

Summary
The gut-associated lymphoid tissue (GALT) is the largest immune organ in the body and is the primary route by which we are exposed to antigens. Tolerance induction is the default immune pathway in the gut, and the type of tolerance induced relates to the dose of antigen fed: anergy/deletion (high dose) or regulatory T-cell (Treg) induction (low dose). Conditioning of gut dendritic cells by gut epithelial cells and the gut flora, which itself has a major influence on gut immunity, induces a CD103+ retinoic acid-dependent dendritic cell that induces Tregs. A number of Tregs are induced at mucosal surfaces. Th3 type Tregs are transforming growth factor-β (TGF-β) dependent and express latency-associated peptide (LAP) on their surface and were discovered in the context of oral tolerance. Tr1 type Tregs (interleukin-10 dependent) are induced by nasal antigen and Foxp3 iTregs are induced by oral antigen and by oral administration of aryl hydrocarbon receptor ligands. Oral or nasal antigen ameliorates autoimmune and inflammatory diseases in animal models by inducing Tregs. Furthermore, anti-CD3 monoclonal antibody is active at mucosal surfaces and oral or nasal anti-CD3 monoclonal antibody induces a LAP+ Tregs that suppresses animal models (experimental autoimmune encephalitis, type 1 and type 2 diabetes, lupus, arthritis, atherosclerosis) and is being tested in humans. Although there is a large literature on treatment of animal models by mucosal tolerance and some positive results in humans, this approach has yet to be translated to the clinic. The successful translation will require defining responsive patient populations, validating biomarkers to measure immunologic effects, and using combination therapy and immune adjuvants to enhance Treg induction. A major avenue being investigated for the treatment of autoimmunity is the induction of Tregs and mucosal tolerance represents a non-toxic, physiologic approach to reach this goal.
Keywords: Tolerance, Tregs, Mucosal, Autoimmunity, Therapy, anti-CD3
Mucosal immune system
The gut-associated lymphoid tissue (GALT) is the largest immune system in the body. The mucosa of the small intestine alone is estimated to be 300 m2 in humans (1), and there are 1012 lymphoid cells per meter of human small intestine (2). Approximately 30 kg of food proteins reach the human intestine during a year, and 130 – 190 g of these proteins are absorbed daily in the gut (3). The microbiota in the small intestine is an additional major source of natural antigenic stimulation and the number of bacteria colonizing the human intestinal mucosa is approximately 1012 microorganisms/g of stool (4). The physiologic role of the GALT is the ingestion of dietary antigens in a manner that does not result in untoward immune reactions and protection of the organism from pathogens. As such, the GALT is primarily a tolerogenic environment and a complex interplay of factors creates the environment.
There are several distinctive features of the gut immune system (5) that participate in the tolerogenic environment. The inductive sites for immune responses in the gut are Peyer's patches which are macroscopic lymphoid aggregates in the submucosa along the length of the small intestine and mesenteric lymph nodes (MLNs), which are the largest lymph nodes in the body. MLNs develop distinct from Peyer's patches and peripheral lymphoid nodes and serve as a crossroads between the peripheral and mucosal recirculation pathways. In addition, there are lymphocytes scattered throughout the epithelium and lamina propria of the mucosa. A single layer of epithelial cells separates the gut microflora from the main elements of the gut immune system. To induce a mucosal immune response, antigen must gain access to antigen-presenting cells by penetrating the mucus layer and then the intestinal epithelial cell barrier. Uptake of antigen occurs through a variety of mechanisms including M cells associated with Peyer's patches and uptake by columnar epithelial cells. In addition, it has been shown that dendritic cells (DCs) themselves sample luminal contents by extending their processes through the epithelium without disruption of tight junctions (6) and that the fetal Fc receptor facilitates vesicular bidirectional transport of immunoglobulin G (IgG) or IgG-antigen complexes across mucosal epithelial cells (7). Another important component of the GALT are intraepithelial lymphocytes (IELs), which serve to regulate intestinal homeostasis, maintain epithelial barrier function, respond to infection and regulate adaptive and innate immune responses (8). In the mouse small intestine, there is one IEL for every 10 intestinal villous epithelial cells. The majority of IELs are CD8+ T cells which express αβ or γδ T-cell receptors (TCRs). Of note, it has been reported that depletion of γδ T cells impairs induction of oral tolerance (9). Thus, the combination of commensals (10), T cells (11), and DCs (6) set up a tolerogenic environment in the gut. Major factors that condition the gut to be a tolerogenic environment are interleukin-10 (IL-10), retinoic acid, and transforming growth factor-β (TGF-β) which serves as a switch factor for IgA, the predominant immunoglobulin of the gut (12).
Oral tolerance refers to physiologic induction of tolerance that occurs in the GALT and more broadly at other mucosal surfaces such as the respiratory tract (13-15). The phenomenon of “oral tolerance” has been known for over a century, viz, hyporesponsiveness to a fed antigen on subsequent challenge with that antigen. Our laboratory has been involved in the study of basic mechanisms of mucosal tolerance, the application of oral tolerance to treat autoimmune and other inflammatory conditions in animals and the attempt to translate oral tolerance to humans. In the current review, we highlight recent advances in our understanding of oral tolerance and its application to treat autoimmune and inflammatory diseases.
Mechanisms of oral tolerance (Fig. 1)
Fig. 1. Mechanisms of oral tolerance induction.
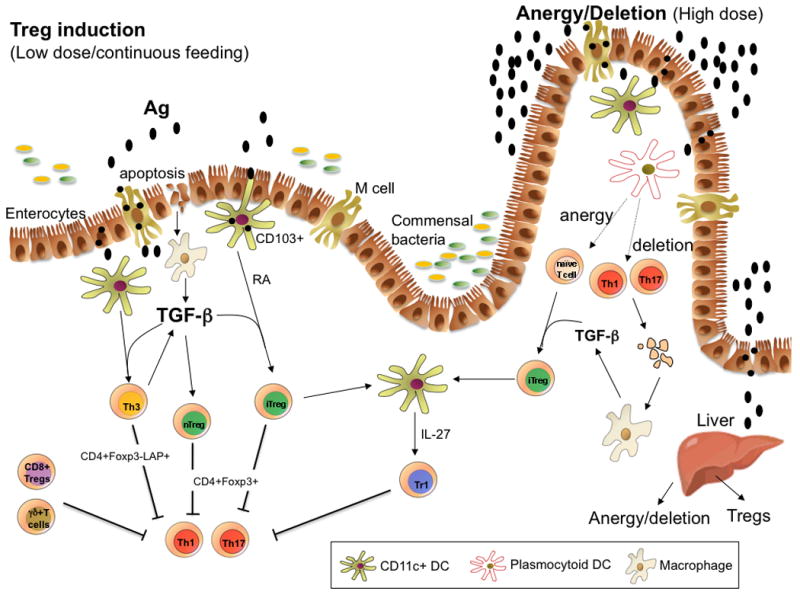
Oral antigen crosses from the intestine into the gut associated lymphoid tissue in a number of ways. It can enter via M cells, be sampled by DC processes that penetrate the lumen, or be taken up by intestinal epithelial cells. DCs in the gut are unique in that they can drive Treg differentiation from Foxp3− cells. These properties of DCs relate to their being conditioned by commensal bacteria, TGFβ and IL-10 from gut epithelial cells, and their expression of retinoic acid, which is provided in the form of vitamin A in the diet and appears to be constitutively expressed by gut DCs. CD11b monocytes may also play a role in the induction of Tregs, and the induction of Tregs occurs in the MLNs and involves both CCR7 and CCR9. Costimulation by PDL1-PDL are also important for the induction of Tregs. Macrophages are stimulated to produce TGF-β after uptaking apoptotic epithelial cells or apoptotic T cells following high dose tolerance. Lower doses of antigen favor the induction of Tregs, whereas higher doses of antigen favor anergy/deletion as a mechanism of tolerance induction. The liver may also play a role in oral tolerance induction and antigen (high dose) may be rapidly taken up by the liver, where it is processed by plasmacytoid DCs that induce anergy and deletion. A number of different types of Tregs may be induced or expanded in the gut including CD4+CD25+Foxp3+ iTregs, nTregs, Tr1 cells, LAP+ Tregs (Th3 cells), CD8+ Tregs, and γδ T cells.
It is now recognized that there are multiple mechanisms of oral tolerance, and one of the prime determinants is the dose of antigen fed. Low doses favor the induction of Tregs, whereas higher doses favor the induction of anergy or deletion. These mechanisms are not exclusive, especially at higher doses. The immunologic mechanisms of oral tolerance have been studied by a many investigators over the past 40 years, and during that time, the immunologic concept of active immune regulation or ‘suppression’ has been in and out of favor. Gershon (16) popularized the concept of ‘suppressor cells’ in the 1970s, and transferable suppression was described by a number of early investigators in the context of oral tolerance (13). We identified suppressor cells in our studies of oral tolerance to myelin antigens in the experimental autoimmune encephalitis (EAE) model and found that they acted by the secretion of TGF-β (17). We were ultimately able to clone these cells and termed them Th3 cells (18). Since then, the field of active cellular regulation has become a mainstream focus of immunologic investigation. Furthermore, with the identification of forkhead box protein 3 (Foxp3) as a key transcription factor for thymus derived natural regulatory T cells, ‘suppressor cells’ are now called ‘Tregs’, and TGF-β is now recognized as a key cytokine in the induction of Foxp3+ Tregs and other T-cell subsets (12). It has also become clear that the GALT is a rich and complex immune network that has evolved to induce immunologic tolerance and Tregs (5).
Role of gut DCs and retinoic acid in the induction of oral tolerance
One of the major advances in our understanding of oral tolerance in recent years has been identification of the role of CD103+ (αE integrin) gut DCs and retinoic acid in the induction of oral tolerance. It has been known that expanding DCs in vivo enhances the induction of oral tolerance (19) and that mucosal antigen-presenting cells are different from splenic DCs. In early studies it was shown that CD11b+ mucosal DCs preferentially produce anti-inflammatory cytokines such as IL-10 and induced Th2 type T cells (20). Differences in DCs were also reported for DCs isolated from the bronchial mucosa, which preferentially induced IL-10 responses whereas those from the gut induced TGF-β responses (21, 22). The gut is a rich source of TGF-β, as TGF-β serves as a switch factor for IgA, the major class of immunoglobulin in the gut. Epithelial cells in the gut produce both TGF-β and IL-10. As discussed above, one of the major mechanisms of oral tolerance is the induction of Treg cells and the mechanism by which this occurs is now better understood. Specifically, the induction of Tregs in the gut is related to gut DCs that have special properties which result in the preferential induction of Tregs and which are linked to both TGF-β and retinoic acid.
The importance of retinoic acid in the gut was first shown in studies which demonstrated that DCs require retinoic acid to trigger the expression of gut-homing receptors such as αEβ7 and CCR9 in T and B cells (14, 15). Subsequently, it was shown that mucosal DCs induce Foxp3 Tregs via the production of TGF-β but that concomitant retinoic acid signaling boosted this process (23). Furthermore, gut DCS could be divided into CD103+ and CD103− cells. It was the CD103+ cells that were able to induce Foxp3 Tregs when provided with exogenous TGF-β, as the CD103+ Tregs themselves produce sufficient amounts of retinoic acid (24). CD103− cells did not have these properties unless both TGF-β and retinoic acid were added. CD103− cells did however produce effector cytokines. It appears that CD103+ DCs may be conditioned by the gut epithelium to serve as ‘tolerogenic’ cells, whereas CD103− cells do not undergo this conditioning. Other groups made similar observations (23-26) during their investigation of the induction of Foxp3 Tregs in the gut and hypothesized that the availability of a precursor of retinoic acid (vitamin A) in food plays an important role in the inherent property of the gut to induce Tregs. Other innate cells in the gut may play a similar role, including macrophages in the lamina propria that produce IL-10 (27), and it has been shown that CD11b plays a role in oral tolerance, as CD11b-deficient animals have a defect in oral tolerance (28). Investigators have shown that CD11b+ DCs are increased during oral tolerance induction and produce both IL-10 and IL-27, which enhance IL-10 production by Tregs. (29) The signaling pathways and mechanisms by which DCs are programmed to become tolerogenic are becoming better understood. It has recently been shown that Wnt-b-catenin signaling in intestinal DCs regulates the balance between inflammatory versus regulatory responses in the gut (30). B-catenin in intestinal DCs was required for the expression of retinoic acid-metabolizing enzymes, IL-10, and TGF-β, and the stimulation of Treg induction while suppressing inflammatory T effector cells. In addition to retinoic acid, it has been shown that gut CD103+ DCs (but not CD103− DCs) express indoleamine-2,3-dioxygenase (IDO) (31), which is involved in the ability to CD103+ DCs to drive Foxp3 Tregs and is required for the development of oral tolerance. In mice, TGF-β may be involved in transforming IDO− DCs into IDO+ DCs and prostaglandin (PG) E2 may also play a role, especially in humans (32).
An important point regarding these studies is the observation that oral antigen induced Foxp3 Tregs in the gut from naive precursors and did not simply expand naturally occurring Tregs that had migrated from the thymus. Thus, these studies established the gut as a major site for the extrathymic induction of Tregs and provided a mechanistic framework for the long-held observation that oral administration of antigen induces Treg cells. Indeed, investigators have demonstrated oral tolerance via the induction of CD4+CD25+Foxp3+CD45RBlow cells in the absence of naturally occurring Tregs that was TGF-β but not IL-10 dependent (33). Interestingly, the same Tregs could be induced by a single intraperitoneal immunization with antigen in alum, but intraperitoneal immunization led to the simultaneous induction of Tregs and effector Th2 cells, whereas oral antigen induced only Tregs.
