

Abstract
Breast tumors expressing estrogen receptor alpha (ER) respond well to therapeutic strategies using SERMs (selective estrogen receptor modulators) such as tamoxifen. However, about thirty percent of invasive breast cancers are hormone independent because they lack ER expression due to hypermethylation of ER promoter. Treatment of ER–negative breast cancer cells with demethylating agents and histone deacetylase inhibitors leads to expression of ER mRNA and functional protein. Additionally, growth factor signaling pathways have also been implicated in ER silencing in ER-negative tumor phenotype. Recently, important role of components of ubiquitin-proteasome pathway has been shown in mediating downregulation of ER. In this article, we will review various mechanisms underlying the silencing of ER in ER negative tumor phenotype and discuss diverse strategies to combat it. Ongoing studies may provide the mechanistic insight to design therapeutic strategies directed towards epigenetic and non-epigenetic mechanisms in the prevention or treatment of ER-negative breast cancer.
Keywords: Breast cancer, Estrogen receptor, Endocrine therapy, Epigenetics, Coregulators
Introduction and Background
Breast cancer is one of the leading cause of cancer and the second leading cause of cancer related mortality in women in the United States. According to the American Cancer Society's most recent estimates for breast cancer in the United States, about 207,090 new cases of invasive breast cancer and about 54,010 new cases of carcinoma in situ (CIS) will be diagnosed in 2010. The lifetime risk of developing invasive breast cancer for a women living in the USA today is approximately a little less than 1 in 8 (12%). Mortality related to breast cancer has been declining since 1990 but still remains at a staggering high level with approximately 1 in 35 (3%) women dying of breast cancer. About 39,840 women will die from breast cancer in 2010.
Breast cancer is a heterogeneous disease consisting of multiple molecular subtypes. Molecular profiling of these subtypes has put forth many prognostic markers that can be used to guide clinical practice for personalized therapy. Despite all the genomic advances, only a few predictive markers are routinely used in the clinic. The presence of estrogen receptor (ER), progesterone receptor (PR) and overexpression of human epidermal growth factor receptor -2/Her-2 play an important role during therapeutic intervention as well as predicting response to therapy. Hormone receptor positive tumors typically present a better prognosis because of their ability to respond to endocrine interventions. Approximately 15– 20% breast tumors exhibit Her2 gene amplification leading to Her2 protein overexpression. Her2 positive tumors are typically associated with a higher rate of relapse and mortality but respond to trastuzumab which significanly improves disease free survival and overall survival (1–4). Tumors lacking ER, PR and Her2 overexpression present yet another biologically and genetically diverse group called triple negative (TN) breast cancer. TN tumors tend to have a poor prognosis partly because of their aggressive phenotype and also because of lack of any targeted therapy unlike their hormone receptor positive and Her2 positive counterparts. Extensive gene expression profiling h a s l e d t o further molecular classification of breast cancer subtypes. The basal like breast cancer shows five distinct gene signatures. Luminal A and luminal B are ER positive while Her2 enriched, basal-like and normal-like are ER negative subtypes (5–7). These subtypes have been used to predict clinical outcomes like relapse free survival and overall survival. Luminal A subtype show a better clinical prognosis than basal-like and Her2 positive, both of which are associated with poorer prognosis (5). Basal-like breast cancer more often occurs in younger, premenopausal women and affects women of African American ethnicity at a disproportionately higher level (8, 9).
While the quest for novel therapeutic options for all molecular subtypes of breast cancer is ongoing, endocrine therapies, first used more than 100 years ago, are the most effective treatment for ER positive tumors. All endocrine therapies are designed to block ER function; selective ER modulators such as tamoxifen bind ER to partially block its transactivation function while selective ER downregulators such as fulvestrant bind ER to completely block its function and inducing degradation. In addition, ovarian ablation, luteinizing hormone-releasing hormone agonists and aromatase inhibitors diminish the levels of estrogen hence inhibiting ligand-dependent ER activation. These endocrine approaches are not only effective in early stage disease; they also benefit advanced metastatic disease. Despite great benefits in a considerable proportion of patients, de novo and acquired resistance remain major problems. Understanding ER biology provides new insight into the molecular mechanisms underlying the development of de novo and acquired resistance as well as new clinically relevant strategies to combat it.
In this article we will review the emerging studies of ER function and epigenetic silencing that reveal the roles of a wide spectrum of chromatin modulators, methyl binding proteins and corepressor complexes as well as components of growth factor signaling. An understanding of the molecular factors that modulate ER can be used for its reactivation and therapeutic targeting using various strategies.
Estrogen Receptor Function: Molecular Mechanism
There are two ERs, ERα and ERβ, encoded by independent genes (10, 11). Both ERα and ERβ belong to a nuclear hormone receptor (NR) superfamily and share similar although not identical modular structures characteristics of the NR superfamily including six functional domains (Figure 1) (12). DNA binding domain (DBD) is the most conserved domain with 97% homology followed by the ligand binding domain (LBD). LBD also contains a dimerization surface and a ligand-dependent activation function-2 (AF-2). Agonist bound receptor adopts a conformation in which alpha helices (3, 5, 12) in the ligand binding domain form a hydrophobic cleft (AF-2) providing a binding surface for NR boxes (LXXLL motifs) in coactivators. Antagonists, like tamoxifen have a bulky side chain that sterically modulates the conformation of the hydrophobic cleft (AF-2) with helix 12 binding to the AF-2 cleft with its own intrinsic NR box, occluding the binding of coactivators. Antagonist-mediated inhibition of receptor is not only a passive process resulting from repositioning of helix 12 thereby blocking the coactivator binding (13), but also involves the active recruitment of corepressors to form repressive receptor complex at target genes. Activation function -1 (AF-1) is located at the N-terminus A/B domain of the receptor and its hormone-independent function is regulated by phosphorylation induced by growth factors. The two activating domains act synergistically to achieve maximal transcriptional activity, although some gene promoters have been shown to be activated independently by AF-1 or AF-2 based on the cellular and promoter context (14).
Figure 1. Schematic representation of the two human estrogen receptors, ERα and ERβ.
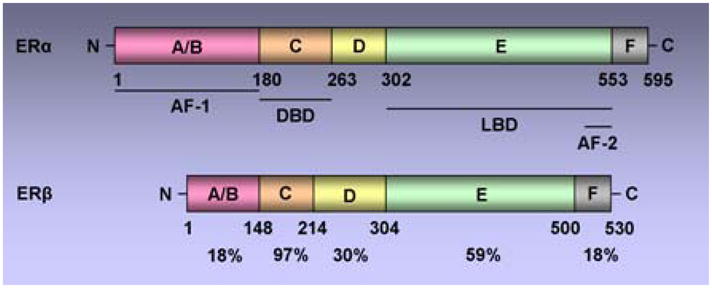
Both receptors contain five functional domains (A-E) as other members of the nuclear hormone receptor superfamily and an additional F domain at C terminal. Functional domains include, the DNA-binding domain (DBD), the Ligand-binding domain (LBD), the ligand-independent activation function AF-1, ligand-dependent activation function AF-2. The percentage identity between the two receptors is indicated.
