

Abstract
The regulation of energy homeostasis integrates diverse biological processes ranging from behavior to metabolism and is linked fundamentally to numerous disease states. To identify new molecules that can bypass homeostatic compensatory mechanisms of energy balance in intact animals, we screened for small molecule modulators of C. elegans fat content. We report on several molecules that modulate fat storage without obvious deleterious effects on feeding, growth, and reproduction. A subset of these compounds also altered fat storage in mammalian and insect cell culture. We found that one of the newly identified compounds exerts its effects in C. elegans through a pathway that requires novel functions of an AMP-activated kinase catalytic subunit and a transcription factor previously unassociated with fat regulation. Thus, our strategy identifies small molecules that are effective within the context of intact animals and reveals relationships between new pathways that operate across phyla to influence energy homeostasis.
INTRODUCTION
In metazoans, energy homeostasis is regulated by cellular and organism-wide networks that control behavior and the intertwined physiologies of nutrient uptake, transport, lipid synthesis, storage and utilization [1]. Excessive fat accumulation and aberrant metabolism underlie susceptibility to numerous pathologies ranging from type II diabetes to certain forms of cancer [2]. Pharmacological agents that modulate energy balance in the context of whole organisms could significantly facilitate the understanding of and treatment of metabolic disorders. However, the homeostatic nature of energy regulatory networks and the sophisticated chemo-protective barriers of intact organisms render many compounds that show excellent in vitro or in cell-based efficacy ineffective when administered to whole animals [3, 4]. One approach to this challenge is to conduct screens on whole animals, rather than pre-determined targets, to identify compounds that elicit desired responses in intact animals [5].
C. elegans offers the possibility for relatively cost-effective whole-animal screening [6–8]. Additionally, the speed and ease with which C. elegans can be genetically manipulated provides a currently unparalleled experimental path for elucidating mechanisms that underlie a compound’s actions [9]. For instance, many fundamental components of neural signaling cascades were first discovered by screening for mutants that were either resistant or hypersensitive to application of various neurotransmitters to whole nematodes [10, 11]. This approach is not restricted to identifying a compound’s binding target, but importantly, can reveal combinations of pathways that mediate the physiological consequences of compound activity.
Since the maintenance of energy homeostasis is central to life, the underlying molecular mechanisms of its regulation are highly conserved across phylogeny. As in mammals, fat in C. elegans is derived from conversion of absorbed nutrients and other internal resources through de novo synthesis as well as the direct uptake of dietary fatty acids [12]. A majority of C. elegans’ extractable fatty acids is derived from dietary uptake of palmitic acid and shorter chain fatty acids [13]. In addition, many of the known mammalian metabolic regulatory pathways operate similarly in C. elegans [12, 14]. These include fat and sugar uptake, transport, synthesis and degradation mechanisms, their transcriptional regulators such as SREBP (sterol response element binding protein) [15], cellular fuel gauge mechanisms such as TOR (target of rapamycin)[16], and neuroendocrine regulators such as insulin [17]. Moreover, in C. elegans as in mammals, serotonin, dopamine and neuropeptide Y-like signaling pathways regulate food-related behaviors [18–20]. Thus, while the mammalian regulatory pathways involving energy homeostasis are more sophisticated than that of nematodes, the core mechanisms are largely conserved.
To generate small molecule tools for probing the complex mechanisms of energy homeostasis, we developed a forward chemical screen for novel compounds that alter fat accumulation in C. elegans. Here, we report on several molecules that either elevate or depress fat storage. By combining compound treatment with systematic gene inactivation, we found that the fat lowering activity of one of the newly identified compounds is ultimately dependent on AMP-activated kinase (AMPK) as well as a transcription factor with no previously identified role in fat metabolism. These results demonstrate the utility of our approach in identifying compounds that exert their effects through evolutionarily conserved pathways as well as revealing new fat regulatory modules.
RESULTS
A whole organism screen for modulators of lipid metabolism
We chose as the basis for our assay the solvatochromatic vital dye Nile Red that has been used extensively in multiple experimental systems to identify and characterize metabolic pathways involved in fat metabolism [15, 21–24]. Nile Red staining of C. elegans intestinal lipid-containing organelles is reproducible, exhibits a large dynamic range, and is sensitive to numerous genetic and environmental manipulations expected to alter fat content [21]. In addition, it allows for the characterization of metabolic regulatory pathways that are impervious to analysis with less sensitive fixed staining or biochemical extraction methods [22]. To validate our approach, we tested whether known pharmacological modifiers of mammalian lipid metabolism altered C. elegans Nile Red staining. Animals treated with 1 mM 5-aminoimidazole-4-carboxyamide ribonucleoside (1, AICAR), an agonist of AMP-activated kinase (AMPK) [25], exhibited decreased staining, as would be predicted when AMPK is activated. The patterns and extents of fat reduction by AICAR were comparable to that seen upon treatment with 100 µM Fluoxetine (2), a serotonin re-uptake inhibitor (Fig. 1a, b) [18]. These experiments indicate that C. elegans fat storage can be modulated at the level of primary signal transduction (AICAR) and neurotransmitter release (Fluoxetine) by pharmacological agents known to modulate mammalian fat.