Other studies have broadened our understanding of the role of DCs and monocytes in the induction of oral tolerance. Plasmacytoid DCs (pDCs) were found to mediate oral tolerance in a delayed type hypersensitivity (DTH) model (34) in which both ovalbumin (OVA) and DNFB were fed; this was observed for both CD8 and CD4 immune responses. pDCs from liver and mesenteric lymph nodes of fed animals could adoptively transfer tolerances whereas pDCs from spleen did not. In vivo depletion of pDCs abrogated oral tolerance. In this model, the pDCs contributed to oral tolerance by inducing anergy/deletion of 70-80% of antigen-specific cells. The authors hypothesize than an immediate phase of oral tolerance relates to antigen that reaches the liver and induces anergy/deletion via pDCs and a secondary phase relates to the induction of Tregs in the MLNs where antigen is presented by CD103+ DCs. In a model of allergic contact dermatitis, protection was first associated with deletion of a large fraction of antigen-specific CD8+ T cells in the liver and MLNs by plasmacytoid DCs followed by triggering of the suppressive function of Tregs in secondary lymphoid organs (35). In a model of asthma, investigators showed that they were able to transfer tolerance from OVA-fed mice, by transferring splenic CD11c+ DCs (36).
Site of induction of oral tolerance
Orally administered antigens are primarily recognized by DCs in the MLN, which require afferent lymph to process oral antigen and induction of oral tolerance is impeded by mesenteric lymphadenectomy (37). In addition, oral tolerance cannot be induced in CCR7-deficient mice that have impaired migration of DCs from the intestine to MLNs, suggesting that immunologically relevant antigen is transported in a cell-bound fashion (37). In studies led by Mora et al. in which we have collaborated, it was found that oral tolerance was abrogated in CCR9−/− mice or when the α4β7 ligand mucosal addressin cell adhesion molecule-1 (MAdCAM-1) was blocked in wildtype mice, indicating that gut-homing receptors are critically required for oral tolerization (unpublished data). Furthermore, DCs in the lamina propria directly contribute to antigen uptake by extending dendrites through the epithelium to sample luminal antigens (38). The importance of Peyer's patches in the induction of mucosal tolerance was investigated in Peyer's patch-deficient ligated small bowel loops. OVA was injected directly into the lumen of the loop prior to footpad immunization. The authors found that high and low dose tolerance could be induced in the absence of Peyer's patches. These results suggest there is a critical role for components of the mucosal immune system other than Peyer's patches in antigen sampling and induction of oral tolerance and are consistent with other studies demonstrating that MLNs are crucial for the induction of oral tolerance (39-41).
Costimulation
Molecules involved in the costimulatory pathway are important in the induction of mucosal tolerance. We were unable to induce either oral or nasal tolerance using myelin oligodendrocyte glycoprotein (MOG) in the EAE model in inducible costimulator (ICOS) −/− animals, whereas EAE was suppressed in these animals by intravenously administered MOG (42). Interestingly, CD4+ T cells from orally treated ICOS−/− mice into wildtype recipients but not ICOS−/− recipients transferred tolerance, suggesting that ICOS may have a key role in controlling the effector functions of Tregs, not in inducing Tregs. We also found that CD86 but not CD80 was required for induction of low dose but not high dose oral tolerance (43) and that CD86 played a role in upregulating TGF-β in the gut (44). Furthermore, we found that cytotoxic T-lymphocyte antigen-4 (CTLA-4) was required for the induction of high dose oral tolerance (45). It has recently been shown that B7-H1 (PD-L1) and B7-DC (PD-L2) is expressed at higher levels on MLN DCs versus systemic DCs, whereas MLN DCs showed similar levels of CD80, CD86, and B7-H2. This has important functional consequences, as oral tolerance and the induction of CD4+Foxp3+ Tregs is deficient in B7-H1−/− and B7-DC−/− mice but not affected in Cd80/CD86−/− and B7H2−/− animals (46). Consistently, blocking B7-H1/CD80 interactions is reported to prevent oral tolerance induction (47). In a model of peanut allergy, oral tolerance was intact in animals treated with CTLA-4Ig, anti-CD86, or anti-CD80 plus anti-CD86, but was impaired in animals treated with anti-CD80 (48). The importance of costimulatory molecules in oral tolerance is consistent with the induction of oral tolerance being an active immunologic event.
Regulatory T cells
It is now well established that mucosal antigen administration induces Tregs, and with a better understanding of Treg biology, it has been shown that all major classes of Tregs, CD4+CD25+Foxp3 induced Tregs, CD25+Foxp3+ natural Tregs, Tr1 cells, Th3 TGF-β dependent (LAP+) Tregs, and CD8+ Tregs can be induced or activated by oral (mucosal) antigen (Table 1). We described Th3 cells in the context of oral tolerance (17, 18), and after the identification of CD4+CD25+ Tregs, we and others demonstrated that oral antigen can induce (activate) CD4+CD25+ Tregs (49, 50). As discussed above, the unique properties of gut DCs leads to the induction of CD4+Foxp3+ Tregs from Foxp3− precursors (induced Tregs). It has also been shown that IL-10-secreting Tr1-like cells may play a role in low dose oral tolerance (51), though Tr1 type Tregs are preferentially induced by nasal antigen or nasal anti-CD3 (see below). Although the in vivo biologic activity of Th3 type Tregs induced by oral antigen is well defined, viz. transferable TGF-β dependent suppression with resultant amelioration of autoimmune and inflammatory diseases, much is unknown about Th3 type Tregs and their relationship to other Treg classes. We described Th3 type Tregs prior to the description of Tr1 cells and the identification of CD4+CD25+Foxp3+ nTregs and distinguished Th3 cells from Th1/Th2 cells by their secretion of TGF-β. It now appears that one of the features of Th3 type Tregs induced in the gut is the expression of LAP on the cell surface, though LAP expression is not limited to orally induced Tregs. LAP is a propeptide that is noncovalently associated to the amino-terminal domain of TGF-β, forming a latent TGF-β complex. We identified a population of CD4+CD25−LAP+ T cells in the spleen which are part of the CD45RBlow cell fraction and suppress CD4+CD45RBhigh-induced colitis in a TGF-β-dependent fashion (52). It is this same population that is expanded by oral anti-CD3 (see below) (53). We have also identified a CD4+LAP+ Treg in the blood of humans that does not express Foxp3 and expresses the activation marker CD69. These human LAP+ Tregs are found in the CD25hi population, secrete IL-8, IL-9, IL-10, IFN-γ, and TGF-β upon activation, express TGF-βRII, suppress in a TGF-β and IL-10-dependent fashion, and are expanded in vitro following activation and amplification by IL-8 (54). A CD4+CD25−CD69+ cell which expresses membrane-bound TGF-β has been reported in mice in association with tumor progression (55). Regarding LAP and natural Tregs, we identified a CD4+CD25+LAP+ Treg population (56) and CD8+LAP+ Tregs (57) in mice, which is consistent with reports of LAP/membrane-bound TGF-β being expressed on natural Tregs both in mice and humans (58-60). It appears that GARP is a major anchoring model for surface LAP (61), though other anchoring molecules may also exist (62). We have also found that TGF-β may induce surface LAP expression on murine CD4+ T cells independent of Foxp3 induction (63). Additionally, activated Foxp3 Tregs express LAP on their surface by a GARP-dependent fashion (60). In summary, membrane bound TGF-β (LAP) is an important component of certain Treg populations and may or may not be expressed on all Foxp3+ Tregs.
Table 1. Classes of Treg induction/expansion following by oral (mucosal) administration of antigen/anti-CD3.
| Thymic derived Foxp3+ Tregs |
| Mucosally induced Foxp3+ Tregs |
| Tr1 type Tregs (IL-10 dependent) |
| Th3 type Tregs (TGFβ dependent, LAP+) |
| CD8 Tregs |
Based on studies to date, we believe a Th3 cell is a CD4+CD25−Foxp3−LAP+ cell that exists in the peripheral immune compartment and that is triggered by TCR signaling in the gut by oral antigen (Fig. 2). Triggering of these cells in the gut enhances their regulatory properties and initiates a regulatory cascade. Following triggering in the gut, the Th3 cell, which is Foxp3−, secretes TGF-β. The secreted TGF-β acts to maintain naturally occurring CD4+CD25+Foxp3+ Tregs, suppress Th1 and Th2 responses, and in concert with IL-6 may induce Th17 responses, though this does not generally occur in the gut milieu. The gut activated Th3 cell is then able to suppress systemic autoimmune and inflammatory responses. The secreted TGF-β from Th3 cells also acts on CD4+Foxp3− cells and converts them to induced Tregs, which now express Foxp3 and are Foxp3+CD25+LAP−. Depending on the milieu, these cells may become Foxp3+CD4+CD25+LAP+. These induced Tregs may also condition DCs to secrete IL-27 and in turn induce IL-10-secreting Tr1 cells (64). Thus by virtue of their TGF-β secretion and LAP expression, Th3 Tregs induced in the gut may have widespread immunoregulatory properties including the triggering of infectious tolerance and enhancing endogenous immune regulation. For example, recovery from EAE is TGF-β dependent and is associated with increases of both CD4+LAP+ and CD4+CD25+ Tregs (65). We are currently investigating the transcription factor profile of Th3 LAP+ Tregs and how they differ from nFoxp3+ Tregs, iFoxp+ Tregs, and Tr1 cells.
Fig. 2. Regulatory T-cell cascade following induction of Th3 type Tregs by oral antigen or oral anti-CD3.
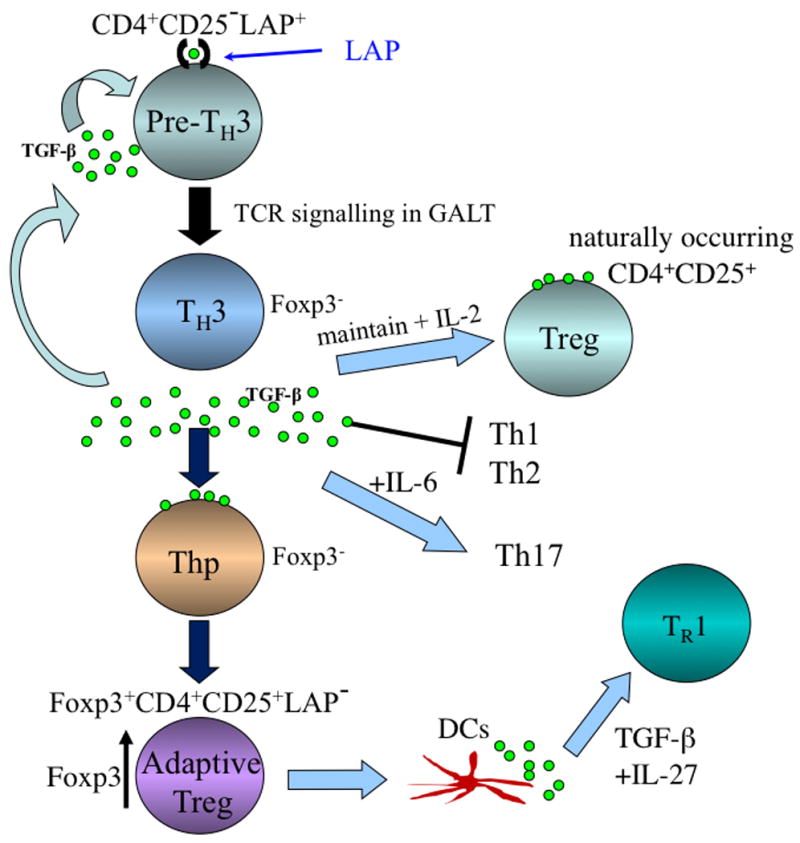
A Th3 cell is a CD4+CD25−Foxp3−LAP+ cell that exists in the peripheral immune compartment and is triggered by TCR signaling in the gut by oral antigen. Following triggering in the gut, the Th3 cell secretes TGF-β. Secreted TGF-β acts to maintain naturally occurring CD4+CD25+Foxp3+ Tregs, suppress Th1 and Th2 responses, and in concert with IL-6 may induce Th17 responses. Secreted TGF-β from Th3 cells also acts on CD4+Foxp3− cells and converts them to iTregs, which are Foxp3+CD25+LAP−. Depending on the milieu, these cells may become Foxp3+CD4+CD25+LAP+. These induced Tregs may also condition DCs to secrete IL-27 and in turn induce IL-10-secreting Tr1 cells.
CD8+ Tregs have also been shown to play a role in oral tolerance (66, 67), and defects in CD8+ T cells were observed in patients with inflammatory bowel disease and correlated with a failure to induce oral tolerance in these patients, suggesting they may play a role in oral tolerance in these patients (68). Intestinal epithelial cells can activate CD8+ T cells with regulatory activity (39). Arnaboldi et al. showed the induction of CD8 Treg by feeding a MHC class I immunodominant peptide of OVA vs. OVA protein and these CD8+ Tregs were found to suppress Th1/Th17 responses but not Th2 responses. Although CD8+ Tregs are induced during oral tolerance, studies have shown that they are not essential for inducing low dose oral tolerance, whereas there is an absolute requirement for CD4+ T cells (66, 69).