Typically, unstimulated estrogen receptor associates with chaperone proteins and resides in the cytoplasm. During ligand-dependent activation, hormone binding to ER activates it through phosphorylation, alters its conformation and dissociates chaperone proteins such as heat-shock protein 90. Alternatively, growth factor signaling networks can induce ER activation via phosphorylation in the absence of ligand (15). This process is termed as ligand-independent activation. Activated ER then dimerizes and binds to estrogen receptor response elements (ERE) in the promoter region of estrogen-responsive genes. Promoter-bound ER induces transactivation function via recruiting various histone acetyltransferase (HAT) activity containing coactivators such as SRC-1, SRC-2, AIB-1. HAT activity containing coactivators induce histone acetylation in a concerted action, leading to open chromatin configuration and recruitment of basal transcription machinery (Figure 2) (16, 17). Some of these coactivator proteins are integral to ER function (18). For example, SRC-3 is overexpressed in 65 % of breast tumors and gene amplified in 5% as compared to normal ductal epithelium (19, 20). Reducing the levels of SRC-3 not only significantly inhibit ER mediated gene activation but also tumor growth in experimental models (21). ER can also mediate repression of certain genes by inducing the binding of histone deacetylase (HDAC) activity containing corepressor complexes which induce histone deacetylation leading to close chromatin conformation. Binding of tamoxifen in the LBD induces a conformation change in AF-2 that poses a steric hindrance to coactivator binding while encouraging binding of corepressors. Tamoxifen-bound ER recruits corepressor complexes and participates in active repression (22). On the other hand, high levels of coactivator proteins may also contribute to endocrine resistance by enhancing estrogen agonist activity of SERMs such as tamoxifen (23, 24). Coregulatory proteins impart more complexity to genomic function of ER. The above described mechanism is referred as the classical genomic activity of ER and is directly related to its ability to regulate the expression of estrogen responsive genes containing an ERE in their promoters. However, different mechanisms of action of ER have been demonstrated.
Figure 2. Genomic classical and non-classical actions of ER.
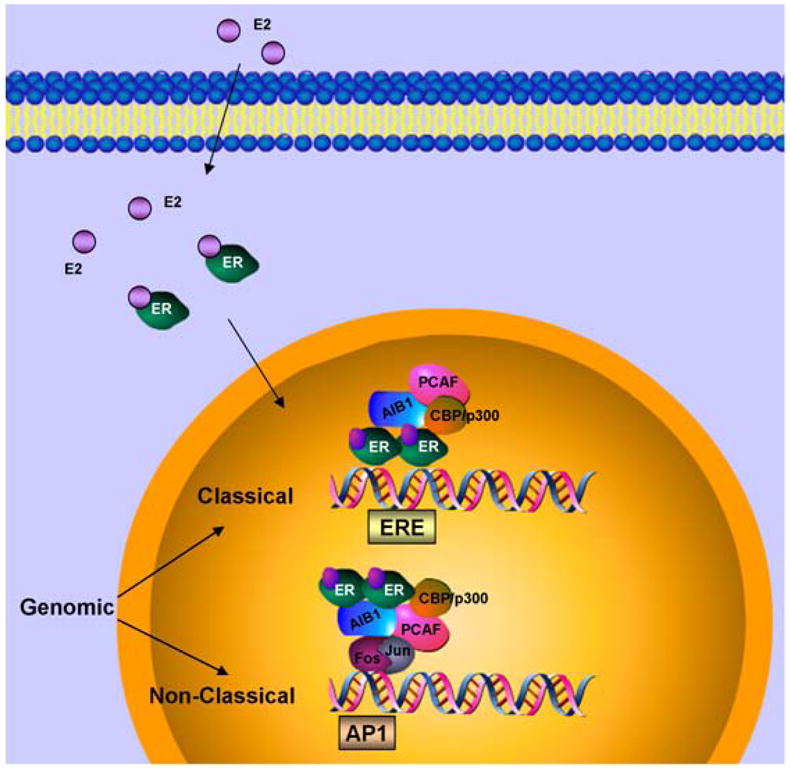
In classical genomic mode of action, estrogen (E2) binds estrogen receptor (ER), induces dimerization of the receptors, nuclear translocation and recruitment to estrogen response element (ERE) in the promoter region of the target genes. Coactivators such as AIB1, CBP/p300, PCAF are recruited to the transcription complex followed by gene transcription. In non-classical mode of action, estrogen bound ER gets recruited to other transcription factors such as Jun/Fos to activate transcription.
In a nonclassical transcriptional regulation mode, ER has been shown to regulate gene expression by interacting with other transcription factors such as the Fos-Jun complex at alternative regulatory DNA sequences such as AP-1, SP-1 and other non-ERE sites (Figure 2). Thus, ER itself functions as a coregulatory protein for the DNA bound transcription factor complex and may also recruit additional coactivators (25–28). ER participates in the regulation of many important genes, such as, cyclin D1, myc, BCl2 and IGF1R via non-classical genomic action. Non-classical genomic action of ER may play an important role not only in breast cancer cell proliferation and survival but also in the development of resistance to endocrine therapy.
Apart from its role as a transcription factor for estrogen-responsive genes and a coactivator for other transcription complexes, ER also functions at the plasma membrane level to elicit rapid action on cells (29). This rapid nongenomic ER activity has been observed in response to estrogen as well as SERMs such as tamoxifen. Presence of full length ER and an alternatively spliced truncated form of ER at the plasma membrane has been observed in some studies (30–33). While the precise cellular localization of these nongenomic ERs and the underlying mechanisms are still not clear, it has been shown that nongenomic action of ER also involves activation of other growth factor receptors, cellular tyrosine kinases (34, 35), mitogen-activated protein kinases (MAPKs) (36), phosphatidylinositol 3 kinase (37), and Akt signaling pathway (Figure 3). Membrane ER directly interacts with the insulin-like growth factor 1 receptor, the p85 regulatory subunit of PI3K, Src and Shc to activate Akt and MAPK pathways (34–37) . These kinases not only induce cell survival and cell proliferation but also phosphorylate ER and its coregulators to influence genomic action of ER. Other proteins such as MNAR/PELP1 (modulators of nongenomic activity of the estrogen receptor) and MTA1 (metastasis associated gene family) also participates in nongenomic activity of ER by facilitating interactions with other membrane components (Figure 3) (38–40). Estrogen receptor, via its nongenomic activity, plays an important role in breast tumors with highly active growth factor signaling pathways such as Her2 amplification. Estrogen activates growth factor signaling via non genomic actions of ER and the growth factor signaling activates ER, hence forming a vicious cycle. Because of multiple mode of actions (classical genomic, non-classical genomic and nongenomic), estrogen receptor has become important for breast tumor progression and have important therapeutic implications.
