptosis.
In a more recent study, we demonstrated that the long acting PDE-5 inhibitor tadalafil also attenuated cardiac dysfunction in DOX-induced cardiomyopathy (22). Although both PDE-5 inhibitors sildenafil and tadalafil, demonstrated protective effects against DOX-induced cardiomyopathy in mice, tadalafil appears to be the preferred choice for long-term management of patients receiving DOX for malignant tumors owing to its favorable pharmacokinetic properties, relatively slow metabolism, and greater selectivity for PDE-5 enzyme. Moreover, the rate and extent of tadalafil absorption is unaffected by food and hence it could be used at a relatively lower dosage regimen during chronic treatment. In a recent study (22), we demonstrated that tadalafil significantly improved the cardiac contractile function impaired by DOX treatment. Similar to sildenafil, tadalafil co-treatment with DOX improved survival of mice, restored the depletion of anti-apoptotic protein Bcl-2 and decreased TUNEL-positive apoptotic cells. GATA-4 is a key transcriptional factor which plays a pivotal role in controlling embryonic development, cardiomyocyte differentiation, and stress responsiveness of the heart. It was recently shown that cardiac-specific deletion of GATA-4 resulted in a progressive and dose-dependent deterioration in cardiac function and dilationin adulthood (32). In response to pressure overload, the GATA-4 deficient mice developed rapid decompensation and heart failure. These detrimental phenotypes were associated with increased cardiomyocyte apoptosis (32). We also observed significant down regulation of GATA-4 expression after DOX treatment, which confirmed the previous reports (33), and GATA-4 expression was significantly preserved by co-treatment with tadalafil (22).
Our study also demonstrated that tadalafil treatment inhibited DOX-induced increase in lipid peroxidation. Antioxidant enzymes including Cu/ZnSOD (cytosolic) and MnSOD (mitochondrial) play a critical role in detoxification of ROS. Tadalafil did not affect the regulation of cytoplasmic Cu/ZnSOD but MnSOD was significantly increased. These data imply that mitochondrial elimination of ROS contribute to the cardioprotective effects of tadalafil during DOX-induced toxicity. Previous studies have also shown that MnSOD overexpression can exert cardioprotection against DOX-induced injury and ischemia-reperfusion injury (34). The antioxidant properties of PDE-5 inhibitors have not yet been well understood. Fernandes et al (35) reported that the physiological concentrations of sildenafil (<50 μmol/L) decreased both H2O2 generation by mitochondria respiring glutamate/malate. Moreover, it was shown that sildenafil decreased superoxide radical generated by hypoxantine/xantine oxidase system, without affecting either mitochondrial bioenergetics or Ca2+-induced mitochondrial permeability transition (35). Most recently, in a rat model of traumatic spinal cord injury Serarslan et al demonstrated that tadalafil reduced the spinal cord injury via increasing tissue/serum levels of nitric oxide and serum activity of SOD (36). Interestingly, our results demonstrated that co-treatment with tadalafil attenuated ROS generation by DOX, suggesting that tadalafil might have potent antioxidant-like effect, either through upregulation of MnSOD or by direct scavenging of ROS. Based on current state of knowledge, we propose that PDE-5 inhibitors could generate therapeutic levels of NO that result in enhanced formation of cGMP. cGMP may activate PKG that can subsequently open mitoKATP channels resulting in protective effect against apoptosis by preservation of GATA-4 and Bcl-2, upregulation of MnSOD, attenuating ROS generation, inhibition of MPTP opening in the mitochondria as outlined in Figure 1.
Figure 1. Proposed mechanism of cardioprotection by PDE-5 inhibitors in DOX-induced cardiomyopathy.
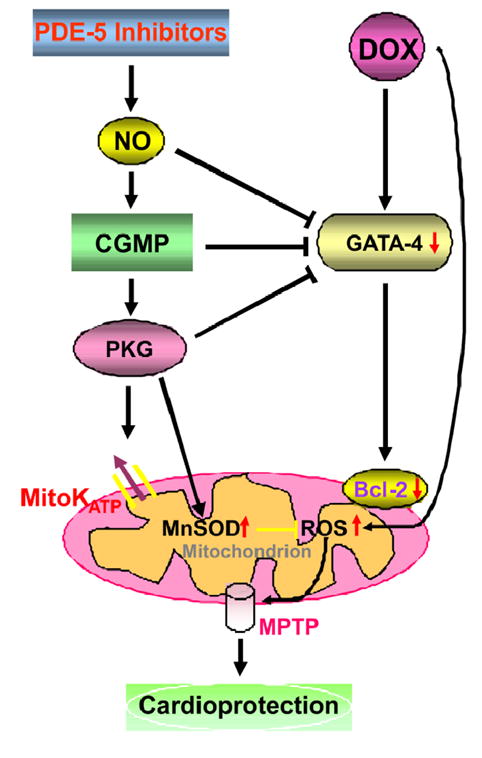
Abbreviations: ROS- reactive oxygen species, NO- nitric oxide, PKG- protein kinase G, mitoKATP-mitochondrial ATP-sensitive potassium channels, mitochondrial MPTP-mitochondrial permeability transition pore.