Anergy
T-cell unresponsiveness or anergy is one of the primary mechanisms by which tolerance is maintained in self-reactive lymphocytes and anergy is induced in high dose oral tolerance. The upregulation of anergy-associated genes is largely nuclear factor of activated T cells (NFAT) dependent (70), and the transmembrane E3 ubiqutin ligase GRAIL is highly upregulated during anergy induction. Related to this, high dose oral tolerance is abrogated in Grail −/− mice (71). Furthermore, investigators have found that although orally tolerized T cells can form conjugates with APCs, they are defective in immunological synapse formation, which appears related to the hyporesponsive state of orally tolerized T cells (72). T cells made anergic in vivo following oral antigen lose the ability to migrate through syngeneic endothelial monolayers and can inhibit the migration of responsive T cells in an antigen independent fashion, demonstrating that hyporesponsive T cells localize at the site of tolerance induction in vivo and amplify tolerogenic signals (73). Whitacre's group (74) examined the role of the thymus in high dose oral tolerance using MBP TCR transgenic mice and found that thymectomized animals were not protected from EAE. The thymus was found to be an important site for the development of CD4+CD25+ Tregs after oral antigen. The thymus did not appear to be a site for clonal deletion of T cells but facilitated clonal deletion in the periphery. Thus, high dose oral tolerance not only induces deletion but may induce/amplify CD4+CD25+ Tregs that resemble natural Foxp3+ Tregs as opposed to Foxp3− Th3 type cells. Of note, Mucida et al. (33) found that oral tolerance could be induced in the absence of thymus-derived Tregs in a mouse Th2 model of asthma. Others reported that low-dose oral tolerance was abrogated when CD25+ cells were depleted prior to feeding but that CD25+ depletion did not affect high dose oral tolerance (75), although anti-TGF-β blocked induction of tolerance in CD25-depleted animals independent of the feeding regimen, suggesting a complementary role of CD4+CD25+ Tregs and TGF-β in oral tolerance. In a model of collagen arthritis, investigators reported that oral administration of type II collagen suppressed IL-17 associated RANKL expression of CD4+ T cells (76).
Oral anti-CD3
The investigation of oral tolerance has classically involved the administration of oral antigen followed by challenge with the homologous antigen (usually in an adjuvant) to demonstrate antigen specific tolerance. An experimental system that others and we have employed for the study of T-cell function in oral tolerance is the use of TCR transgenic (Tg) mice in which all T cells have a common TCR. Using TCR Tg mice, we administered the cognate antigens myelin basic protein (MBP) and ovalbumin (OVA) and investigated how oral administration of an antigen affected specific T-cell subsets. In these studies we demonstrated the dose-dependent induction of Tregs in MBP TCR Tg mice (77) and deletion following high dose oral administration of OVA in OVA TCR Tg mice (78).
During the course of our experiments, we found that feeding OVA to OVA TCR Tg mice induced CD4+CD25+ Treg cells (26, 49). Other investigators also showed that oral antigen induced CD4+CD25+ Tregs (50). The CD4+ cells from OVA TCR Tg fed animals had greater suppressive properties in vitro than natural Tregs, mediated suppression in part by both TGF-β and IL-10, and induced increased expression of CTLA-4, a molecule known to be involved in Treg activity (79, 80). Although these findings demonstrated that oral antigen could induce/expand Tregs, administration of OVA to OVA TCR Tg mice is dependent on TCR Tg mice and not translatable to humans. We thus asked whether it was possible to trigger the TCR in wildtype mice in the gut and induced Tregs without using cognate antigen. It is known that anti-CD3 binds to the ε chain of the TCR and given intravenously deletes T cells and has been shown to be an effective treatment for type 1 diabetes in the nonobese diabetic (NOD) mouse (81). We hypothesized that oral administration of anti-CD3 monoclonal antibody would replace the use of a cognate antigen to trigger the TCR and would thus induce Tregs when given orally. Monoclonal antibodies have not been given orally on the assumption that they would be degraded in the gut and thus would not be biologically active. Nonetheless, it is known that orally administered cytokines (82) and peptides (42, 83) are biologically active, demonstrating that orally administered proteins are not completely degraded in the gut.
Thus to test this hypotheses, we administered hamster anti-mouse CD3 (2C-11) to SJL mice and immunized mice with PLP/CFA to induce EAE. We found that oral anti-CD3 suppressed both clinical and pathologic features of EAE both in the PLP and MOG EAE model (53). There was a dose effect observed with EAE suppression by oral anti-CD3 seen at lower (5μg), but not higher doses (50μg, 500μg). These findings were consistent with the classic paradigm of oral tolerance in which induction of Tregs is seen at lower but not higher doses (Fig. 1). Furthermore, it demonstrates that induction of Tregs by oral anti-CD3 is not simply related to administering large amounts of antibody to overcome degradation of the antibody in the gut. Indeed, we isolated biologically active anti-CD3 from intestinal eluates of animals orally dosed with anti-C3 (84) and could visualize anti-CD3 being taken up by gut epithelial cells and binding to gut DCs in intestinal loop experiments (Fig. 3). Of note, the Fc portion of anti-CD3 was not required, as Fab'2 anti-CD3 antibody is active orally and induces Tregs.
Fig. 3. Intestinal closed loop experiments demonstrating binding of anti-CD3 to DCs in the gut.

Mice with a targeted deletion of chemokine receptor CX3CR1 with eGFP insertion (HC. Reinecker, MGH) have all monocytes and DCs in the gut labeled with eGFP (green). Mice were anesthetized, stomach opened, and a closed loop was created in a part of the small intestine. Alexa Fluor 700 labeled anti-CD3 (2c11) antibody (blue) was injected into the loop and the intestinal loop harvested 45 min later. The intestinal content was cleared with PBS before imaging with confocal microscopy using multi-tracking for 2-color imaging. Image acquisition was carried out with Volocity software. DCs are labeled green and anti-CD3 antibody labeled blue. Green = DCs; Blue = anti-CD3 monoclonal antibody.
How is oral anti-CD3 different mechanistically from intravenous anti-CD3? It is known that intravenous anti-CD3 enters the blood stream, modulates CD3 from the cell surface, and lyses CD3+ T cells. Oral anti-CD3, on the other hand, does not enter the blood stream or modulate CD3 from the cell surface but acts locally in the gut to induce Th3 type CD4+CD25−LAP+ Tregs in the MLNs. Because oral anti-CD3 does not enter the bloodstream, there is no cytokine release syndrome. In the EAE model, intravenous anti-CD3 is effective when given after disease manifests but not when given prior to disease induction. Oral anti-CD3, on the other hand, ameliorates EAE both when given prior to EAE induction and at the height of disease. The explanation for this difference is a follows: intravenous anti-CD3 acts primarily by lysing disease effector cells (present only after disease induction), whereas oral anti-CD3 acts by inducing Tregs (Fig. 2). Intravenous anti-CD3 has been reported to induce Tregs that act in a TGF-β-dependent fashion, but only after lysis of T cells. (85).
We found that oral anti-CD3 ameliorates disease in other autoimmune and inflammatory diseases (Table 2) including streptozocin-induced (84) and NOD autoimmune diabetes (unpublished data), type 2 diabetes in the Ob/Ob mouse (86), lupus prone SNF1mice (87), and collagen-induced arthritis (88). Other investigators reported that oral anti-CD3 suppresses atherosclerosis in ApoE−/− mice. (89). In all these models, disease amelioration was related to the induction of TGF-β-dependent Tregs that express LAP on their surface. We also found that nasal anti-CD3 ameliorates lupus but does so by inducing an IL-10-dependent CD4+CD25−LAP+ Treg as opposed to the TGF-β-dependent LAP+ Treg induced by oral anti-CD3 (90). This is consistent with the observation that GALT DCs induce TGF-β-dependent Tregs versus IL-10-dependent Tregs induced in the bronchial associated lymphoid tissue (22). Furthermore, these results demonstrate that mucosally administered anti-CD3 appears to act in a fashion analogous to mucosally administered cognate antigen (22).
Table 2. Animal models treated by oral (nasal) anti-CD3.
| Experimental autoimmune encephalomyelitis (SJL, NOD) |
| Streptozocin induced autoimmune diabetes (AKR) |
| Autoimmune diabetes in the NOD mouse (NOD) |
| Collagen-induced arthritis (DBA1) |
| Type II diabetes (Ob/Ob mouse) |
| Lupus (NZB) |
| Atherosclerosis (ApoE -/- mice) |
Investigators have reported that feeding mice with intravenous immunoglobulin (IVIg) reduced the antibody response against β2 glycoprotein-1 and significantly attenuated clinical symptoms in a murine model of experimental anti-phospholipid syndrome (91). Other investigators induced immune tolerance by oral IVIg in a model related to rheumatoid factor autoantibodies. They reported that the effect of human IgG was mediated by the Fc portion and not the Fab portion if the IgG (92).
The effects of oral anti-CD3 raise the question whether it is more advantageous to induce antigen-specific versus antigen non-specific Tregs for the treatment of autoimmune and inflammatory diseases. It is assumed that the induction of antigen specific Tregs is preferable, as one would have specific immune modulation with less potential side effects. Furthermore, because of the phenomenon of bystander suppression which we first described in association with oral tolerance (93), cytokines such as TGF-β released from antigen specific Tregs at the target organ would suppress reactivity to other autoantigens that developed in the course of epitope spreading. Of note, there may be target organ specificity even when antigen non-specific Tregs are induced with oral anti-CD3 as we observed increased numbers of Th3 type LAP+ Tregs in the pancreatic lymph nodes of autoimmune diabetic mice (84), and this has been suggested for atherosclerosis models (89). Furthermore, in conditions such as type 2 diabetes, lupus, and atherosclerosis, there are not well-defined target antigens and in these conditions induction of antigen non-specific Tregs by anti-CD3 may be preferable. As discussed below, oral anti-CD3 is currently being tested in humans.
Induction of Tregs by oral administration of aryl hydrocarbon receptor ligands
The induction of FoxP3+ Tregs is viewed as a promising approach for the treatment of human autoimmune disorders (94). Several methods have been described to differentiate and expand human FoxP3+ Treg in vitro, but the ability to produce significant numbers of functional FoxP3+ Tregs in a consistent manner is limited (95). Thus, strategies aimed at the induction of functional FoxP3+ Tregs in vivo are more likely to be translated into clinical practice. We and others have shown that aryl hydrocarbon receptor (AHR) activation induces functional FoxP3+ Tregs that suppress the development of experimental models of autoimmunity and transplant rejection (96-99); thus, AHR is an attractive target for the induction of functional FoxP3+ Tregs. However, studies on the effect of AHR ligands in models of autoimmunity have mostly focused on TCDD (2,3,7,8- tetrachlorodibenzo-p-dioxin), a synthetic toxin which is an environmental pollutant. To investigate an AHR ligand that lacked toxicity (100), we investigated the endogenous non-toxic mucosal AHR ligand 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) as method to induce functional FoxP3+ Tregs in vivo following oral administration.
To investigate the role of AHR ligands in the induction of FoxP3+ Tregs in the gut and the relationship of AHR ligands to oral tolerance, we used mice carrying a green fluorescence protein (GFP) reporter in foxp3 and a mutant AHR protein which displays a significant reduction in its affinity for AHR ligands (AHR-d Foxp3gfp mice) (101). We first analyzed the frequency of Foxp3+ Tregs in the thymus, MLNs, and spleen. We found a significant reduction of the frequency of Foxp3+ Tregs in the MLN of AHR-d Foxp3gfp mice but no difference in the thymus or spleen. We also found an impaired ability of AHR-d CD4+ FoxP3− T cells to differentiate into FoxP3+ iTregs both in vitro and in vivo, suggesting that AHR activation in T cells by endogenous ligands plays a physiological role in the differentiation of FoxP3+ iTregs. This is in agreement with the impaired differentiation of AHR-deficient CD4+ T cells into FoxP3+ iTregs described by Kimura et al. (102) and suggests that endogenous AHR ligands in the gut modulate T-cell differentiation in vivo.
Our observation of decreased Foxp3+ Tregs in the MLNs but not the thymus of AHR-d mice is consistent with the importance of AHR ligands in the gut and their effect on iTreg as opposed to nTregs. Consistently, the biologic properties of AHR limits the activation of Stat1 during Th17 and Treg differentiation (102, 103), and Stat1 activation antagonizes the differentiation of FoxP3+ iTregs (104) but not nTregs (105); indeed several cytokines, signaling pathways and genomic elements have differential contributions to the generation of iTregs versus nTregs (106, 107).
As discussed above, DCs play a central role in the induction of Tregs in the gut. AHR activation has been shown to modulate the function and maturation of DCs (96, 108, 109) and macrophages (110, 111). Indeed, AHR in macrophages limits lipopolysaccharide (LPS)-induced inflammation (110, 111), and AHR in DCs mediates the anti-inflammatory activities of lipoxin A4 (108). We thus investigated the effect of ITE on DCs and found that the AHR ligand ITE induces DCs that promote the differentiation of FoxP3+ Tregs in a RA-dependent manner (103). Mucosal CD103+ DCs promote the differentiation of FoxP3+ Tregs via RA (23, 24, 26), and RA (112) and IL-10 (113) also have autocrine anti-inflammatory effects on DCs. Thus, our results support a model in which AHR activation induces tolerogenic DCs that promote the generation of FoxP3+ Tregs via the production of RA and the concomitant downregulation of pro-inflammatory cytokines that interfere with FoxP3+ Treg differentiation (114, 115). Moreover, it is possible that under physiological conditions, endogenous AHR ligands participate in the development of the mucosal CD103+ DCs that promote the differentiation of FoxP3+ iTregs.
We have found that the endogenous AHR ligand ITE, given orally, acts on DCs and T cells to promote the induction of functional FoxP3+ Tregs that suppress EAE (103). We recently reported that AHR also interacts with c-Maf to promote the differentiation of Tr1 type Tregs induced by IL-27 (116) in mice, and AHR activation promotes the differentiation of Tr1 suppressive human Tregs in vitro (117). Thus, non-toxic endogenous AHR ligands such as ITE are potential new compounds that can be given orally to induce the Treg component of oral tolerance for the treatment of autoimmune disorders. In addition to showing that oral ITE suppresses EAE via the induction of Tregs (103) we found that oral ITE suppresses type 1 diabetes in the NOD mouse and given together with oral MOG enhances the induction of low dose oral tolerance to MOG in the EAE model (unpublished). Thus, as shown in Fig. 4, both exogenous ITE or natural AHR ligands lead to the induction of Tregs in the gut by acting on both T cells and DCs to induce Foxp3+ Treg and Tr1 cells.