Figure 3. Nongenomic ER activity. Estrogen activates ER in or near membrane.
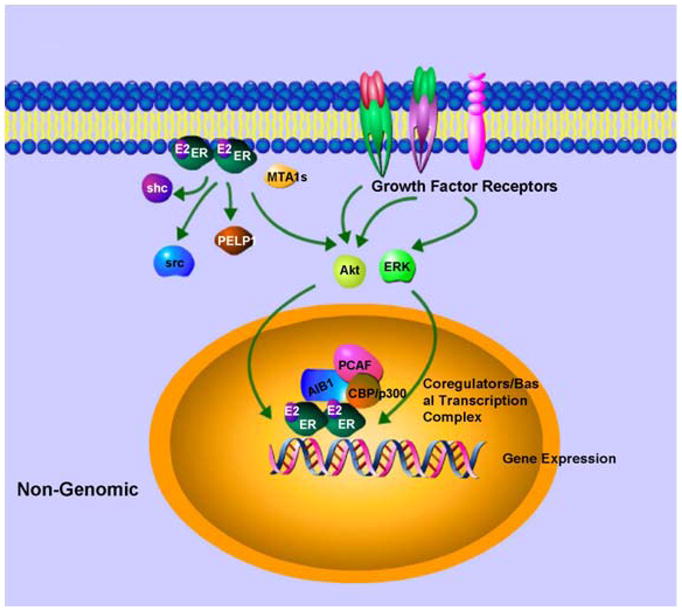
Membrane ER binds to growth factors signaling elements and activates key molecules of growth factor signaling which can further activate ER and its coregulators to enhance nuclear effects.
Estrogen Receptor Silencing: Role of Epigenetics
Aberrant cytosine methylation of promoter regions of numerous cancer-related and tumor suppressor genes is one of the mechanism leading to gene silencing. It is known that 3–5% of the cytosine residues in mammalian genomic DNA occur as 5-methylcytosines (41). A major number (approximately 70–80%) of 5-methylcytosines residues are found within CpG dinucleotides which accumulate to form CpG islands (42). CpG islands normally remain unmethylated but may reversibly regulate gene expression. Cytosine methylation and transcription levels are inversely related for a large number of genes. Two major epigenetic modifications are DNA methylation and histone acetylation that act in concert to regulate gene silencing.
ER promoter is hypermethylated and ER mRNA is absent in some ER-negative breast cancer cells. Treatment of ER negative breast cancer cells with DNA methyltransferase (DNMT) and/or histone deacetylase (HDAC) inhibitors leads to the reactivation of expression of ER mRNA and functional protein, underscoring the importance of DNMTs and HDACs in maintaining the repressive environment at target genes like ER (43, 44). Hypermethylation of CpG island may inhibit transcription by interfering with the recruitment and function of basal transcription factors or transcriptional coactivators. Also, hypermethylation of CpG dinucleotides near the transcriptional regulatory region may initiate the recruitment of the methyl-CpG binding domain (MBD) family proteins that mediate silencing of genes via facilitation of a repressive chromatin environment (45). Five methyl-CpG binding proteins including MeCP2, MBD1, MBD2, MBD3 and MBD4 have been identified (46–50). It has been shown that MeCP2, MBD1, MBD2 and MBD3 can all recruit HDAC-containing repressor complex but with distinctive functional features (48, 51–53). MBD1, MBD2 and MBD4 have been reported to bind specifically to a variety of DNA sequences containing methyl CpG whereas MBD3 does not directly bind DNA either in vitro or in vivo (53–55).
Many tissue-specific or ubiquitous DNMTs that initiate methylation at position 5 of cytosines of CpG dinucleotides have been identified (56). DNMT1, the chief enzyme responsible for maintenance of mammalian DNA methylation during DNA replication using hemimethylated DNA, can also bind HDAC2 and DMAP1 (DNMT associated protein) to mediate transcriptional repression (57). The de novo methylases, DNMT3a and DNMT3b (58, 59), can act as transcriptional repressors by using their ATRX domain to recruit HDAC1 (60, 61). Heterochromatic structure characterized by differential modifications of histones is another facet of the complex machinery influencing repression. Amino terminal tails of the core histones undergo modifications such as acetylation at lysine, methylation at lysine or arginine and phosphorylation at serine to evolve a histone code for transcriptional activation and repression (62, 63). These posttranslational modifications modulate the chromatin structure by altering the electrostatic interactions between histone proteins and DNA and modifying the recruitment of various non-histone proteins such as coactivators and corepressors to chromatin.
CpG methylation of the ER promoter results in transcriptional silencing (64) and inhibition of HDAC and/or DNMT activity reactivates ER (43, 44). These findings support a model in which methyl-CpG binding proteins, DNMTs, and HDACs might be involved in transcriptional control of ER. It has been shown that the unmethylated active ER promoter in ER-positive MCF-7 cells is enriched for H3 and H4 acetylation and H3-K4 methylation and shows little binding of any methyl binding protein or DNMT. In ER-negative MDA-MB-231 cells, the ER promoter is silenced by DNA hypermethylation, histone hypoacetylation, H3-K9 methylation and the recruitment of MeCP2, MBD1, MBD2, DNMT1, DNMT3b and HDAC1 proteins (Figure 4). HDAC inhibitor, TSA, causes histone hyperacetylation and a low level of ER mRNA reexpression in ER negative breast cancer cells as methyl binding proteins (DNMT1, DNMT3b, MeCP2, MBD1 and MBD2) remain bound to the methylated ER promoter. DNMT inhibitor, 5-aza-dC, also induces ER mRNA expression as it facilitates promoter demethylation and partial dissociation of MeCP2, MBD1, MBD2, DNMT1, DNMT3b, and DNMT1. ER negative breast cancer cells also display a relative depletion of acetylated H3 and H4 and methylated K9 H3. Thus, both HDAC and DNMT inhibitors lead to reexpression of ER but strikingly different protein complexes are associated with the ER promoter in each case. The combination facilitates the release of a repressor complex containing various MBD proteins (MeCP2, MBD1 and MBD2), DNMTs (DNMT1 and DNMT3b) and HDAC1 from the ER promoter. Release of corepressor complex leads to concomitant enrichment of acetyl-H4, acetyl-H3, and K4-dimethylated H3 and diminished methylation at K9-H3 (65) (Figure 5). Thus the epigenetically reactivated ER promoter in ER negative breast cancer cells treated with both drugs acquires a chromatin profile similar to that of the innately active ER promoter in ER positive cells.
Figure 4. Differential recruitment of coregulatory complexes to the promoter region of un/hypomethylatedvshypermethylated ER.