Figure 1. Pharmacological modulation of Nile Red staining in C. elegans.

(a) Young adult animals were cultured from first larval stage of development with Nile Red and either 1 mM AICAR, 100 µM Fluoxetine or 0.1% DMSO as a control. A representative image for each case is shown. Animals are orientated anterior (left) to posterior (right). The anterior half of each intestine is shown for Fluoxetine and AICAR treated animals. Scale bar = 40 µm (b) The integrated fluorescence intensity of the two anterior-most intestinal cells was measured for 5–7 animals. Error bars represent the standard error of the mean. (c) An outline of screen used to identify new small molecule modulators of fat metabolism.
We next screened previously uncharacterized small molecules for those that altered Nile Red staining. Developmentally synchronized nematodes were cultured in 384-well plates with individual compounds (5 µM average concentration), E. coli (food) and Nile Red for two days; then the content of each well was imaged by automated fluorescence microscopy (Fig. 1c). Nematodes were scored for growth and Nile Red fluorescence levels. We found that of 3200 compounds of a commercially available diversity library whose compounds’ physical-chemical properties were similar to most orally available drugs, 1% lowered and 0.4% elevated Nile Red staining more than two-fold relative to control treated animals. An additional 1.2% of compounds caused lethality or inhibited development. While the hit rate of this screen is high relative to in vitro high-throughput screens, it is comparable to other whole organism phenotypic screens [8].
Of the identified compounds, we further characterized 10 that caused highly reproducible Nile Red phenotypes without obvious developmental abnormalities. Compounds H17 (3), F21 (4), F17 (5), A15 (6), A13 (7), I13 (8), C5 (9) and F14(10) (Fig. 2a) reduced Nile Red staining, while H6 (11) and E8 (12) (Fig. 2b) caused increased staining. Highlighting the dynamic range of Nile Red, exposure of animals to 10 µM H17 reduced staining by >20-fold, while that of E8 elevated staining by >4-fold relative to control treated animals (Fig. 2c). Compounds that increased or decreased Nile Red staining similarly affected staining of the animals by BODIPY-conjugated fatty acids (Supplementary Fig. S1a, b), a structurally unrelated vital stain for fat. In addition, we measured the bulk ratio of triglycerides to protein extractable from nematodes that were treated with each of the compounds that reduced Nile Red staining. We found that compounds H17, F21, F17, A13 and I13 significantly decreased this ratio in a manner that correlated with their activity for reducing Nile Red staining (Supplementary Fig. S1c). No significant changes in total fat content were detected for compounds A15, C5 and F14, which may reflect the sensitivity differences or compartment/tissue specificity of the vital dye assays compared to the whole animal extraction assay.
Figure 2. Fat metabolism in C. elegans is modulated by diverse compounds.

(a,b) Structures of fat-reducing (a) and fat-increasing (b) compounds and associated Nile Red phenotypes. All images were acquired from C. elegans that were treated with 10 µM compound or 0.1% DMSO as a vehicle control. Scale bar, 20 µm. Because of the large dynamic range of the effects of compounds, shown images for the series of fat reducing compounds were acquired at different exposure intensities than those of fat increasing compounds to facilitate visualization. (c) Quantification of the integrated intensity of Nile Red-stained lipid droplets in the anterior-most 3-pairs of intestinal cells of 10 animals per condition cultured as in a and b. Images used for quantification were all acquired with the same exposure time to allow direct comparison between all conditions. ** p < 0.01: two-tailed t-test. (d) The integrated intensities of the anterior intestinal cells of 7–10 animals per serial dilution of compound. Error bars: ± s.d. (e) An overlay of green fluorescence and bright field images (top panel) showing fat-7::gfp expression in the first anterior pair of intestinal cells. Scale bar, 20 µm. Green fluorescence images of the first pair of intestinal cells from transgenic animals cultured with either 0.1% DMSO or with 10 µM compound. E8 and adjacent control images were acquired at different exposure time to facilitate visualization. (f) The mean fluorescence intensities of the first anterior intestinal cells of were quantified. n = 5–7 animals per condition *** p < 0.05: two-tailed t-test.
In the 10 compounds, eight different core scaffolds are represented, reflecting the structurally diverse library from which they were derived. The 4-hydroxy-2-oxo quinoline carboxamides F17 and F21 are structural analogs and induced similar fat phenotypes (Fig. 2a, c). In contrast, the 2-aryl substituted quinoline 4-carboxamides F14 and H6 share a similar scaffold but induced moderately low fat and high fat phenotypes, respectively. The effects of compounds F21, F17, A13, H17, F14 and E8 were dose-dependent and saturable at levels that were compound-dependent (F21, F17, A13: Fig. 2d; H17, F14, E8: Supplementary Fig. S1d), with EC50’s between 0.1–2 µM. These data are consistent with the hypothesis that the compounds induce fat reduction or elevation through specific, saturable interactions with the fat regulatory machinery and not as the result of non-specific toxicological responses to xenobiotics.