Fig. 4. Immune regulatory pathways in the gut induced by AHR ligands.
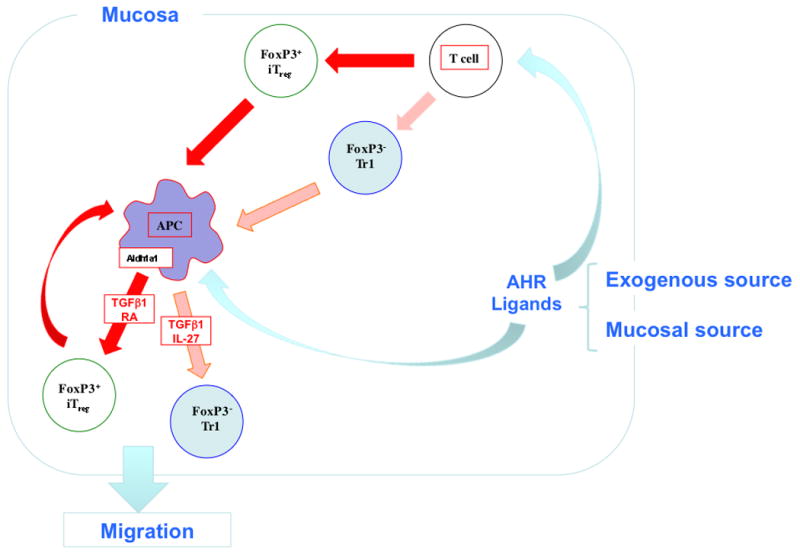
AHR ligands such as ITE in the gut either from commensal bacteria or administered orally act directly on T cells and DCs in the gut to induce Foxp3iTregs and Foxp3-Tr1 cells. Both Tregs and AHR ligands condition DCs to amplify Treg induction through the production of IL-27 and retinoic acid.
Other investigators have studied the effect of the AHR ligand TCDD on oral tolerance and have reported that TCDD impairs the stable establishment of oral tolerance in mice (118, 119). Immunotoxicological studies in mice exposed to TCDD show changes in thymic lineage and changes in immune cells including cytokine profiles (120-122). Chmill et al. showed that AHR is expressed in the small intestine, especially in intestinal epithelial cells, and that the AHR marker gene cyp1a1 is induced in intestinal epithelial cells by oral TCDD exposure. They studied a high dose oral tolerance regimen (20mg OVA given three times) and found that TCDD impaired stable oral tolerance in mice as measured by OVA-specific IgG1 antibodies and that this was associated with a increase in IL-6 producing CD103+ DCs in the MLNs and a small increase in the frequency of Th17 cells. Thus, it appears that AHR ligands may differentially affect oral tolerance depending on the AHR ligand and the type of oral tolerance induced. In humans, the primary environmental exposure to TCDD is via food (121, 123) and many food constituents, such as plant flavonoids and indoles, or bacterial tryptophan products are agonists of AHR and may activate the AHR pathway in the gut (124).
The microbiota is a crucial component of the immunologic milieu that conditions the gut and creates the substrate for oral tolerance (10, 125-127). Animal studies have provided evidence of a link between the gut and autoimmunity. In the EAE model, probiotic administration of lactobacillus results in reduction of disease activity (128). Depletion of microflora using broad spectrum antibiotics in inbred SJL and C57BL/6 mice impairs EAE development, and this effect is associated with a reduction of proinflammatory cytokines, suggesting a role of gut commensals in induction of peripheral tolerance (129). These investigators also reported positive effects on EAE in animals treated with gut components orally (129, 130). In the NOD model, NOD mice lacking myeloid differentiation factor 88 (MyD88) protein (an adapter for multiple innate immune receptors that recognize microbial stimuli) do not develop type 1 diabetes (131). The effect is dependent on commensal microbes because germ-free MyD88− NOD mice develop robust diabetes, whereas colonization of germ-free MyD88− NOD mice with a defined microbial consortium (representing bacterial phyla normally present in human gut) attenuates diabetes. Of note, microbiota-derived metabolites, including ATP, and filamentous bacteria have been shown to drive the development of Th17 polarizing DCs (132, 133) and gut-residing segmented filamentous bacteria drive autoimmune arthritis via Th17 cells (134).
T-cell-mediated oral tolerance is intact in germ-free mice using a feeding of both high and low dose OVA (135), and the susceptibility to nasal and oral tolerance induction to the major birch pollen allergen, BET v 1, is not dependent on the presence of the microflora (136). Others found that intestinal commensal bacteria promote T-cell hypo-responsiveness and downregulate serum antibody responses induced by dietary antigen (137).
Role of the liver in oral tolerance
Blood draining the intestine via the portal vein reaches the liver, where oral antigen metabolism occurs and intestinal venous drainage through the liver is important for oral tolerance. Diverting blood that drains from the intestine to the liver by a portocaval shunt impairs oral tolerance (138). Oral antigens are presented within the liver by DCs which leads to the generation of IL-4 -expressing CD4+ T cells (139). Antigen uptake is performed most efficiently by liver sinusoidal endothelial cells, which leads to CD8+ T-cell tolerance. (140). Early studies demonstrated that the portal administration of transplantation antigens was very effective in inducing graft tolerance (141). Tregs develop in liver-draining celiac lymph nodes during induction of oral tolerance to OVA, and expression of CD103 on OVA specific T cells in the celiac lymph nodes may favor homing to the epithelium of the intestine (142). To study the role of the liver and IL-10 in oral tolerance, Safadi et al. (143) generated transgenic mice with sustained hepatocyte-specific expression of rat IL-10. They found that transgenically expressed IL-10 in the liver enhanced oral tolerance, whereas systemic administration of IL-10 had only a modest effect. Ectopic expression of MBP in mouse liver induces antigen-specific Tregs and suppresses EAE (144). Interestingly, antibody titers to intestinal flora are elevated in humans with chronic liver disease who undergo portocaval shunts (145), suggesting a role of the liver in tolerance to the intestinal microbiota.
NKT cells
NKT cells are distinct from conventional T cells, and NKT cells in mice express a single invariant chain which recognizes glycolipid antigens presented by non-polymorphic MHC proteins. Upon activation, NKT cells rapidly produce large amounts of IL-4 and IFNγ. It has been reported that NK T cells may play an important role in oral tolerance by modulating DCs (146) or by inducing regulatory T cells that produce IL-10 and TGF-β and by clonally deleting antigen specific cells (147). Induction of oral tolerance activates NKT cells, which is linked to suppression of colitis and hepatitis (148, 149). In human studies, oral administration of a mixture of autologous colonic extracted proteins activated of NKT cells and demonstrated positive effects on colitis in an initial double-blind trial (150).
Nasal tolerance
The nasal cavity and respiratory tract comprise one of the major mucosal surfaces of the body, and as described above, nasal antigen preferentially induces IL-10-dependent Tregs. We have studied nasal tolerance in models of atherosclerosis, stroke, cardiac ischemia, and lupus (13). We found that nasal anti-CD3 induces Tr1 cells and this induction is dependent on upper airway resident DC -derived IL-27 signaling. Furthermore, the in vivo induction of Tr1 cells by nasal anti-CD3 involves activation of transcription factors AHR and cMAF. Subsequently, IL-21 acts in an autocrine fashion in the expansion and maintenance of Tr1 cell in vivo (unpublished data). Recently, investigators reported nasally and orally induced Tregs suppress arthritis and proliferation of arthritogenic T cells in joint-draining lymph nodes. The effect was seen not only prophylactically but also inhibiting established pathogenic B and T-cell responses and was associated with IL-10 producing Tregs cells and enhanced expression of Foxp3 and TGFβ (151). Others reported hierarchical suppression depending on whether oral or nasal antigen was given in a model of asthma (152). Airway eosinophilia, airway hyperactivity, mucous hypersecretion, and cytokine production were suppressed with both oral and nasal OVA, but nasal OVA was not as effective in suppressing IgG1 antibodies. In other studies, both oral and nasal antigen were found to induce CD8+ T-cell tolerance (153). In addition to nasal administration, investigators are studying the effect of sublingual administration of antigen. Sublingual immunotherapy has been used for the treatment of various allergic conditions and has been shown to be more effective than subcutaneous therapy (154, 155). Sublingual tolerance with antigen or antigen conjugated to cholera-toxin B subunit induces strong tolerance involving both regulatory T cells and apoptosis and depletion of effector T cells. The degree to which sublingual tolerance compares directly to oral or nasal tolerance remains to be determined (156, 157).
Neonatal tolerance
Allergic asthma appears to result from inappropriate Th2-type responses to environmental airborne antigens. Its prevalence has increased markedly, and exposure to environmental antigens during infancy appears crucial in the development of asthma. Investigators reported that airborne antigens can be transferred from mother to neonate through milk and that tolerance induction did not require the transfer of immunoglobulins (158). Interestingly, this breastfeeding-induced tolerance relied on the presence of TGF-β during lactation and was mediated by regulatory CD4+ T cells. The mechanisms and maternal influences of neonatal tolerance by breastfeeding are not well understood (159). Neonatal exposure to staphylococcal superantigen (SEA) in animals improves oral tolerance in a mouse model of airway allergy (160). These findings correspond to a reduced incidence of food allergy in infants who neonatally colonized by Staphylococcus aureus in the gut (161). In a mouse model, oral exposure to non-inherited maternal antigens during pregnancy had an impact on transplantation performed later in life (162). The tolerogenic milk effects disappeared when donor mice were injected with CD5 monoclonal antibody during the lactation period, suggesting a regulatory T-cell-dependent mechanism. We previously reported differential effects of neonatal oral tolerance in EAE: neonatal oral administration of myelin antigens enhanced EAE in adult animals, whereas oral administration of such antigens in adults were protected (163). This was not true for all autoantigens, as oral insulin given to neonates did not enhance diabetes in NOD mice (164).
Animal models
The use of oral tolerance to treat animal models of autoimmunity was first reported in collagen arthritis (165) and EAE (166, 167). Since then, oral and nasal administration of autoantigens has been reported to ameliorate a large number of conditions (Table 3). These results were the impetus for trials of oral and nasal tolerance to treat human disease states (see below). In general, the primary immune mechanism in these studies has been the induction of Tregs. An important feature of the induction of Tregs by oral or nasal antigen is bystander suppression (93). Because Tregs induced by mucosal antigen secrete TGF-β or IL-10 at the target organ after antigen specific triggering, knowledge of the autoantigen is not required and bystander suppression obviates the need to deal with different epitopes that may be targeted by epitope spreading. This also creates the possibility to use Tregs for the treatment of nonimmun-mediated diseases that have an inflammatory component. This is illustrated in our studies of stroke and myocardial reperfusion injury in which nasal administration of myelin oligodenderocyte protein or troponin induced brain or cardiac specific IL-10-secreting Tregs that decreased infarct size (168, 169). In a model of Sjögren's syndrome induced by immunization with Ro6o derived peptides, epitope spreading was prevented, lymphocytic infiltration was blocked, and saliva flow was restored by feeding Ro60 or Ro274 (170).
Table 3. Experimental Disease Models Amelioratedby Oral Tolerance.
| Disease Model | Protein Fed |
|---|---|
| Airway Eosinophilia | OVA |
| Allergy | Derp1, cedar pollen |
| Anti-phospholipid Syndrome | β2-glycoprotein |
| Arthritis (CIA, AA, AIA, PIA, SCW) | Collagen II, Hsp65, BSA |
| Atherosclerosis | Hsp65 |
| Cardiac reperfusion injury | Troponin |
| Colitis | Colonic proteins, OVA |
| Diabetes (NOD mouse) | Insulin, GAD, OVA |
| Encephalomyelitis (EAE) | MBP, PLP, MOG, GA |
| Food Hypersensitivity | aS1-casein |
| Myasthenia Gravis | AchR |
| Neuritis | PNS-myelin |
| Nickel Sensitization | Nickel |
| Sogren's syndrome | Rho peptides |
| Stroke | MOG |
| Thyroiditis | Thyroglobulin |
| Transplantation | Alloantigen, MHC peptide |
| Uveitis | S-Ag, IRBP |
| Nerve Injury | MBP |
Mucosal adjuvants to enhance oral tolerance
It is likely that the translation of oral tolerance to humans will ultimately involve a mucosal adjuvant to enhance the induction of Tregs (13). Recent studies have shown that Lactacoccus lactis constructed to secrete ovalbumin and/or IL-10 enhances oral tolerance (171, 172), and in a model of gluten sensitivity, Lactococcus lactis delivered immunodominant Dq8-restricted gliadin peptide in sensitized NOD Ab0Dq8 transgenic mice induced IL-10, TGF-β, and Foxp3 Tregs (173). Holmgren, Czerkinsky, and Sun (reviewed in 174) have performed an extensive series of investigations demonstrating that oral, nasal, or sublingual administration of antigen coupled to the cholera toxin B subunit (CTB) enhances mucosal tolerance. CTB increases mucosal antigen uptake and presentation to APCs by binding to GM1 ganglioside and the induction of Foxp3− T cells that express LAP or IL-10. The use of CTB was applied to humans in initial trials of Behçet's disease. In other studies, CTB with allergen-specific T-cell epitopes accumulated in rice seed were given orally, and the levels of allergen-specific CD4+ T-cell-derived cytokines IL-4, IL-5, and IL-13, and histamine release were significantly decreased. (175). Another group targeted allergen to DCs with muco-adhesive chitosan particles and found that this enhanced tolerance induction (176). These authors used the sublingual approach to induce oral tolerance. Probiotic mixtures have been used as mucosal adjuvants and suppress EAE by induction of IL-10-producing T cells (177). Orally administered cytokines are biologically active in the intestinal mucosal including TGF-β (178) and IL-10. In other studies, the sigma 1 protein of reovirus targets M cells (179) and coupled with antigen facilitates oral tolerance with reduction of antigen-specific CD4+ cells (180). Poly(lactic-co-glycolic acid) nanoparticles entrapping type II collagen were effective in enhancing oral tolerance in a collagen arthritis model (181). The immunomodulatory drug cyclosporine given with oral type V collagen prevented rejection of MHC class I and II incompatible lung allographs (182). This combination of type V collagen and cyclosporine was associated with alloantigen-induced expression of IL-10 in mediastinal lymph nodes and spleen plus intragraft expression of IL-10 and Foxp3. Others have shown that intravenous anti-CD3 boosts the induction of mucosal tolerance triggered by nasal proinsulin (183).