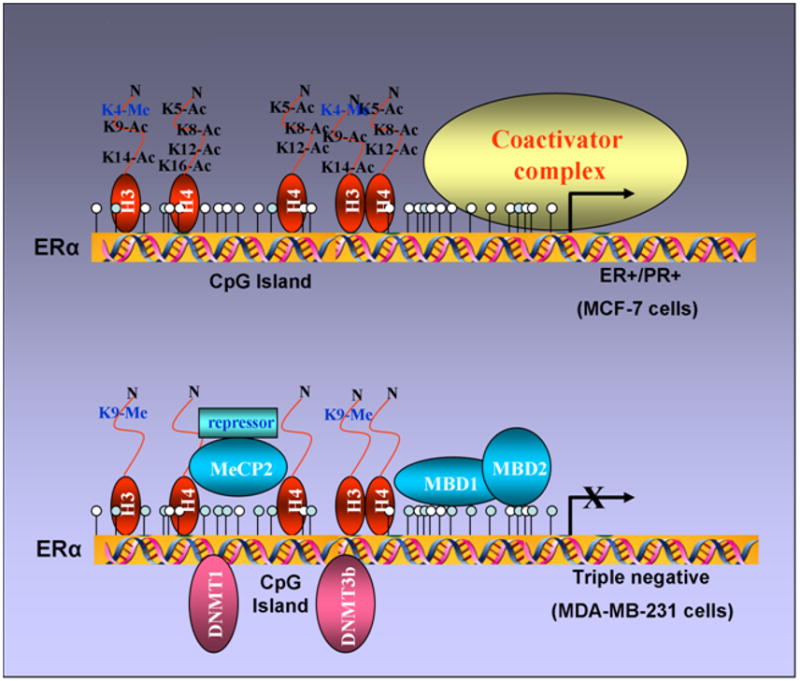
ER promoter is un/hypomethylated in ER-positive breast cancer cells with acetylated histones and binding of coactivator complexes. In contrast, ER promoter is hypermethylated in ER-negative breast cancer cells with deacetylated histones and binding of various methyl-binding proteins (MBD1, MBD2 and MeCP2) and DNA methyltransferases (DNMT1 and DNMT3b).
Figure 5. Reactivation of ER in ER-negative breast cancer cells.
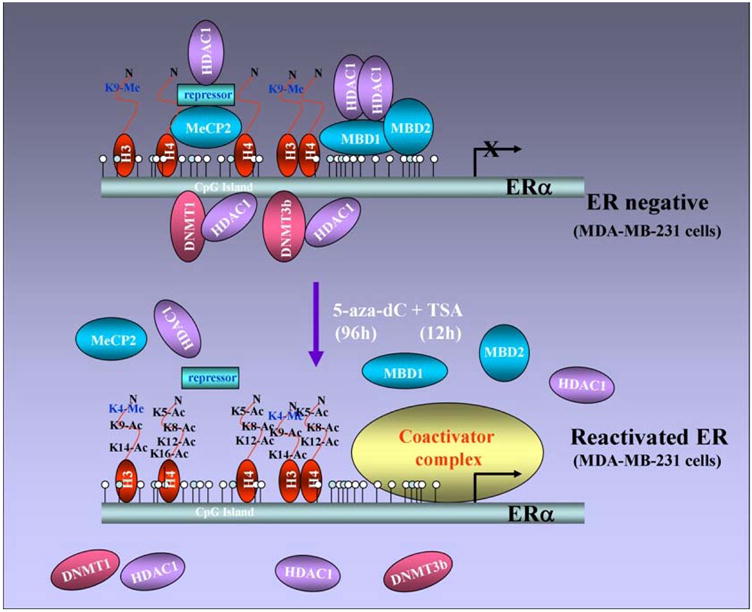
ER-negative cells can be treated with a combination of DNMT and HDAC inhibitors resulting in demethylation and release of the repression complex consisting of various methyl-binding proteins and DNA methyltransferases. Demethylation and release of repression complex paves the way for histone acetylation and coactivator binding resulting in ER reexpression in ER-negative breast cancer cells.
Estrogen Receptor Reactivation: Therapeutic targeting
The effects of endocrine therapy are primarily mediated through the estrogen receptor therefore ER expression is a strong predictor of response to SERM treatment. Indeed, lack of ER expression is the dominant mechanism of de novo resistance to SERMs such as tamoxifen (66–68). Also, during breast cancer progression, many initially ER positive tumors lose ER expression and attain hormone unresponsiveness (69, 70). ER negative tumors are more aggressive and considering the ability of these tumors to metastasize and their heterogeneity, new therapies or strategies for sensitization of ER negative tumors to endocrine treatment are required.
A number of enzymatic inhibitors targeting HDACs have been developed with good in vivo bioavailability and intracellular capability to inhibit HDAC. Preclinical studies and initial clinical trials indicate that HDAC inhibitors from different structural classes are very well tolerated and exhibit clinical activity against a variety of human cancers (71, 72). The hydroxamatetrichostatin A has been shown to have an in vivo antitumor activity with daily parenteral dosing associated with little systemic toxicity (73). The greatest potential of HDAC inhibitors lies in their ability to modulate the activity of other therapeutic agents. Demethylating agents such as 5-aza-dC are particularly interesting candidates owing to the interaction of DNA methylation with histone deacetylation in gene silencing of tumor suppressor genes. Combined treatment of TSA or depsipeptide with 5-aza-dC has been shown to synergistically reactivate silenced tumor suppressor genes in human cancer cells, including MLH1, TIMP3, CDKN2B, CDKN2A, gelsolin and maspin (74, 75). ER negative breast cancer cells can be sensitized to anti-tumor effects of tamoxifen by combined treatment with 5-aza-dC/TSA, underscoring the importance of drugs having the potential to derepress the expression of epigenetically silenced key genes in cancer therapeutics. Reactivation of ER directs tamoxifen-dependent repression of endogenous ER target genes indicating that 5-Aza-dC/TSA reactivated ER is able to interact with both agonists and antagonists to modulate transcription.
The molecular basis of repression of ER responsive genes by tamoxifen bound reactivated ER in ER-negative breast cancer cells can be comprehended by deciphering the nature of corepressor complex involved in these antagonistic actions. Tamoxifen-bound reactivated ER show the formation of a distinct complex containing HDAC3, NCoR and TBL1 on promoter regions of ER responsive genes (Figure 6). HDAC3 has been shown as the major HDAC associated with NCoR/SMRT complexes and NCoR interacts directly with HDAC3 through a deacetylase-activating domain (DAD) activating HDAC3 activity (76, 77). TBL1 then recognizes and binds the resultant deacetylated histone tails further stabilizing the binding of this multiprotein complex le ading to repression . TBL1 and TBLR1 are not required for HDAC3 activity or initial binding of the NCoR/SMRT complex to nuclear receptors, but they can interact with core histones to stabilize the binding. This is similar to the role of RbAp46 and RbAp48 in NuRD complex. While RbAp48 binds to H2A, H3 and H4, TBL1 bind preferentially to H2B and H4. Binding of NuRD complex to the ER responsive promoters has also been observed in ER-negative breast cancer cells reexpressing functional estrogen receptor in response to tamoxifen.