Concluding Remarks
Over the past decade, experimental and clinical studies have reported on the efficacy of PDE-5 inhibition to treat various forms of heart disease including myocardial infarction (25, 26), heart failure (37) and hypertrophy (38). These studies have helped in the creation of ongoing NIH multicenter trial (RELAX: Evaluating the Effectiveness of Sildenafil at Improving Health Outcomes and Exercise Ability in People with Diastolic Heart Failure; NCT00763867) in patients with heart failure and a preserved ejection fraction (i.e. EF>50%). Our studies suggest that PDE-5 inhibitors like sildenafil and tadalafil attenuated DOX-induced apoptosis, depletion of pro-survival proteins including Bcl-2 and dissipation of mitochondrial membrane potential (ΔΨm) and improved cardiac contractile function which was impaired by DOX. Cardiac oxidative stress was attenuated by tadalafil via upregulation of MnSOD and by direct scavenging of ROS. Moreover, tadalafil treatment increased cardiac cGMP level and PKG activity and restored the depletion of GATA-4 in the DOX treated myocardium. Furthermore, considering the specificity of tadalafil, these studies suggest that PDE-5 is the molecular target for attenuating DOX cardiotoxicity. Overall, our studies provide valuable new information about the efficacy of tadalafil in attenuation of DOX-induced cardiac dysfunction. Tadalafil exhibits dual mechanisms (Figure 1) i.e. it acts both via mitochondrial antioxidative and cGMP/PKG dependent mechanisms that do not interfere with antineoplastic activity of DOX (22). Currently there is no optimal therapeutic intervention for protecting the heart against the DOX-cytotoxicity. We believe that PDE-5 inhibitors have great potential in the management of the DOX-induced cardiotoxicity and therefore we propose that the class of PDE-5 inhibitors can represent attractive novel therapeutic approach for the treatment of DOX-induced cardiotoxicity in patients.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL51045, HL79424, HL93685) to Dr. Rakesh C Kukreja.
Footnotes
Conflicts of Interest
No potential conflicts of interest to disclose.
References
- 1.Bristow MR, Billingham ME, Mason JW, Daniels JR. Clinical spectrum of anthracycline antibiotic cardiotoxicity. Cancer Treat Rep. 1978;62:873–9. [PubMed] [Google Scholar]
- 2.Hortobagyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs. 1997;54 (Suppl 4):1–7. doi: 10.2165/00003495-199700544-00003. [DOI] [PubMed] [Google Scholar]
- 3.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32:302–14. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 4.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 5.Singal PK, Li T, Kumar D, Danelisen I, Iliskovic N. Adriamycin-induced heart failure: mechanism and modulation. Mol Cell Biochem. 2000;207:77–86. doi: 10.1023/a:1007094214460. [DOI] [PubMed] [Google Scholar]
- 6.Fu LX, Waagstein F, Hjalmarson A. A new insight into adriamycin-induced cardiotoxicity. Int J Cardiol. 1990;29:15–20. doi: 10.1016/0167-5273(90)90267-9. [DOI] [PubMed] [Google Scholar]
- 7.Arai M, Yoguchi A, Takizawa T, et al. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum Ca(2+)-ATPase gene transcription. Circ Res. 2000;86:8–14. doi: 10.1161/01.res.86.1.8. [DOI] [PubMed] [Google Scholar]
- 8.Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–92. [PubMed] [Google Scholar]
- 9.Billingham ME, Mason JW, Bristow MR, Daniels JR. Anthracycline cardiomyopathy monitored by morphologic changes. Cancer Treat Rep. 1978;62:865–72. [PubMed] [Google Scholar]
- 10.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261:3068–74. [PubMed] [Google Scholar]
- 11.Myers CE, McGuire WP, Liss RH, Ifrim I, Grotzinger K, Young RC. Adriamycin: the role of lipid peroxidation in cardiac toxicity and tumor response. Science. 1977;197:165–7. doi: 10.1126/science.877547. [DOI] [PubMed] [Google Scholar]
- 12.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–5. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 13.Jiang B, Zhang L, Wang Y, et al. Tanshinone IIA sodium sulfonate protects against cardiotoxicity induced by doxorubicin in vitro and in vivo. Food Chem Toxicol. 2009;47:1538–44. doi: 10.1016/j.fct.2009.03.038. [DOI] [PubMed] [Google Scholar]
- 14.Kumar D, Kirshenbaum LA, Li T, Danelisen I, Singal PK. Apoptosis in adriamycin cardiomyopathy and its modulation by probucol. Antioxid Redox Signal. 2001;3:135–45. doi: 10.1089/152308601750100641. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Chen Z, Chua CC, et al. Melatonin as an effective protector against doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol. 2002;283:H254–H263. doi: 10.1152/ajpheart.01023.2001. [DOI] [PubMed] [Google Scholar]