Translation of oral tolerance to human disease
Despite the extensive literature on the effectiveness of oral tolerance to treat diseases in animals, and some positive reports in phase II trials, this approach has yet to successfully translate to the clinic in phase III trials. Nonetheless, with a better understanding of the mucosal immunology and the biology of Tregs, it appears that the time is appropriate for the next phase of human studies of mucosal tolerance. The establishment of immunologic markers will provide the basis for dosing and measuring the effect of immune adjuvants (Table 4). One of the major goals of immunotherapy is to induce Tregs and to date there are no specific methods to do this in vivo. Mucosal induction would appear to be a very attractive avenue.
Table 4. Factors associated with translation of oral tolerance to human disease.
| Dose |
| Immune marker of immunologic effect |
| Route (oral vs. nasal) |
| Mucosal adjuvant |
| Protein preparation |
| Combination therapy |
| Early therapy |
| Immunologic subgroups |
Studies of oral tolerance in humans have been reviewed previously (13). Recent studies on oral tolerance in humans are as follows. In rheumatoid arthritis (RA) (184), investigators fed dnaJP1 which is a 15-mer dominant epitope heat-shock protein is thought to be involved in RA pathogenesis, though independent from the primary trigger of RA (185). One hundred sixty active RA patients with immune reactivity to the HSP received 25 mg of oral dnaJP1 or placebo for six months in a phase II study. There was a significant reduction in T cells producing TNF and a trend towards an increase of T cells producing IL-10. Some positive clinical effects were observed and there were no side effects. In another human trial, 186 patients with diffuse cutaneous, systemic sclerosis (186) received oral type I collagen at 500μ per day or placebo for 12 months. There were no positive findings in the defined clinical parameters, though sub analysis identified positive results in the collagen type I treated group with late phase DCSSC that may identify a group for future studies. In a study of oral KLH, feeding normal subjects high or low doses of KLH followed by immunization modulated systemic KLH specific immune responses (187). Mucosal tolerance to KLH has been reported previously (188). Other investigators studied oral tolerance to KLH to patients with ulcerative colitis and first degree relatives and reported a genetic defect in oral tolerance in this group (189).
An important result in humans has been subgroup analysis of the oral insulin trial for prevention of type 1 diabetes (DPT-10). Although there were no differences between the oral insulin and placebo groups in the primary outcome, a subset of individuals in the oral insulin prevention trial with high levels of insulin autoantibodies (baseline IAA ≥ 300) had an apparent several year delay in progression to diabetes (p= 0.01), and a follow-up study is planned (190) (Fig. 5). An important implication of the results from the oral insulin trial is that there may be responsive immunologic subgroups to oral tolerance therapy. Success of specific immunotherapy may thus relate to a better understanding of the immune status of individual patients.
Fig. 5. Oral insulin effect most evident in subjects with baseline IAA ≥ 300.
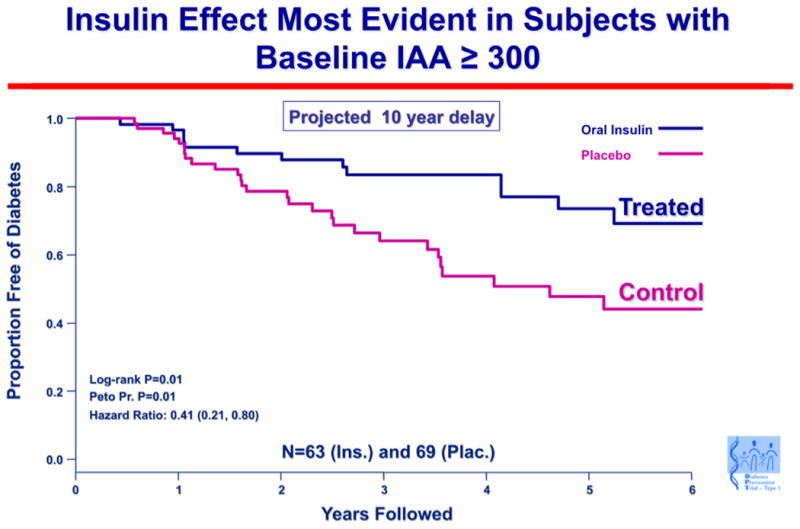
A subgroup of islet autoantibody relatives with the highest level of insulin autoantibodies showed delayed progression to diabetes when treated with oral insulin to induce mucosal tolerance. Survival curve depicting time to diagnosis of type 1 diabetes in the DPT-1 oral insulin trial, for the subset of subjects with baseline-confirmed insulin autoantibody level of 300 nU/mL or above. Skyler, JSS Ann NY Acad Sci 2008. 1150: p 194.
Oral tolerance has also been investigated in food (191) and cow's milk allergy. Caminiti (192) reported oral desensitization programs in children with IgE-mediated cow's milk allergy in a pilot study. New food allergy models have been developed by impairing oral tolerance and administering Staphylococcus aureus-derived enterotoxins with OVA or peanut extract. These studies demonstrate mechanisms by which oral tolerance can be impaired and permit allergic responses (193).
Summary
One of the gold standards for immunotherapy of autoimmune inflammatory diseases is antigen or organ-specific non-toxic therapy. Oral tolerance provides such an avenue, and it represents a clinically applicable physiologic manner in which to suppress inflammation through the induction of regulatory T cells.
Acknowledgments
The investigations in our laboratory are supported by grants from the NIH, MS Society, and Juvenile Diabetes Society. Dr. Weiner is a consultant to NasVax Ltd, a company developing oral anti-CD3 for human disease.
Literature Cited
- 1.Moog F. The lining of the small intestine. Sci Am. 1981 Nov;245(5):154–8. 60, 62. doi: 10.1038/scientificamerican1181-154. et passiom. [DOI] [PubMed] [Google Scholar]
- 2.Mestecky J, McGhee JR. Immunoglobulin A (IgA): molecular and cellular interactions involved in IgA biosynthesis and immune response. Adv Immunol. 1987;40:153–245. doi: 10.1016/s0065-2776(08)60240-0. [DOI] [PubMed] [Google Scholar]
- 3.Brandtzaeg P. Development and basic mechanisms of human gut immunity. Nutr Rev. 1998 Jan;56(1 Pt 2):S5–18. doi: 10.1111/j.1753-4887.1998.tb01645.x. [DOI] [PubMed] [Google Scholar]
- 4.Macfarlane GT, Macfarlane S. Human colonic microbiota: ecology, physiology and metabolic potential of intestinal bacteria. Scand J Gastroenterol Suppl. 1997;222:3–9. doi: 10.1080/00365521.1997.11720708. [DOI] [PubMed] [Google Scholar]
- 5.Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol. 2003 Apr;3(4):331–41. doi: 10.1038/nri1057. [DOI] [PubMed] [Google Scholar]
- 6.Rescigno M. Intestinal dendritic cells. Adv Immunol. 2010;107:109–38. doi: 10.1016/B978-0-12-381300-8.00004-6. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida M, Kobayashi K, Kuo TT, Bry L, Glickman JN, Claypool SM, et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J Clin Invest. 2006 Aug;116(8):2142–51. doi: 10.1172/JCI27821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheridan BS, Lefrancois L. Intraepithelial lymphocytes: to serve and protect. Curr Gastroenterol Rep. 2010 Dec;12(6):513–21. doi: 10.1007/s11894-010-0148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ke Y, Pearce K, Lake JP, Ziegler HK, Kapp JA. Gamma delta T lymphocytes regulate the induction and maintenance of oral tolerance. J Immunol. 1997 Apr 15;158(8):3610–8. [PubMed] [Google Scholar]
- 10.Strober W. The multifaceted influence of the mucosal microflora on mucosal dendritic cell responses. Immunity. 2009 Sep 18;31(3):377–88. doi: 10.1016/j.immuni.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev. 2006 Aug;212:256–71. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 12.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008 Aug 8;134(3):392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faria AM, Weiner HL. Oral tolerance. Immunol Rev. 2005 Aug;206:232–59. doi: 10.1111/j.0105-2896.2005.00280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004 Oct;21(4):527–38. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006 Nov 17;314(5802):1157–60. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 16.Gershon RK. A disquisition on suppressor T cells. Transplant Rev. 1975;26:170–85. doi: 10.1111/j.1600-065x.1975.tb00179.x. [DOI] [PubMed] [Google Scholar]
- 17.Miller A, Lider O, Roberts AB, Sporn MB, Weiner HL. Suppressor T cells generated by oral tolerization to myelin basic protein suppress both in vitro and in vivo immune responses by the release of transforming growth factor beta after antigen-specific triggering. Proc Natl Acad Sci U S A. 1992 Jan 1;89(1):421–5. doi: 10.1073/pnas.89.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994 Aug 26;265(5176):1237–40. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 19.Viney JL, Mowat AM, O'Malley JM, Williamson E, Fanger NA. Expanding dendritic cells in vivo enhances the induction of oral tolerance. J Immunol. 1998 Jun 15;160(12):5815–25. [PubMed] [Google Scholar]
- 20.Iwasaki A, Kelsall BL. Freshly isolated Peyer's patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J Exp Med. 1999 Jul 19;190(2):229–39. doi: 10.1084/jem.190.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, et al. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat Med. 2002 Sep;8(9):1024–32. doi: 10.1038/nm745. [DOI] [PubMed] [Google Scholar]
- 22.Weiner HL. The mucosal milieu creates tolerogenic dendritic cells and T(R)1 and T(H)3 regulatory cells. Nat Immunol. 2001 Aug;2(8):671–2. doi: 10.1038/90604. [DOI] [PubMed] [Google Scholar]
- 23.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007 Jul 13;317(5835):256–60. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 24.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007 Aug 6;204(8):1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007 Aug 6;204(8):1765–74. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007 Aug 6;204(8):1775–85. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007 Oct;8(10):1086–94. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 28.Ehirchiou D, Xiong Y, Xu G, Chen W, Shi Y, Zhang L. CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J Exp Med. 2007 Jul 9;204(7):1519–24. doi: 10.1084/jem.20062292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiokawa A, Tanabe K, Tsuji NM, Sato R, Hachimura S. IL-10 and IL-27 producing dendritic cells capable of enhancing IL-10 production of T cells are induced in oral tolerance. Immunol Lett. 2009 Jun 30;125(1):7–14. doi: 10.1016/j.imlet.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 30.Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. 2010 Aug 13;329(5993):849–53. doi: 10.1126/science.1188510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matteoli G, Mazzini E, Iliev ID, Mileti E, Fallarino F, Puccetti P, et al. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut. 2010 May;59(5):595–604. doi: 10.1136/gut.2009.185108. [DOI] [PubMed] [Google Scholar]
- 32.Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood. 2005 Oct 1;106(7):2375–81. doi: 10.1182/blood-2005-03-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest. 2005 Jul;115(7):1923–33. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, et al. Plasmacytoid dendritic cells mediate oral tolerance. Immunity. 2008 Sep 19;29(3):464–75. doi: 10.1016/j.immuni.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubois B, Joubert G, Gomez de Aguero M, Gouanvic M, Goubier A, Kaiserlian D. Sequential role of plasmacytoid dendritic cells and regulatory T cells in oral tolerance. Gastroenterology. 2009 Sep;137(3):1019–28. doi: 10.1053/j.gastro.2009.03.055. [DOI] [PubMed] [Google Scholar]
- 36.Nagatani K, Dohi M, To Y, Tanaka R, Okunishi K, Nakagome K, et al. Splenic dendritic cells induced by oral antigen administration are important for the transfer of oral tolerance in an experimental model of asthma. J Immunol. 2006 Feb 1;176(3):1481–9. doi: 10.4049/jimmunol.176.3.1481. [DOI] [PubMed] [Google Scholar]
- 37.Worbs T, Bode U, Yan S, Hoffmann MW, Hintzen G, Bernhardt G, et al. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J Exp Med. 2006 Mar 20;203(3):519–27. doi: 10.1084/jem.20052016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001 Apr;2(4):361–7. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 39.Kraus TA, Brimnes J, Muong C, Liu JH, Moran TM, Tappenden KA, et al. Induction of mucosal tolerance in Peyer's patch-deficient, ligated small bowel loops. J Clin Invest. 2005 Aug;115(8):2234–43. doi: 10.1172/JCI19102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spahn TW, Fontana A, Faria AM, Slavin AJ, Eugster HP, Zhang X, et al. Induction of oral tolerance to cellular immune responses in the absence of Peyer's patches. Eur J Immunol. 2001 Apr;31(4):1278–87. doi: 10.1002/1521-4141(200104)31:4<1278::aid-immu1278>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 41.Spahn TW, Weiner HL, Rennert PD, Lugering N, Fontana A, Domschke W, et al. Mesenteric lymph nodes are critical for the induction of high-dose oral tolerance in the absence of Peyer's patches. Eur J Immunol. 2002 Apr;32(4):1109–13. doi: 10.1002/1521-4141(200204)32:4<1109::AID-IMMU1109>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 42.Miyamoto K, Kingsley CI, Zhang X, Jabs C, Izikson L, Sobel RA, et al. The ICOS molecule plays a crucial role in the development of mucosal tolerance. J Immunol. 2005 Dec 1;175(11):7341–7. doi: 10.4049/jimmunol.175.11.7341. [DOI] [PubMed] [Google Scholar]