Figure 6. Sensitizing ER-negative breast cancer cells to endocrine therapy.
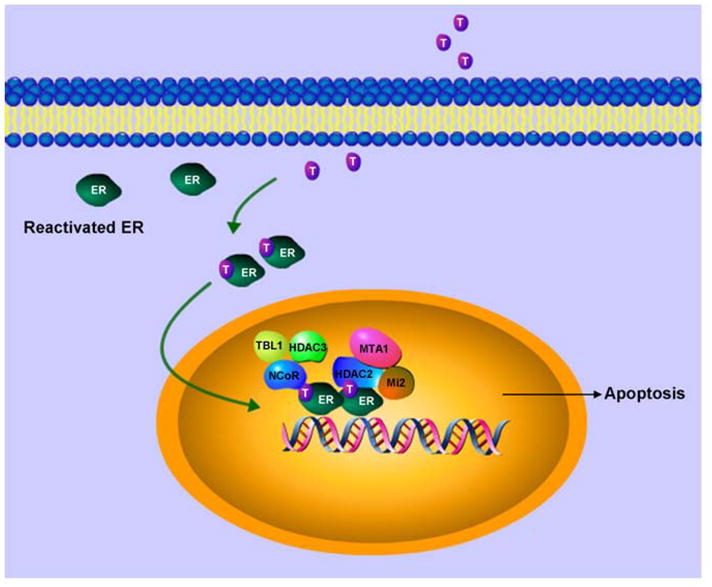
ER can be reactivated in ER-negative breast cancer cells using a combination of therapies. Reactivated ER can be targeted with tamoxifen. Tamoxifen bound reactivated ER recruits compressor complexes resulting in modulation of ER-responsive gene expression.
Combinatorial utilization of multiple corepressor complexes may be required to achieve physiologic levels of repression on some promoters whereas on other promoters different complexes might get recruited independent of each other. NCoR directly interacts with nuclear receptors via its NR box-related conserved bipartite NR interaction domain (NRID) containing L/IXXI/VI sequence (77), anchoring NCoR/HDAC3 multiprotein complex. NCoR can also interact with components of both the SAP (Sin-associated protein) and the NuRD complexes (78), suggesting that NCoR and NuRD complexes may be co-recruited to ER or other nuclear receptor gene targets. NCoR/HDAC3 and NuRD complex bind to ER responsive promoters containing either classical or non-classical EREs in a mutually exclusive manner in ChIP/Re-ChIP experiments. Mutually exclusive binding of both NCoR and NuRD corepressor complexes rules out the possibility of NCoR mediated recruitment of NuRD complex, at least in the case of tamoxifen-bound reactivated ER. Since human NuRD complex is a multi-subunit protein complex, it is possible that it gets recruited using one of its own subunits as the anchoring protein. Biochemical and immunofluorescence studies have shown that MTA1 interacts directly with the estrogen receptor (53). However whether MTA1 targets the NuRD complex to ER-responsive promoter has not been elucidated. Other candidate subunits of the NuRD complex are methyl binding proteins such as MBD2 and MBD3. While human MBD3 does not recognize methylated DNA (54), MBD2 might direct the recruitment of NuRD complex to methylated loci at target gene promoters (79). It has been suggested that a DNA methylation mediated mechanism is unlikely as NuRD complex components bind at the EBAG9 promoter within 40 minutes of tamoxifen treatment. In addition, NuRD complex purified with HDAC1 contains MBD2 (80), whereas a similar immunoaffinity purification of HDAC2 generated a NuRD complex with no detectable MBD2 (80). The recruitment of HDAC2 but not HDAC1 containing NuRD complex at the ER-responsive promoters suggests that MBD2 is not involved in tamoxifen mediated repression by reactivated ER. An ordered recruitment of NCoR complex followed by NuRD complex at distinct ER target promoters in ER-negative breast cancer cells via tamoxifen-bound reexpressed ER has also been shown. Sequential recruitment of various cofactors has been reported for regulation of various mammalian genes (81, 82). Given the ordered recruitment of corepressor complexes, a multistep model of tamoxifen- mediated repression by reactivated ER has been suggested. NCoR complex can directly interact with tamoxifen-bound reactivated ER resulting in deacetylation of local histones through recruitment of HDAC activity (76, 77). One possibility is that removal of the acetyl groups from K9 and K14 of histone H3 (83) creates an environment that promotes the binding of Suv39H1/Clr4. The methylation of H3-K9 by Suv39H1/Clr4 after histone deacetylases remove the acetyl groups from K9 and K14 of histone H3 (83) then serves as a binding site for the chromodomain of HP1/Swi6 (84, 85). NuRD complex contains Mi2/CHD family proteins which have a chromodomain (86) and biochemical analysis have shown that the NuRD complex associates with histone H3 when lysine 9 is methylated (87). This model is in accordance with the histone code hypothesis as the pattern of histone tail modifications serves as a recognition code for the recruitment of cofactors resulting in modulation of chromatin structure and function.
Additionally, DNMT inhibitor, Aza has been used in combination with scriptaid, a HDAC inhibitor, leading to reactivation of ER (88). Combination of DNMT and HDAC inhibitors can restore response to endocrine therapy in ER negative tumors in a xenograft model in nude mice (89). ER can also be reexpressed using clinically relevant HDAC inhibitor LBH589 without demethylation of the CpG island within the ER promoter (90). Similar to Aza-TSA combination treatment, LBH589 treatment also results in release of DNMT1 and HDAC1. Additional studies using suberoylanilidehydroxamic acid (SAHA) have shown reexpression of ER as well as an inhibition of EGFR expression via disruption of the EGFR mRNA stability. EGFR inhibition further decreases EGF-initiated pathways including PAK1, p38MAPK and Akt (91). Collectively, these studies show clinical relevance of HDAC and DNMT inhibitors.