- 16.Nazeyrollas P, Prevost A, Baccard N, Manot L, Devillier P, Millart H. Effects of amifostine on perfused isolated rat heart and on acute doxorubicin-induced cardiotoxicity. Cancer Chemother Pharmacol. 1999;43:227–32. doi: 10.1007/s002800050888. [DOI] [PubMed] [Google Scholar]
- 17.Granger CB. Prediction and prevention of chemotherapy-induced cardiomyopathy: can it be done? Circulation. 2006;114:2432–3. doi: 10.1161/CIRCULATIONAHA.106.666248. [DOI] [PubMed] [Google Scholar]
- 18.Kukreja RC, Ockaili R, Salloum F, Xi L. Sildenafil-induced cardioprotection in rabbits. Cardiovasc Res. 2003;60:700–1. doi: 10.1016/j.cardiores.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 19.Ockaili R, Salloum F, Hawkins J, Kukreja RC. Sildenafil (Viagra) induces powerful cardioprotective effect via opening of mitochondrial K(ATP) channels in rabbits. Am J Physiol Heart Circ Physiol. 2002;283:H1263–H1269. doi: 10.1152/ajpheart.00324.2002. [DOI] [PubMed] [Google Scholar]
- 20.Salloum F, Yin C, Xi L, Kukreja RC. Sildenafil induces delayed preconditioning through inducible nitric oxide synthase-dependent pathway in mouse heart. Circ Res. 2003;92:595–7. doi: 10.1161/01.RES.0000066853.09821.98. [DOI] [PubMed] [Google Scholar]
- 21.Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005;111:1601–10. doi: 10.1161/01.CIR.0000160359.49478.C2. [DOI] [PubMed] [Google Scholar]
- 22.Koka S, Das A, Zhu SG, Durrant D, Xi L, Kukreja RC. Long-acting phosphodiesterase-5 inhibitor tadalafil attenuates doxorubicin-induced cardiomyopathy without interfering with chemotherapeutic effect. J Pharmacol Exp Ther. 2010;334:1023–30. doi: 10.1124/jpet.110.170191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuan J, Brock G. Selective phosphodiesterase type 5 inhibition using tadalafil for the treatment of erectile dysfunction. Expert Opin Investig Drugs. 2002;11:1605–13. doi: 10.1517/13543784.11.11.1605. [DOI] [PubMed] [Google Scholar]
- 24.Kouvelas D, Goulas A, Papazisis G, Sardeli C, Pourzitaki C. PDE5 inhibitors: in vitro and in vivo pharmacological profile. Curr Pharm Des. 2009;15:3464–75. doi: 10.2174/138161209789206971. [DOI] [PubMed] [Google Scholar]
- 25.Kukreja RC, Ockaili R, Salloum F, et al. Cardioprotection with phosphodiesterase-5 inhibition--a novel preconditioning strategy. J Mol Cell Cardiol. 2004;36:165–73. doi: 10.1016/j.yjmcc.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Kukreja RC, Salloum F, Das A, et al. Pharmacological preconditioning with sildenafil: Basic mechanisms and clinical implications. Vascul Pharmacol. 2005;42:219–32. doi: 10.1016/j.vph.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Das A, Xi L, Kukreja RC. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem. 2005;280:12944–55. doi: 10.1074/jbc.M404706200. [DOI] [PubMed] [Google Scholar]
- 28.Childs AC, Phaneuf SL, Dirks AJ, Phillips T, Leeuwenburgh C. Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2:Bax ratio. Cancer Res. 2002;62:4592–8. [PubMed] [Google Scholar]
- 29.Heling A, Zimmermann R, Kostin S, et al. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ Res. 2000;86:846–53. doi: 10.1161/01.res.86.8.846. [DOI] [PubMed] [Google Scholar]
- 30.Weisleder N, Taffet GE, Capetanaki Y. Bcl-2 overexpression corrects mitochondrial defects and ameliorates inherited desmin null cardiomyopathy. Proc Natl Acad Sci USA. 2004;101:769–74. doi: 10.1073/pnas.0303202101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Osinska H, Dorn GW, et al. Mouse model of desmin-related cardiomyopathy. Circulation. 2001;103:2402–7. doi: 10.1161/01.cir.103.19.2402. [DOI] [PubMed] [Google Scholar]
- 32.Oka T, Maillet M, Watt AJ, et al. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–45. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 33.Aries A, Paradis P, Lefebvre C, Schwartz RJ, Nemer M. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc Natl Acad Sci USA. 2004;101:6975–80. doi: 10.1073/pnas.0401833101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest. 1996;98:1253–60. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandes MA, Marques RJ, Vicente JA, et al. Sildenafil citrate concentrations not affecting oxidative phosphorylation depress H2O2 generation by rat heart mitochondria. Mol Cell Biochem. 2008;309:77–85. doi: 10.1007/s11010-007-9645-9. [DOI] [PubMed] [Google Scholar]
- 36.Serarslan Y, Yonden Z, Ozgiray E, et al. Protective effects of tadalafil on experimental spinal cord injury in rats. J Clin Neurosci. 2010;17:349–52. doi: 10.1016/j.jocn.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 37.Salloum FN, Abbate A, Das A, et al. Sildenafil (Viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. Am J Physiol Heart Circ Physiol. 2008;294:H1398–H1406. doi: 10.1152/ajpheart.91438.2007. [DOI] [PubMed] [Google Scholar]
- 38.Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–22. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]