- 43.Liu L, Kuchroo VK, Weiner HL. B7.2 (CD86) but not B7.1 (CD80) costimulation is required for the induction of low dose oral tolerance. J Immunol. 1999 Aug 15;163(4):2284–90. [PubMed] [Google Scholar]
- 44.Gonnella PA, Chen YH, Waldner H, Weiner HL. Induction of oral tolerization in CD86 deficient mice: a role for CD86 and B cells in the up-regulation of TGF-beta. J Autoimmun. 2006 Mar;26(2):73–81. doi: 10.1016/j.jaut.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Samoilova EB, Horton JL, Zhang H, Khoury SJ, Weiner HL, Chen Y. CTLA-4 is required for the induction of high dose oral tolerance. Int Immunol. 1998 Apr;10(4):491–8. doi: 10.1093/intimm/10.4.491. [DOI] [PubMed] [Google Scholar]
- 46.Fukaya T, Takagi H, Sato Y, Sato K, Eizumi K, Taya H, et al. Crucial roles of B7-H1 and B7-DC expressed on mesenteric lymph node dendritic cells in the generation of antigen-specific CD4+Foxp3+ regulatory T cells in the establishment of oral tolerance. Blood. 2010 Sep 30;116(13):2266–76. doi: 10.1182/blood-2009-10-250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood. 2010 Aug 26;116(8):1291–8. doi: 10.1182/blood-2010-01-265975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Wijk F, Nierkens S, de Jong W, Wehrens EJ, Boon L, van Kooten P, et al. The CD28/CTLA-4-B7 signaling pathway is involved in both allergic sensitization and tolerance induction to orally administered peanut proteins. J Immunol. 2007 Jun 1;178(11):6894–900. doi: 10.4049/jimmunol.178.11.6894. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Izikson L, Liu L, Weiner HL. Activation of CD25(+)CD4(+) regulatory T cells by oral antigen administration. J Immunol. 2001 Oct 15;167(8):4245–53. doi: 10.4049/jimmunol.167.8.4245. [DOI] [PubMed] [Google Scholar]
- 50.Thorstenson KM, Khoruts A. Generation of anergic and potentially immunoregulatory CD25+CD4 T cells in vivo after induction of peripheral tolerance with intravenous or oral antigen. J Immunol. 2001 Jul 1;167(1):188–95. doi: 10.4049/jimmunol.167.1.188. [DOI] [PubMed] [Google Scholar]
- 51.Tsuji NM, Mizumachi K, Kurisaki J. Interleukin-10-secreting Peyer's patch cells are responsible for active suppression in low-dose oral tolerance. Immunology. 2001 Aug;103(4):458–64. doi: 10.1046/j.1365-2567.2001.01265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oida T, Zhang X, Goto M, Hachimura S, Totsuka M, Kaminogawa S, et al. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J Immunol. 2003 Mar 1;170(5):2516–22. doi: 10.4049/jimmunol.170.5.2516. [DOI] [PubMed] [Google Scholar]
- 53.Ochi H, Abraham M, Ishikawa H, Frenkel D, Yang K, Basso AS, et al. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+ CD25- LAP+ T cells. Nat Med. 2006 Jun;12(6):627–35. doi: 10.1038/nm1408. [DOI] [PubMed] [Google Scholar]
- 54.Gandhi R, Farez MF, Wang Y, Kozoriz D, Quintana FJ, Weiner HL. Cutting edge: human latency-associated peptide+ T cells: a novel regulatory T cell subset. J Immunol. 2010 May 1;184(9):4620–4. doi: 10.4049/jimmunol.0903329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han Y, Guo Q, Zhang M, Chen Z, Cao X. CD69+ CD4+ CD25- T cells, a new subset of regulatory T cells, suppress T cell proliferation through membrane-bound TGF-beta 1. J Immunol. 2009 Jan 1;182(1):111–20. doi: 10.4049/jimmunol.182.1.111. [DOI] [PubMed] [Google Scholar]
- 56.Chen ML, Yan BS, Bando Y, Kuchroo VK, Weiner HL. Latency-associated peptide identifies a novel CD4+CD25+ regulatory T cell subset with TGFbeta-mediated function and enhanced suppression of experimental autoimmune encephalomyelitis. J Immunol. 2008 Jun 1;180(11):7327–37. doi: 10.4049/jimmunol.180.11.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen ML, Yan BS, Kozoriz D, Weiner HL. Novel CD8+ Treg suppress EAE by TGF-beta- and IFN-gamma-dependent mechanisms. Eur J Immunol. 2009 Dec;39(12):3423–35. doi: 10.1002/eji.200939441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001 Sep 3;194(5):629–44. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, et al. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol. 2004 Jan 15;172(2):834–42. doi: 10.4049/jimmunol.172.2.834. [DOI] [PubMed] [Google Scholar]
- 60.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A. 2009 Aug 11;106(32):13445–50. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Battaglia M, Roncarolo MG. The Tregs' world according to GARP. Eur J Immunol. 2009 Dec;39(12):3296–300. doi: 10.1002/eji.200940117. [DOI] [PubMed] [Google Scholar]
- 62.Oida T, Weiner HL. Overexpression of TGF-beta1 gene induces cell surface localized glucose-regulated protein 78-associated latency-associated peptide/TGF-beta. J Immunol. 2010 Sep 15;185(6):3529–35. doi: 10.4049/jimmunol.0904121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oida T, Weiner HL. TGF-beta Induces Surface LAP Expression on Murine CD4 T Cells Independent of Foxp3 Induction. PLoS One. 2010;5(11):e15523. doi: 10.1371/journal.pone.0015523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007 Dec;8(12):1380–9. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- 65.Zhang X, Reddy J, Ochi H, Frenkel D, Kuchroo VK, Weiner HL. Recovery from experimental allergic encephalomyelitis is TGF-beta dependent and associated with increases in CD4+LAP+ and CD4+CD25+ T cells. Int Immunol. 2006 Apr;18(4):495–503. doi: 10.1093/intimm/dxh390. [DOI] [PubMed] [Google Scholar]
- 66.Chen Y, Inobe J, Weiner HL. Induction of oral tolerance to myelin basic protein in CD8-depleted mice: both CD4+ and CD8+ cells mediate active suppression. J Immunol. 1995 Jul 15;155(2):910–6. [PubMed] [Google Scholar]
- 67.Lider O, Santos LM, Lee CS, Higgins PJ, Weiner HL. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein. II. Suppression of disease and in vitro immune responses is mediated by antigen-specific CD8+ T lymphocytes. J Immunol. 1989 Feb 1;142(3):748–52. [PubMed] [Google Scholar]
- 68.Brimnes J, Allez M, Dotan I, Shao L, Nakazawa A, Mayer L. Defects in CD8+ regulatory T cells in the lamina propria of patients with inflammatory bowel disease. J Immunol. 2005 May 1;174(9):5814–22. doi: 10.4049/jimmunol.174.9.5814. [DOI] [PubMed] [Google Scholar]
- 69.Garside P, Steel M, Liew FY, Mowat AM. CD4+ but not CD8+ T cells are required for the induction of oral tolerance. Int Immunol. 1995 Mar;7(3):501–4. doi: 10.1093/intimm/7.3.501. [DOI] [PubMed] [Google Scholar]
- 70.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002 Jun 14;109(6):719–31. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 71.Kriegel MA, Rathinam C, Flavell RA. E3 ubiquitin ligase GRAIL controls primary T cell activation and oral tolerance. Proc Natl Acad Sci U S A. 2009 Sep 29;106(39):16770–5. doi: 10.1073/pnas.0908957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ise W, Nakamura K, Shimizu N, Goto H, Fujimoto K, Kaminogawa S, et al. Orally tolerized T cells can form conjugates with APCs but are defective in immunological synapse formation. J Immunol. 2005 Jul 15;175(2):829–38. doi: 10.4049/jimmunol.175.2.829. [DOI] [PubMed] [Google Scholar]
- 73.Mirenda V, Millington O, Lechler RI, Scott D, Hernandez-Fuentes MP, Read J, et al. Tolerant T cells display impaired trafficking ability. Eur J Immunol. 2005 Jul;35(7):2146–56. doi: 10.1002/eji.200425823. [DOI] [PubMed] [Google Scholar]
- 74.Song F, Guan Z, Gienapp IE, Shawler T, Benson J, Whitacre CC. The thymus plays a role in oral tolerance in experimental autoimmune encephalomyelitis. J Immunol. 2006 Aug 1;177(3):1500–9. doi: 10.4049/jimmunol.177.3.1500. [DOI] [PubMed] [Google Scholar]
- 75.Chung Y, Lee SH, Kim DH, Kang CY. Complementary role of CD4+CD25+ regulatory T cells and TGF-beta in oral tolerance. J Leukoc Biol. 2005 Jun;77(6):906–13. doi: 10.1189/jlb.1004599. [DOI] [PubMed] [Google Scholar]
- 76.Ju JH, Cho ML, Jhun JY, Park MJ, Oh HJ, Min SY, et al. Oral administration of type-II collagen suppresses IL-17-associated RANKL expression of CD4+ T cells in collagen-induced arthritis. Immunol Lett. 2008 Apr 15;117(1):16–25. doi: 10.1016/j.imlet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 77.Chen Y, Inobe J, Kuchroo VK, Baron JL, Janeway CA, Jr, Weiner HL. Oral tolerance in myelin basic protein T-cell receptor transgenic mice: suppression of autoimmune encephalomyelitis and dose-dependent induction of regulatory cells. Proc Natl Acad Sci U S A. 1996 Jan 9;93(1):388–91. doi: 10.1073/pnas.93.1.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Y, Inobe J, Marks R, Gonnella P, Kuchroo VK, Weiner HL. Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature. 1995 Jul 13;376(6536):177–80. doi: 10.1038/376177a0. [DOI] [PubMed] [Google Scholar]
- 79.Oida T, Xu L, Weiner HL, Kitani A, Strober W. TGF-beta-mediated suppression by CD4+CD25+ T cells is facilitated by CTLA-4 signaling. J Immunol. 2006 Aug 15;177(4):2331–9. doi: 10.4049/jimmunol.177.4.2331. [DOI] [PubMed] [Google Scholar]
- 80.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008 Oct 10;322(5899):271–5. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 81.Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol. 2007 Aug;7(8):622–32. doi: 10.1038/nri2134. [DOI] [PubMed] [Google Scholar]
- 82.Slavin AJ, Maron R, Weiner HL. Mucosal administration of IL-10 enhances oral tolerance in autoimmune encephalomyelitis and diabetes. Int Immunol. 2001 Jun;13(6):825–33. doi: 10.1093/intimm/13.6.825. [DOI] [PubMed] [Google Scholar]
- 83.Peron JP, Yang K, Chen ML, Brandao WN, Basso AS, Commodaro AG, et al. Oral tolerance reduces Th17 cells as well as the overall inflammation in the central nervous system of EAE mice. J Neuroimmunol. 2010 Oct 8;227(1-2):10–7. doi: 10.1016/j.jneuroim.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 84.Ishikawa H, Ochi H, Chen ML, Frenkel D, Maron R, Weiner HL. Inhibition of autoimmune diabetes by oral administration of anti-CD3 monoclonal antibody. Diabetes. 2007 Aug;56(8):2103–9. doi: 10.2337/db06-1632. [DOI] [PubMed] [Google Scholar]
- 85.Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003 Sep;9(9):1202–8. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- 86.Ilan Y, Maron R, Tukpah AM, Maioli TU, Murugaiyan G, Yang K, et al. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci U S A. 2010 May 25;107(21):9765–70. doi: 10.1073/pnas.0908771107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu HY, Center EM, Tsokos GC, Weiner HL. Suppression of murine SLE by oral anti-CD3: inducible CD4+CD25-LAP+ regulatory T cells control the expansion of IL-17+ follicular helper T cells. Lupus. 2009 Jun;18(7):586–96. doi: 10.1177/0961203308100511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu HY, Maron R, Tukpah AM, Weiner HL. Mucosal anti-CD3 monoclonal antibody attenuates collagen-induced arthritis that is associated with induction of LAP+ regulatory T cells and is enhanced by administration of an emulsome-based Th2-skewing adjuvant. J Immunol. 2010 Sep 15;185(6):3401–7. doi: 10.4049/jimmunol.1000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sasaki N, Yamashita T, Takeda M, Shinohara M, Nakajima K, Tawa H, et al. Oral anti-CD3 antibody treatment induces regulatory T cells and inhibits the development of atherosclerosis in mice. Circulation. 2009 Nov 17;120(20):1996–2005. doi: 10.1161/CIRCULATIONAHA.109.863431. [DOI] [PubMed] [Google Scholar]
- 90.Wu HY, Quintana FJ, Weiner HL. Nasal anti-CD3 antibody ameliorates lupus by inducing an IL-10-secreting CD4+ CD25- LAP+ regulatory T cell and is associated with down-regulation of IL-17+ CD4+ ICOS+ CXCR5+ follicular helper T cells. J Immunol. 2008 Nov 1;181(9):6038–50. doi: 10.4049/jimmunol.181.9.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krause I, Blank M, Sherer Y, Gilburd B, Kvapil F, Shoenfeld Y. Induction of oral tolerance in experimental antiphospholipid syndrome by feeding with polyclonal immunoglobulins. Eur J Immunol. 2002 Dec;32(12):3414–24. doi: 10.1002/1521-4141(200212)32:12<3414::AID-IMMU3414>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 92.Maier E, Reipert BM, Novy-Weiland T, Auer W, Baumgartner B, Muchitsch EM, et al. Induction of immune tolerance by oral IVIG. Int Immunopharmacol. 2007 Mar;7(3):351–9. doi: 10.1016/j.intimp.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 93.Miller A, Lider O, Weiner HL. Antigen-driven bystander suppression after oral administration of antigens. J Exp Med. 1991 Oct 1;174(4):791–8. doi: 10.1084/jem.174.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bluestone JA, Thomson AW, Shevach EM, Weiner HL. What does the future hold for cell-based tolerogenic therapy? Nat Rev Immunol. 2007 Aug;7(8):650–4. doi: 10.1038/nri2137. [DOI] [PubMed] [Google Scholar]