Additional approaches for Estrogen Receptor Reactivation and Therapeutic Targeting
Silencing of estrogen receptor because of promoter methylation occurs in approximately 25% of ER negative tumors. This suggests the existence of additional pathways that may contribute to ER silencing. Overexpression of EGFR has been inversely correlated with ER expression (92). Stable transfection of growth factor signaling components like EGFR, Her2, Ras, Raf and MEK1 results in both estrogen independent growth and down-regulation of ER expression in ER positive cells (93–99). MAPK has emerged as a pivotal component of these upstream growth factor pathway as cells stably expressing EGFR (97), Her2 (95), Raf (93) and MEK1 (100), exhibit MAPK hyperactivation. Hyperactivation of MAPK results in the down-regulation of ER expression and inhibition of this hyperactive MAPK results in restoration of functional ER protein (100). Molecular profiling of hyperactive MAPK cells show down-regulation of ER as well as a large number of ER responsive genes (101). MAPK inhibition restores ER expression in both ex vivo tissues and primary cultures from breast tumors as well as restores response to endocrine treatment (102). Hence, some ER negative tumors exhibiting hyperactive MAPK may benefit from a combined MAPK inhibition and hormonal therapy. Ubiquitylation and proteolysis have recently been shown as another possible mechanism leading to ER down-regulation. In genomic action of ER, estrogen binding to ER rapidly stimulates ER ubiquitylation and proteolysis (103, 104). E6-AP acts as a coactivator and ubiquitin ligase for ER and is a component of ubiquitin-proteasome pathway. E6-AP expression has recently been shown to have an inverse correlation with ER expression in breast cancer. E6-AP is upregulated in ER negative breast cancer. It is possible that E6-AP may induce down-regulation of ER. Some ER negative tumors do express ER mRNA indicating the role of proteasomal degradation of ER. Recently, crosstalk between Src and ER has been shown to increase ER degradation. Transfection of Src in ER positive breast cancer cells leads to decreased levels of ER which can be prevented by a Src inhibitor (105). Hence a subset of ER negative tumors expressing activated Src may benefit from Src inhibition.
Endocrine therapy targeting ER has proven its efficacy with the development of anti-estrogens and aromatase inhibitors. Sensitizing hormone-resistant ER-negative breast cancer cells to endocrine therapy by combined treatment with DNA methyltransferase inhibitors and histone deacetylase inhibitors or MAPK inhibitors and Src inhibitors, provide new treatment options for patients with de novo resistance. In addition, the elucidation of the specific corepressor complexes and components of important upstream regulators as well as proteasome-ubiquitin pathway involved in the ER mediated repression of endogenous ER-responsive genes might help in designing more combined therapies using other therapeutic agents and innovative drug delivery strategies.
Acknowledgments
This work was supported by the USPHS grants DK077137 and DK089130 (to NKS), CA131294 and CA155686(to DS).
Footnotes
Conflicts of Interest
No potential conflicts of interest to disclose.
References
- 1.Mackey J, McLeod D, Ragaz J, et al. Adjuvant targeted therapy in early breast cancer. Cancer. 2009;115:1154–68. doi: 10.1002/cncr.24114. [DOI] [PubMed] [Google Scholar]
- 2.Martin M, Pienkowski T, Mackey J, et al. Adjuvant docetaxel for node-positive breast cancer. N Engl J Med. 2005;352:2302–13. doi: 10.1056/NEJMoa043681. [DOI] [PubMed] [Google Scholar]
- 3.Smith I, Procter M, Gelber RD, et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet. 2007;369:29–36. doi: 10.1016/S0140-6736(07)60028-2. [DOI] [PubMed] [Google Scholar]
- 4.Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–84. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 5.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 7.Fan C, Oh DS, Wessels L, et al. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med. 2006;355:560–9. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- 8.Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492–502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 9.Millikan RC, Newman B, Tse CK, et al. Epidemiology of basal-like breast cancer. Breast Cancer Res Treat. 2008;109:123–39. doi: 10.1007/s10549-007-9632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green S, Walter P, Greene G, et al. Cloning of the human oestrogen receptor cDNA. J Steroid Biochem. 1986;24:77–83. doi: 10.1016/0022-4731(86)90035-x. [DOI] [PubMed] [Google Scholar]
- 11.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93:5925–30. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–51. doi: 10.1016/0092-8674(87)90581-2. [DOI] [PubMed] [Google Scholar]
- 13.Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–37. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 14.Gronemeyer H. Transcription activation by estrogen and progesterone receptors. Annu Rev Genet. 1991;25:89–123. doi: 10.1146/annurev.ge.25.120191.000513. [DOI] [PubMed] [Google Scholar]
- 15.Osborne CK, Schiff R, Fuqua SA, Shou J. Estrogen receptor: current understanding of its activation and modulation. Clin Cancer Res. 2001;7:4338s–42s. discussion 411s–412s. [PubMed] [Google Scholar]
- 16.McKenna NJ, Nawaz Z, Tsai SY, Tsai MJ, O'Malley BW. Distinct steady-state nuclear receptor coregulator complexes exist in vivo. Proc Natl Acad Sci U S A. 1998;95:11697–702. doi: 10.1073/pnas.95.20.11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–44. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 18.Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L. Nuclear receptor coactivators and corepressors. Mol Endocrinol. 1996;10:1167–77. doi: 10.1210/mend.10.10.9121485. [DOI] [PubMed] [Google Scholar]
- 19.Anzick SL, Kononen J, Walker RL, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–8. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 20.Bouras T, Southey MC, Venter DJ. Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu. Cancer Res. 2001;61:903–7. [PubMed] [Google Scholar]
- 21.List HJ, Lauritsen KJ, Reiter R, Powers C, Wellstein A, Riegel AT. Ribozyme targeting demonstrates that the nuclear receptor coactivator AIB1 is a rate-limiting factor for estrogen-dependent growth of human MCF-7 breast cancer cells. J Biol Chem. 2001;276:23763–8. doi: 10.1074/jbc.M102397200. [DOI] [PubMed] [Google Scholar]
- 22.Smith CL, Nawaz Z, O'Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11:657–66. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- 23.Takimoto GS, Graham JD, Jackson TA, et al. Tamoxifen resistant breast cancer: coregulators determine the direction of transcription by antagonist-occupied steroid receptors. J Steroid Biochem Mol Biol. 1999;69:45–50. doi: 10.1016/s0960-0760(98)00148-4. [DOI] [PubMed] [Google Scholar]