- 95.Tran DQ, Shevach EM. Therapeutic potential of FOXP3(+) regulatory T cells and their interactions with dendritic cells. Hum Immunol. 2009 May;70(5):294–9. doi: 10.1016/j.humimm.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, Woisetschlager M, et al. Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell-mediated effects on regulatory T cells. Blood. 2008 Aug 15;112(4):1214–22. doi: 10.1182/blood-2007-08-109843. [DOI] [PubMed] [Google Scholar]
- 97.Kerkvliet NI, Steppan LB, Vorachek W, Oda S, Farrer D, Wong CP, et al. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy. 2009;1(4):539–47. doi: 10.2217/imt.09.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453(7191):65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 99.Zhang L, Ma J, Takeuchi M, Usui Y, Hattori T, Okunuki Y, et al. Suppression of experimental autoimmune uveoretinitis by inducing differentiation of regulatory T cells via activation of aryl hydrocarbon receptor. Invest Ophthalmol Vis Sci. 2010 Dec 10;51(4):2109–17. doi: 10.1167/iovs.09-3993. [DOI] [PubMed] [Google Scholar]
- 100.Henry EC, Bemis JC, Henry O, Kende AS, Gasiewicz TA. A potential endogenous ligand for the aryl hydrocarbon receptor has potent agonist activity in vitro and in vivo. Arch Biochem Biophys. 2006 Jun 1;450(1):67–77. doi: 10.1016/j.abb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 101.Okey AB, Vella LM, Harper PA. Detection and characterization of a low affinity form of cytosolic Ah receptor in livers of mice nonresponsive to induction of cytochrome P1-450 by 3-methylcholanthrene. Mol Pharmacol. 1989 Jun;35(6):823–30. [PubMed] [Google Scholar]
- 102.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008 Jul 15;105(28):9721–6. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, et al. From the Cover: An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010 Nov 30;107(48):20768–73. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wei J, Duramad O, Perng OA, Reiner SL, Liu YJ, Qin FX. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2007 Nov 13;104(46):18169–74. doi: 10.1073/pnas.0703642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chang JH, Kim YJ, Han SH, Kang CY. IFN-gamma-STAT1 signal regulates the differentiation of inducible Treg: potential role for ROS-mediated apoptosis. Eur J Immunol. 2009 May 1;39(5):1241–51. doi: 10.1002/eji.200838913. [DOI] [PubMed] [Google Scholar]
- 106.Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? 2009 doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 107.Control of Regulatory T Cell Lineage Commitment and Maintenance. 2009 doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Machado FS, Johndrow JE, Esper L, Dias A, Bafica A, Serhan CN, et al. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat Med. 2006 Mar;12(3):330–4. doi: 10.1038/nm1355. [DOI] [PubMed] [Google Scholar]
- 109.Platzer B, Richter S, Kneidinger D, Waltenberger D, Woisetschlager M, Strobl H. Aryl hydrocarbon receptor activation inhibits in vitro differentiation of human monocytes and Langerhans dendritic cells. J Immunol. 2009 Jul 1;183(1):66–74. doi: 10.4049/jimmunol.0802997. [DOI] [PubMed] [Google Scholar]
- 110.Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med. 2009 Aug 31;206(9):2027–35. doi: 10.1084/jem.20090560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sekine H, Mimura J, Oshima M, Okawa H, Kanno J, Igarashi K, et al. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol Cell Biol. 2009 Dec;29(24):6391–400. doi: 10.1128/MCB.00337-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wada Y, Hisamatsu T, Kamada N, Okamoto S, Hibi T. Retinoic acid contributes to the induction of IL-12-hypoproducing dendritic cells. Inflamm Bowel Dis. 2009 Oct;15(10):1548–56. doi: 10.1002/ibd.20934. [DOI] [PubMed] [Google Scholar]
- 113.Kawakami Y, Inagaki N, Salek-Ardakani S, Kitaura J, Tanaka H, Nagao K, et al. Regulation of dendritic cell maturation and function by Bruton's tyrosine kinase via IL-10 and Stat3. Proc Natl Acad Sci U S A. 2006 Jan 3;103(1):153–8. doi: 10.1073/pnas.0509784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006 May 11;441(7090):235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 115.Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2008 Nov 25;105(47):18460–5. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010 Sep;11(9):854–61. doi: 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol. 2010 Sep;11(9):846–53. doi: 10.1038/ni.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chmill S, Kadow S, Winter M, Weighardt H, Esser C. 2,3,7,8-Tetrachlorodibenzo-p-dioxin impairs stable establishment of oral tolerance in mice. Toxicol Sci. 2010 Nov;118(1):98–107. doi: 10.1093/toxsci/kfq232. [DOI] [PubMed] [Google Scholar]
- 119.Kinoshita H, Abe J, Akadegawa K, Yurino H, Uchida T, Ikeda S, et al. Breakdown of mucosal immunity in gut by 2,3,7,8-tetraclorodibenzo-p-dioxin (TCDD) Environmental Health and Preventive Medicine. 2006;11(5):256–63. doi: 10.1007/BF02898015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kerkvliet NI. AHR-mediated immunomodulation: the role of altered gene transcription. Biochem Pharmacol. 2009 Feb 15;77(4):746–60. doi: 10.1016/j.bcp.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schecter A, Birnbaum L, Ryan JJ, Constable JD. Dioxins: an overview. Environ Res. 2006 Jul;101(3):419–28. doi: 10.1016/j.envres.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 122.Kerkvliet NI. Recent advances in understanding the mechanisms of TCDD immunotoxicity. Int Immunopharmacol. 2002 Feb;2(2-3):277–91. doi: 10.1016/s1567-5769(01)00179-5. [DOI] [PubMed] [Google Scholar]
- 123.van Leeuwen FX, Feeley M, Schrenk D, Larsen JC, Farland W, Younes M. Dioxins: WHO's tolerable daily intake (TDI) revisited. Chemosphere. 2000 May-Jun;40(9-11):1095–101. doi: 10.1016/s0045-6535(99)00358-6. [DOI] [PubMed] [Google Scholar]
- 124.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008 Jan;21(1):102–16. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008 Jun;8(6):411–20. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 126.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005 Jul 15;122(1):107–18. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 127.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008 May 29;453(7195):620–5. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 128.Maassen CB, Claassen E. Strain-dependent effects of probiotic lactobacilli on EAE autoimmunity. Vaccine. 2008 Apr 16;26(17):2056–7. doi: 10.1016/j.vaccine.2008.02.035. [DOI] [PubMed] [Google Scholar]
- 129.Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque-Begum S, et al. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. 2009 Nov 15;183(10):6041–50. doi: 10.4049/jimmunol.0900747. [DOI] [PubMed] [Google Scholar]
- 130.Ochoa-Reparaz J, Mielcarz DW, Wang Y, Begum-Haque S, Dasgupta S, Kasper DL, et al. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 2010 Sep;3(5):487–95. doi: 10.1038/mi.2010.29. [DOI] [PubMed] [Google Scholar]
- 131.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008 Oct 23;455(7216):1109–13. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008 Oct 9;455(7214):808–12. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 133.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008 Oct 16;4(4):337–49. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010 Jun 25;32(6):815–27. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Walton KL, Galanko JA, Balfour Sartor R, Fisher NC. T cell-mediated oral tolerance is intact in germ-free mice. Clin Exp Immunol. 2006 Mar;143(3):503–12. doi: 10.1111/j.1365-2249.2006.03019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Repa A, Kozakova H, Hudcovic T, Stepankova R, Hrncir T, Tlaskalova-Hogenova H, et al. Susceptibility to nasal and oral tolerance induction to the major birch pollen allergen Bet v 1 is not dependent on the presence of the microflora. Immunol Lett. 2008 Apr 15;117(1):50–6. doi: 10.1016/j.imlet.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 137.Tsuda M, Hosono A, Yanagibashi T, Kihara-Fujioka M, Hachimura S, Itoh K, et al. Intestinal commensal bacteria promote T cell hyporesponsiveness and down-regulate the serum antibody responses induced by dietary antigen. Immunol Lett. 2010 Aug 16;132(1-2):45–52. doi: 10.1016/j.imlet.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 138.Yang R, Liu Q, Grosfeld JL, Pescovitz MD. Intestinal venous drainage through the liver is a prerequisite for oral tolerance induction. J Pediatr Surg. 1994 Aug;29(8):1145–8. doi: 10.1016/0022-3468(94)90297-6. [DOI] [PubMed] [Google Scholar]
- 139.Watanabe T, Katsukura H, Shirai Y, Yamori M, Nishi T, Chiba T, et al. A liver tolerates a portal antigen by generating CD11c+ cells, which select Fas ligand+ Th2 cells via apoptosis. Hepatology. 2003 Aug;38(2):403–12. doi: 10.1053/jhep.2003.50343. [DOI] [PubMed] [Google Scholar]
- 140.Limmer A, Ohl J, Wingender G, Berg M, Jungerkes F, Schumak B, et al. Cross-presentation of oral antigens by liver sinusoidal endothelial cells leads to CD8 T cell tolerance. Eur J Immunol. 2005 Oct;35(10):2970–81. doi: 10.1002/eji.200526034. [DOI] [PubMed] [Google Scholar]
- 141.May AG, Bauer S, Leddy JP, Panner B, Vaughan J, Russell PS. Survival of allografts after hepatic portal venous administration of specific transplantation antigen. Ann Surg. 1969 Nov;170(5):824–32. doi: 10.1097/00000658-196911000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hultkrantz S, Ostman S, Telemo E. Induction of antigen-specific regulatory T cells in the liver-draining celiac lymph node following oral antigen administration. Immunology. 2005 Nov;116(3):362–72. doi: 10.1111/j.1365-2567.2005.02236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Safadi R, Alvarez CE, Ohta M, Brimnes J, Kraus T, Mehal W, et al. Enhanced oral tolerance in transgenic mice with hepatocyte secretion of IL-10. J Immunol. 2005 Sep 15;175(6):3577–83. doi: 10.4049/jimmunol.175.6.3577. [DOI] [PubMed] [Google Scholar]
- 144.Luth S, Huber S, Schramm C, Buch T, Zander S, Stadelmann C, et al. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J Clin Invest. 2008 Oct;118(10):3403–10. doi: 10.1172/JCI32132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yu S, Nakafusa Y, Flye MW. Portal vein administration of donor cells promotes peripheral allospecific hyporesponsiveness and graft tolerance. Surgery. 1994 Aug;116(2):229–34. discussion 34-5. [PubMed] [Google Scholar]
- 146.Roelofs-Haarhuis K, Wu X, Gleichmann E. Oral tolerance to nickel requires CD4+ invariant NKT cells for the infectious spread of tolerance and the induction of specific regulatory T cells. J Immunol. 2004 Jul 15;173(2):1043–50. doi: 10.4049/jimmunol.173.2.1043. [DOI] [PubMed] [Google Scholar]
- 147.Kim HJ, Hwang SJ, Kim BK, Jung KC, Chung DH. NKT cells play critical roles in the induction of oral tolerance by inducing regulatory T cells producing IL-10 and transforming growth factor beta, and by clonally deleting antigen-specific T cells. Immunology. 2006 May;118(1):101–11. doi: 10.1111/j.1365-2567.2006.02346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Trop S, Samsonov D, Gotsman I, Alper R, Diment J, Ilan Y. Liver-associated lymphocytes expressing NK1.1 are essential for oral immune tolerance induction in a murine model. Hepatology. 1999 Mar;29(3):746–55. doi: 10.1002/hep.510290334. [DOI] [PubMed] [Google Scholar]
- 149.Ilan Y, Weksler-Zangen S, Ben-Horin S, Diment J, Sauter B, Rabbani E, et al. Treatment of experimental colitis by oral tolerance induction: a central role for suppressor lymphocytes. Am J Gastroenterol. 2000 Apr;95(4):966–73. doi: 10.1111/j.1572-0241.2000.01935.x. [DOI] [PubMed] [Google Scholar]