- 24.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–8. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 25.Kushner PJ, Agard DA, Greene GL, et al. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74:311–7. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 26.Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001;62:231–52. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- 27.Xing W, Archer TK. Upstream stimulatory factors mediate estrogen receptor activation of the cathepsin D promoter. Mol Endocrinol. 1998;12:1310–21. doi: 10.1210/mend.12.9.0159. [DOI] [PubMed] [Google Scholar]
- 28.Ray P, Ghosh SK, Zhang DH, Ray A. Repression of interleukin-6 gene expression by 17 beta-estradiol: inhibition of the DNA-binding activity of the transcription factors NF-IL6 and NF-kappa B by the estrogen receptor. FEBS Lett. 1997;409:79–85. doi: 10.1016/s0014-5793(97)00487-0. [DOI] [PubMed] [Google Scholar]
- 29.Losel RM, Falkenstein E, Feuring M, et al. Nongenomic steroid action: controversies, questions, and answers. Physiol Rev. 2003;83:965–1016. doi: 10.1152/physrev.00003.2003. [DOI] [PubMed] [Google Scholar]
- 30.Filardo EJ. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol. 2002;80:231–8. doi: 10.1016/s0960-0760(01)00190-x. [DOI] [PubMed] [Google Scholar]
- 31.Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc Natl Acad Sci U S A. 2003;100:4807–12. doi: 10.1073/pnas.0831079100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figtree GA, McDonald D, Watkins H, Channon KM. Truncated estrogen receptor alpha 46-kDa isoform in human endothelial cells: relationship to acute activation of nitric oxide synthase. Circulation. 2003;107:120–6. doi: 10.1161/01.cir.0000043805.11780.f5. [DOI] [PubMed] [Google Scholar]
- 33.Levin ER. Cellular functions of plasma membrane estrogen receptors. Steroids. 2002;67:471–5. doi: 10.1016/s0039-128x(01)00179-9. [DOI] [PubMed] [Google Scholar]
- 34.Levin ER. Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol Endocrinol. 2003;17:309–17. doi: 10.1210/me.2002-0368. [DOI] [PubMed] [Google Scholar]
- 35.Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;275:18447–53. doi: 10.1074/jbc.M910345199. [DOI] [PubMed] [Google Scholar]
- 36.Song RX, McPherson RA, Adam L, et al. Linkage of rapid estrogen action to MAPK activation by ERalpha-Shc association and Shc pathway activation. Mol Endocrinol. 2002;16:116–27. doi: 10.1210/mend.16.1.0748. [DOI] [PubMed] [Google Scholar]
- 37.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–41. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vadlamudi RK, Wang RA, Mazumdar A, et al. Molecular cloning and characterization of PELP1, a novel human coregulator of estrogen receptor alpha. J Biol Chem. 2001;276:38272–9. doi: 10.1074/jbc.M103783200. [DOI] [PubMed] [Google Scholar]
- 39.Balasenthil S, Vadlamudi RK. Functional interactions between the estrogen receptor coactivator PELP1/MNAR and retinoblastoma protein. J Biol Chem. 2003;278:22119–27. doi: 10.1074/jbc.M212822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar R, Wang RA, Mazumdar A, et al. A naturally occurring MTA1 variant sequesters oestrogen receptor-alpha in the cytoplasm. Nature. 2002;418:654–7. doi: 10.1038/nature00889. [DOI] [PubMed] [Google Scholar]
- 41.Ehrlich M, Gama-Sosa MA, Huang LH, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709–21. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A. 1993;90:11995–9. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang X, Ferguson AT, Nass SJ, et al. Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res. 2000;60:6890–4. [PubMed] [Google Scholar]
- 44.Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001;61:7025–9. [PubMed] [Google Scholar]
- 45.Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999;99:451–4. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 46.Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–9. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 47.Wade PA, Jones PL, Vermaak D, Wolffe AP. A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr Biol. 1998;8:843–6. doi: 10.1016/s0960-9822(98)70328-8. [DOI] [PubMed] [Google Scholar]
- 48.Ng HH, Jeppesen P, Bird A. Active repression of methylated genes by the chromosomal protein MBD1. Mol Cell Biol. 2000;20:1394–406. doi: 10.1128/mcb.20.4.1394-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ng HH, Zhang Y, Hendrich B, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23:58–61. doi: 10.1038/12659. [DOI] [PubMed] [Google Scholar]
- 50.Snape A. MBDs mediate methylation, deacetylation and transcriptional repression. Trends Genet. 2000;16:20. doi: 10.1016/s0168-9525(99)01925-3. [DOI] [PubMed] [Google Scholar]
- 51.Meehan RR, Lewis JD, McKay S, Kleiner EL, Bird AP. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell. 1989;58:499–507. doi: 10.1016/0092-8674(89)90430-3. [DOI] [PubMed] [Google Scholar]
- 52.Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21:4886–92. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–47. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–35. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saito M, Ishikawa F. The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J Biol Chem. 2002;277:35434–9. doi: 10.1074/jbc.M203455200. [DOI] [PubMed] [Google Scholar]
- 56.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 57.Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–77. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- 58.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 59.Xie S, Wang Z, Okano M, et al. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene. 1999;236:87–95. doi: 10.1016/s0378-1119(99)00252-8. [DOI] [PubMed] [Google Scholar]
- 60.Bachman KE, Rountree MR, Baylin SB. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem. 2001;276:32282–7. doi: 10.1074/jbc.M104661200. [DOI] [PubMed] [Google Scholar]
- 61.Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 2001;20:2536–44. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–8. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 63.Dillon N, Festenstein R. Unravelling heterochromatin: competition between positive and negative factors regulates accessibility. Trends Genet. 2002;18:252–8. doi: 10.1016/s0168-9525(02)02648-3. [DOI] [PubMed] [Google Scholar]
- 64.Lapidus RG, Nass SJ, Butash KA, et al. Mapping of ER gene CpG island methylation-specific polymerase chain reaction. Cancer Res. 1998;58:2515–9. [PubMed] [Google Scholar]
- 65.Sharma D, Blum J, Yang X, Beaulieu N, Macleod AR, Davidson NE. Release of methyl CpG binding proteins and histone deacetylase 1 from the Estrogen receptor alpha (ER) promoter upon reactivation in ER-negative human breast cancer cells. Mol Endocrinol. 2005;19:1740–51. doi: 10.1210/me.2004-0011. [DOI] [PubMed] [Google Scholar]
- 66.Platet N, Cathiard AM, Gleizes M, Garcia M. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol. 2004;51:55–67. doi: 10.1016/j.critrevonc.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 67.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–19. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnston SR, Dowsett M, Smith IE. Towards a molecular basis for tamoxifen resistance in breast cancer. Ann Oncol. 1992;3:503–11. doi: 10.1093/oxfordjournals.annonc.a058251. [DOI] [PubMed] [Google Scholar]
- 69.Roodi N, Bailey LR, Kao WY, et al. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J Natl Cancer Inst. 1995;87:446–51. doi: 10.1093/jnci/87.6.446. [DOI] [PubMed] [Google Scholar]
- 70.Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 1994;54:2552–5. [PubMed] [Google Scholar]
- 71.Gray SG, Ekstrom TJ. The human histone deacetylase family. Exp Cell Res. 2001;262:75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- 72.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–49. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vigushin DM, Ali S, Pace PE, et al. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin Cancer Res. 2001;7:971–6. [PubMed] [Google Scholar]
- 74.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–7. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 75.Primeau M, Gagnon J, Momparler RL. Synergistic antineoplastic action of DNA methylation inhibitor 5-AZA-2'-deoxycytidine and histone deacetylase inhibitor depsipeptide on human breast carcinoma cells. Int J Cancer. 2003;103:177–84. doi: 10.1002/ijc.10789. [DOI] [PubMed] [Google Scholar]
- 76.Li J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000;19:4342–50. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoon HG, Chan DW, Huang ZQ, et al. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 2003;22:1336–46. doi: 10.1093/emboj/cdg120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li J, Lin Q, Wang W, Wade P, Wong J. Specific targeting and constitutive association of histone deacetylase complexes during transcriptional repression. Genes Dev. 2002;16:687–92. doi: 10.1101/gad.962502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Humphrey GW, Wang Y, Russanova VR, et al. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J Biol Chem. 2001;276:6817–24. doi: 10.1074/jbc.M007372200. [DOI] [PubMed] [Google Scholar]
- 80.Mazumdar A, Wang RA, Mishra SK, et al. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol. 2001;3:30–7. doi: 10.1038/35050532. [DOI] [PubMed] [Google Scholar]
- 81.Sharma D, Fondell JD. Temporal formation of distinct thyroid hormone receptor coactivator complexes in HeLa cells. Mol Endocrinol. 2000;14:2001–9. doi: 10.1210/mend.14.12.0567. [DOI] [PubMed] [Google Scholar]
- 82.Sharma D, Fondell JD. Ordered recruitment of histone acetyltransferases and the TRAP/Mediator complex to thyroid hormone-responsive promoters in vivo. Proc Natl Acad Sci U S A. 2002;99:7934–9. doi: 10.1073/pnas.122004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rea S, Eisenhaber F, O'Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–9. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 84.Bannister AJ, Zegerman P, Partridge JF, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 85.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 86.Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–61. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 87.Zegerman P, Canas B, Pappin D, Kouzarides T. Histone H3 lysine 4 methylation disrupts binding of nucleosome remodeling and deacetylase (NuRD) repressor complex. J Biol Chem. 2002;277:11621–4. doi: 10.1074/jbc.C200045200. [DOI] [PubMed] [Google Scholar]
- 88.Keen JC, Yan L, Mack KM, et al. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2'-deoxycytidine. Breast Cancer Res Treat. 2003;81:177–86. doi: 10.1023/A:1026146524737. [DOI] [PubMed] [Google Scholar]
- 89.Fan J, Yin WJ, Lu JS, et al. ER alpha negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J Cancer Res Clin Oncol. 2008;134:883–90. doi: 10.1007/s00432-008-0354-x. [DOI] [PubMed] [Google Scholar]
- 90.Zhou Q, Atadja P, Davidson NE. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol Ther. 2007;6:64–9. doi: 10.4161/cbt.6.1.3549. [DOI] [PubMed] [Google Scholar]
- 91.Zhou Q, Shaw PG, Davidson NE. Inhibition of histone deacetylase suppresses EGF signaling pathways by destabilizing EGFR mRNA in ER-negative human breast cancer cells. Breast Cancer Res Treat. 2009;117:443–51. doi: 10.1007/s10549-008-0148-5. [DOI] [PubMed] [Google Scholar]
- 92.Sainsbury JR, Farndon JR, Harris AL, Sherbet GV. Epidermal growth factor receptors on human breast cancers. Br J Surg. 1985;72:186–8. doi: 10.1002/bjs.1800720309. [DOI] [PubMed] [Google Scholar]
- 93.El-Ashry D, Miller DL, Kharbanda S, Lippman ME, Kern FG. Constitutive Raf-1 kinase activity in breast cancer cells induces both estrogen-independent growth and apoptosis. Oncogene. 1997;15:423–35. doi: 10.1038/sj.onc.1201198. [DOI] [PubMed] [Google Scholar]
- 94.Kasid A, Lippman ME, Papageorge AG, Lowy DR, Gelmann EP. Transfection of v-rasH DNA into MCF-7 human breast cancer cells bypasses dependence on estrogen for tumorigenicity. Science. 1985;228:725–8. doi: 10.1126/science.4039465. [DOI] [PubMed] [Google Scholar]
- 95.Liu Y, el-Ashry D, Chen D, Ding IY, Kern FG. MCF-7 breast cancer cells overexpressing transfected c-erbB-2 have an in vitro growth advantage in estrogen-depleted conditions and reduced estrogen-dependence and tamoxifen-sensitivity in vivo. Breast Cancer Res Treat. 1995;34:97–117. doi: 10.1007/BF00665783. [DOI] [PubMed] [Google Scholar]
- 96.McLeskey SW, Zhang L, El-Ashry D, et al. Tamoxifen-resistant fibroblast growth factor-transfected MCF-7 cells are cross-resistant in vivo to the antiestrogen ICI 182,780 and two aromatase inhibitors. Clin Cancer Res. 1998;4:697–711. [PubMed] [Google Scholar]
- 97.Miller DL, el-Ashry D, Cheville AL, Liu Y, McLeskey SW, Kern FG. Emergence of MCF-7 cells overexpressing a transfected epidermal growth factor receptor (EGFR) under estrogen-depleted conditions: evidence for a role of EGFR in breast cancer growth and progression. Cell Growth Differ. 1994;5:1263–74. [PubMed] [Google Scholar]
- 98.Pietras RJ, Arboleda J, Reese DM, et al. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995;10:2435–46. [PubMed] [Google Scholar]
- 99.Tang CK, Perez C, Grunt T, Waibel C, Cho C, Lupu R. Involvement of heregulin-beta2 in the acquisition of the hormone-independent phenotype of breast cancer cells. Cancer Res. 1996;56:3350–8. [PubMed] [Google Scholar]
- 100.Oh AS, Lorant LA, Holloway JN, Miller DL, Kern FG, El-Ashry D. Hyperactivation of MAPK induces loss of ERalpha expression in breast cancer cells. Mol Endocrinol. 2001;15:1344–59. doi: 10.1210/mend.15.8.0678. [DOI] [PubMed] [Google Scholar]
- 101.Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Activation of mitogen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res. 2006;66:3903–11. doi: 10.1158/0008-5472.CAN-05-4363. [DOI] [PubMed] [Google Scholar]
- 102.Bayliss J, Hilger A, Vishnu P, Diehl K, El-Ashry D. Reversal of the estrogen receptor negative phenotype in breast cancer and restoration of antiestrogen response. Clin Cancer Res. 2007;13:7029–36. doi: 10.1158/1078-0432.CCR-07-0587. [DOI] [PubMed] [Google Scholar]
- 103.Lonard DM, Nawaz Z, Smith CL, O'Malley BW. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell. 2000;5:939–48. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- 104.Nawaz Z, Lonard DM, Dennis AP, Smith CL, O'Malley BW. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci U S A. 1999;96:1858–62. doi: 10.1073/pnas.96.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chu I, Arnaout A, Loiseau S, et al. Src promotes estrogen-dependent estrogen receptor alpha proteolysis in human breast cancer. J Clin Invest. 2007;117:2205–15. doi: 10.1172/JCI21739. [DOI] [PMC free article] [PubMed] [Google Scholar]