- 150.Margalit M, Israeli E, Shibolet O, Zigmond E, Klein A, Hemed N, et al. A double-blind clinical trial for treatment of Crohn's disease by oral administration of Alequel, a mixture of autologous colon-extracted proteins: a patient-tailored approach. Am J Gastroenterol. 2006 Mar;101(3):561–8. doi: 10.1111/j.1572-0241.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 151.Broere F, Wieten L, Klein Koerkamp EI, van Roon JA, Guichelaar T, Lafeber FP, et al. Oral or nasal antigen induces regulatory T cells that suppress arthritis and proliferation of arthritogenic T cells in joint draining lymph nodes. J Immunol. 2008 Jul 15;181(2):899–906. doi: 10.4049/jimmunol.181.2.899. [DOI] [PubMed] [Google Scholar]
- 152.Keller AC, Mucida D, Gomes E, Faquim-Mauro E, Faria AM, Rodriguez D, et al. Hierarchical suppression of asthma-like responses by mucosal tolerance. J Allergy Clin Immunol. 2006 Feb;117(2):283–90. doi: 10.1016/j.jaci.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 153.van den Berg H, Greuter M, Kraal G, den Haan JM. Different mechanisms regulate CD4(+) T cell independent induction of oral and nasal tolerance of CD8(+) T cells. Immunobiology. 2010;215(2):163–71. doi: 10.1016/j.imbio.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 154.Cox LS, Larenas Linnemann D, Nolte H, Weldon D, Finegold I, Nelson HS. Sublingual immunotherapy: a comprehensive review. J Allergy Clin Immunol. 2006 May;117(5):1021–35. doi: 10.1016/j.jaci.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 155.Ciprandi G, Fenoglio D, Cirillo I, Tosca MA, La Rosa M, Licari A, et al. Sublingual immunotherapy: an update on immunologic and functional effects. Allergy Asthma Proc. 2007 Jan-Feb;28(1):40–3. doi: 10.2500/aap.2007.28.2974. [DOI] [PubMed] [Google Scholar]
- 156.Sun JB, Cuburu N, Blomquist M, Li BL, Czerkinsky C, Holmgren J. Sublingual tolerance induction with antigen conjugated to cholera toxin B subunit induces Foxp3+CD25+CD4+ regulatory T cells and suppresses delayed-type hypersensitivity reactions. Scand J Immunol. 2006 Sep;64(3):251–9. doi: 10.1111/j.1365-3083.2006.01823.x. [DOI] [PubMed] [Google Scholar]
- 157.Sun JB, Czerkinsky C, Holmgren J. Sublingual ‘oral tolerance’ induction with antigen conjugated to cholera toxin B subunit generates regulatory T cells that induce apoptosis and depletion of effector T cells. Scand J Immunol. 2007 Aug-Sep;66(2-3):278–86. doi: 10.1111/j.1365-3083.2007.01975.x. [DOI] [PubMed] [Google Scholar]
- 158.Verhasselt V, Milcent V, Cazareth J, Kanda A, Fleury S, Dombrowicz D, et al. Breast milk-mediated transfer of an antigen induces tolerance and protection from allergic asthma. Nat Med. 2008 Feb;14(2):170–5. doi: 10.1038/nm1718. [DOI] [PubMed] [Google Scholar]
- 159.Verhasselt V. Oral tolerance in neonates: from basics to potential prevention of allergic disease. Mucosal Immunol. 2010 Jul;3(4):326–33. doi: 10.1038/mi.2010.25. [DOI] [PubMed] [Google Scholar]
- 160.Lonnqvist A, Ostman S, Almqvist N, Hultkrantz S, Telemo E, Wold AE, et al. Neonatal exposure to staphylococcal superantigen improves induction of oral tolerance in a mouse model of airway allergy. Eur J Immunol. 2009 Feb;39(2):447–56. doi: 10.1002/eji.200838418. [DOI] [PubMed] [Google Scholar]
- 161.Lundell AC, Adlerberth I, Lindberg E, Karlsson H, Ekberg S, Aberg N, et al. Increased levels of circulating soluble CD14 but not CD83 in infants are associated with early intestinal colonization with Staphylococcus aureus. Clin Exp Allergy. 2007 Jan;37(1):62–71. doi: 10.1111/j.1365-2222.2006.02625.x. [DOI] [PubMed] [Google Scholar]
- 162.Aoyama K, Koyama M, Matsuoka K, Hashimoto D, Ichinohe T, Harada M, et al. Improved outcome of allogeneic bone marrow transplantation due to breastfeeding-induced tolerance to maternal antigens. Blood. 2009 Feb 19;113(8):1829–33. doi: 10.1182/blood-2008-05-155283. [DOI] [PubMed] [Google Scholar]
- 163.Miller A, Lider O, Abramsky O, Weiner HL. Orally administered myelin basic protein in neonates primes for immune responses and enhances experimental autoimmune encephalomyelitis in adult animals. Eur J Immunol. 1994 May;24(5):1026–32. doi: 10.1002/eji.1830240503. [DOI] [PubMed] [Google Scholar]
- 164.Maron R, Guerau-de-Arellano M, Zhang X, Weiner HL. Oral administration of insulin to neonates suppresses spontaneous and cyclophosphamide induced diabetes in the NOD mouse. J Autoimmun. 2001 Feb;16(1):21–8. doi: 10.1006/jaut.2000.0471. [DOI] [PubMed] [Google Scholar]
- 165.Nagler-Anderson C, Bober LA, Robinson ME, Siskind GW, Thorbecke GJ. Suppression of type II collagen-induced arthritis by intragastric administration of soluble type II collagen. Proc Natl Acad Sci U S A. 1986 Oct;83(19):7443–6. doi: 10.1073/pnas.83.19.7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Higgins PJ, Weiner HL. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein and its fragments. J Immunol. 1988 Jan 15;140(2):440–5. [PubMed] [Google Scholar]
- 167.Bitar DM, Whitacre CC. Suppression of experimental autoimmune encephalomyelitis by the oral administration of myelin basic protein. Cell Immunol. 1988 Apr 1;112(2):364–70. doi: 10.1016/0008-8749(88)90305-x. [DOI] [PubMed] [Google Scholar]
- 168.Frenkel D, Huang Z, Maron R, Koldzic DN, Hancock WW, Moskowitz MA, et al. Nasal vaccination with myelin oligodendrocyte glycoprotein reduces stroke size by inducing IL-10-producing CD4+ T cells. J Immunol. 2003 Dec 15;171(12):6549–55. doi: 10.4049/jimmunol.171.12.6549. [DOI] [PubMed] [Google Scholar]
- 169.Frenkel D, Pachori AS, Zhang L, Dembinsky-Vaknin A, Farfara D, Petrovic-Stojkovic S, et al. Nasal vaccination with troponin reduces troponin specific T-cell responses and improves heart function in myocardial ischemia-reperfusion injury. Int Immunol. 2009 Jul;21(7):817–29. doi: 10.1093/intimm/dxp051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kurien BT, Asfa S, Li C, Dorri Y, Jonsson R, Scofield RH. Induction of oral tolerance in experimental Sjogren's syndrome autoimmunity. Scand J Immunol. 2005 May;61(5):418–25. doi: 10.1111/j.1365-3083.2005.01593.x. [DOI] [PubMed] [Google Scholar]
- 171.Huibregtse IL, Snoeck V, de Creus A, Braat H, De Jong EC, Van Deventer SJ, et al. Induction of ovalbumin-specific tolerance by oral administration of Lactococcus lactis secreting ovalbumin. Gastroenterology. 2007 Aug;133(2):517–28. doi: 10.1053/j.gastro.2007.04.073. [DOI] [PubMed] [Google Scholar]
- 172.Frossard CP, Steidler L, Eigenmann PA. Oral administration of an IL-10-secreting Lactococcus lactis strain prevents food-induced IgE sensitization. J Allergy Clin Immunol. 2007 Apr;119(4):952–9. doi: 10.1016/j.jaci.2006.12.615. [DOI] [PubMed] [Google Scholar]
- 173.Huibregtse IL, Marietta EV, Rashtak S, Koning F, Rottiers P, David CS, et al. Induction of antigen-specific tolerance by oral administration of Lactococcus lactis delivered immunodominant DQ8-restricted gliadin peptide in sensitized nonobese diabetic Abo Dq8 transgenic mice. J Immunol. 2009 Aug 15;183(4):2390–6. doi: 10.4049/jimmunol.0802891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sun JB, Czerkinsky C, Holmgren J. Mucosally induced immunological tolerance, regulatory T cells and the adjuvant effect by cholera toxin B subunit. Scand J Immunol. 2010 Jan;71(1):1–11. doi: 10.1111/j.1365-3083.2009.02321.x. [DOI] [PubMed] [Google Scholar]
- 175.Takagi H, Hiroi T, Yang L, Takamura K, Ishimitsu R, Kawauchi H, et al. Efficient induction of oral tolerance by fusing cholera toxin B subunit with allergen-specific T-cell epitopes accumulated in rice seed. Vaccine. 2008 Nov 11;26(48):6027–30. doi: 10.1016/j.vaccine.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 176.Saint-Lu N, Tourdot S, Razafindratsita A, Mascarell L, Berjont N, Chabre H, et al. Targeting the allergen to oral dendritic cells with mucoadhesive chitosan particles enhances tolerance induction. Allergy. 2009 Jul;64(7):1003–13. doi: 10.1111/j.1398-9995.2009.01945.x. [DOI] [PubMed] [Google Scholar]
- 177.Lavasani S, Dzhambazov B, Nouri M, Fak F, Buske S, Molin G, et al. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS One. 2010;5(2):e9009. doi: 10.1371/journal.pone.0009009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ando T, Hatsushika K, Wako M, Ohba T, Koyama K, Ohnuma Y, et al. Orally administered TGF-beta is biologically active in the intestinal mucosa and enhances oral tolerance. J Allergy Clin Immunol. 2007 Oct;120(4):916–23. doi: 10.1016/j.jaci.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 179.Rynda A, Maddaloni M, Mierzejewska D, Ochoa-Reparaz J, Maslanka T, Crist K, et al. Low-dose tolerance is mediated by the microfold cell ligand, reovirus protein sigma1. J Immunol. 2008 Apr 15;180(8):5187–200. doi: 10.4049/jimmunol.180.8.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Suzuki H, Sekine S, Kataoka K, Pascual DW, Maddaloni M, Kobayashi R, et al. Ovalbumin-protein sigma 1 M-cell targeting facilitates oral tolerance with reduction of antigen-specific CD4+ T cells. Gastroenterology. 2008 Sep;135(3):917–25. doi: 10.1053/j.gastro.2008.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kim WU, Lee WK, Ryoo JW, Kim SH, Kim J, Youn J, et al. Suppression of collagen-induced arthritis by single administration of poly(lactic-co-glycolic acid) nanoparticles entrapping type II collagen: a novel treatment strategy for induction of oral tolerance. Arthritis Rheum. 2002 Apr;46(4):1109–20. doi: 10.1002/art.10198. [DOI] [PubMed] [Google Scholar]
- 182.Yamada Y, Sekine Y, Yoshida S, Yasufuku K, Petrache I, Benson HL, et al. Type V collagen-induced oral tolerance plus low-dose cyclosporine prevents rejection of MHC class I and II incompatible lung allografts. J Immunol. 2009 Jul 1;183(1):237–45. doi: 10.4049/jimmunol.0804028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, et al. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006 May;116(5):1371–81. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Park KS, Park MJ, Cho ML, Kwok SK, Ju JH, Ko HJ, et al. Type II collagen oral tolerance; mechanism and role in collagen-induced arthritis and rheumatoid arthritis. Mod Rheumatol. 2009;19(6):581–9. doi: 10.1007/s10165-009-0210-0. [DOI] [PubMed] [Google Scholar]
- 185.Koffeman EC, Genovese M, Amox D, Keogh E, Santana E, Matteson EL, et al. Epitope-specific immunotherapy of rheumatoid arthritis: clinical responsiveness occurs with immune deviation and relies on the expression of a cluster of molecules associated with T cell tolerance in a double-blind, placebo-controlled, pilot phase II trial. Arthritis Rheum. 2009 Nov;60(11):3207–16. doi: 10.1002/art.24916. [DOI] [PubMed] [Google Scholar]
- 186.Postlethwaite AE, Wong WK, Clements P, Chatterjee S, Fessler BJ, Kang AH, et al. A multicenter, randomized, double-blind, placebo-controlled trial of oral type I collagen treatment in patients with diffuse cutaneous systemic sclerosis: I. oral type I collagen does not improve skin in all patients, but may improve skin in late-phase disease. Arthritis Rheum. 2008 Jun;58(6):1810–22. doi: 10.1002/art.23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kapp K, Maul J, Hostmann A, Mundt P, Preiss JC, Wenzel A, et al. Modulation of systemic antigen-specific immune responses by oral antigen in humans. Eur J Immunol. 2010 Nov;40(11):3128–37. doi: 10.1002/eji.201040701. [DOI] [PubMed] [Google Scholar]
- 188.Husby S, Mestecky J, Moldoveanu Z, Holland S, Elson CO. Oral tolerance in humans. T cell but not B cell tolerance after antigen feeding. J Immunol. 1994 May 1;152(9):4663–70. [PubMed] [Google Scholar]
- 189.Kraus TA, Cheifetz A, Toy L, Meddings JB, Mayer L. Evidence for a genetic defect in oral tolerance induction in inflammatory bowel disease. Inflamm Bowel Dis. 2006 Feb;12(2):82–8. doi: 10.1097/01.MIB.0000200343.61707.52. discussion 1. [DOI] [PubMed] [Google Scholar]
- 190.Skyler JS. Update on worldwide efforts to prevent type 1 diabetes. Ann N Y Acad Sci. 2008 Dec;1150:190–6. doi: 10.1196/annals.1447.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Vickery BP, Burks AW. Immunotherapy in the treatment of food allergy: focus on oral tolerance. Curr Opin Allergy Clin Immunol. 2009 Aug;9(4):364–70. doi: 10.1097/ACI.0b013e32832d9add. [DOI] [PubMed] [Google Scholar]
- 192.Caminiti L, Passalacqua G, Barberi S, Vita D, Barberio G, De Luca R, et al. A new protocol for specific oral tolerance induction in children with IgE-mediated cow's milk allergy. Allergy Asthma Proc. 2009 Jul-Aug;30(4):443–8. doi: 10.2500/aap.2009.30.3221. [DOI] [PubMed] [Google Scholar]
- 193.Ganeshan K, Neilsen CV, Hadsaitong A, Schleimer RP, Luo X, Bryce PJ. Impairing oral tolerance promotes allergy and anaphylaxis: a new murine food allergy model. J Allergy Clin Immunol. 2009 Jan;123(1):231–8 e4. doi: 10.1016/j.jaci.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]