To ascertain further that the newly identified compounds alter fat metabolism in the nematode, we examined their effects on transcriptional expression of fat-7, a C. elegans ortholog of mammalian stearoyl-CoA desaturase-1 (SCD-1), which catalyzes the conversion of saturated stearoyl-CoA into mono-unsaturated oleoyl-CoA [26]. These Δ−9 desaturases regulate the ratio of unsaturated to saturated fats and are transcriptionally sensitive to the metabolic state of the cell, being induced during periods of anabolic activity and repressed during periods of fat utilization (i.e., starvation) [23, 27, 28]. We therefore expected that many small molecules that affect fat storage should in turn alter expression of this enzyme. Transgenic C. elegans carrying a fat-7::gfp reporter express FAT-7:GFP in intestinal cells, where it is most strongly visible in the first anterior pair of cells (Fig. 2e, top panel). Nematodes expressing FAT-7:GFP were cultured with 10 µM of each of the compounds shown in Fig. 2a and b or DMSO as a control. Induced changes in Nile Red intensity generally paralleled the extent of induced change in fat-7::gfp expression. Nematodes cultured on Nile Red-reducing compounds H17, A13, A15, F17, F21, and C5 exhibited reduced levels of fat-7 expression (Fig. 2e, f), while the Nile Red-increasing compound E8 caused increased expression (Fig. 2e, f). Taken together, these findings indicate that the screening strategy succeeded in identifying compounds that alter C. elegans fat levels.
Properties of newly identified fat regulatory compounds
To investigate whether the observed changes in fat could be attributed to non-specific deleterious consequences of compound treatment, we examined growth rates, fecundity, and feeding rates of compound treated animals. In all 10 cases, compound treatments induced only very slight developmental delays with no correlation between severity of the fat phenotype and growth rate (Supplementary Fig. S2a). None of the compounds significantly altered fecundity of animals (Supplementary Fig. S2b). Compounds that induced a low fat phenotype caused either normal (H17, F21, F14, I13 and C5) or elevated (F17, A15 and A13) feeding rates (Supplementary Fig. S2c). The two compounds that increased fat storage (H6, E8) did not alter feeding rates. Taken together, these results suggest that the observed fat phenotypes are not merely due to general sickness or altered feeding behavior. Our results echo recent genetic studies in C. elegans [18], in that fat content is not merely a passive consequence of feeding behavior. The excess feeding rate of animals with reduced fat levels may be indicative of compensatory mechanisms.
For an initial assessment of whether the newly identified fat regulatory compounds may have utility in the study of fat metabolism other than in C. elegans, we examined consequences of treatment of the mouse 3T3-L1 cell culture model of adipogenesis [29] and the macrophage-like Drosophila Schneider S2 cells [30] with each of the 10 compounds. Upon induction of adipogenesis, 3T3-L1 cells developed a rounded morphology and contained numerous intracellular lipid droplets (Fig. 3a). Concomitant treatment of pre-adipocyte 3T3-L1 cells with the adipogenic cocktail and each of A13, I13 or F21 compounds that reduce fat in C. elegans resulted in a dose-dependent reduction in lipid droplet formation (Fig. 3a, b). While lipid droplets in 3T3-L1 cells are derived from de novo fatty acid synthesis, S2 cells store lipids in droplets after uptake of lipoprotein complexes [30]. Of the 10 compounds tested, only A15 induced a significant alteration in the appearance of fat depots of these cells. A15 treated cells exhibited abnormally large lipid droplets (Fig. 3c, d), a phenotype similar to that observed by RNAi of genes involved in phospholipid biosynthesis [30]. Thus, the C. elegans Nile Red screen identified compounds that also modulate fat storage in mammalian and insect cell-based assays of fat metabolism.
Figure 3. Compounds that lower Nile Red staining in C. elegans modulate lipid accumulation in mammalian and insect cell culture models of adipogenesis and lipid uptake.

(a) NIH 3T3-L1 pre-adipocytes were differentiated in the presence of either DMSO or 20 µM of each compound, then fixed, stained with Oil Red O and counterstained with hematoxylin. Scale bar = 40 µm. (b) NIH 3T3-L1 cells were differentiated in the presence of different concentrations of compound then fixed and stained with Oil Red O. The amount of bound dye was extracted and quantified by measuring absorbance at 520 nm. (c) Drosophila Schneider S2 cells were cultured overnight in medium supplemented with oleate-albumin complexes. The cells were then fixed and stained with green LipidTOX neutral lipid stain to visualize lipid droplets then counter-stained with DAPI. Confocal microscopic images are overlaid. Example cells in each inset are enlarged to illustrate the droplet size phenotype. Scale bars are 10 µM. (d) The maximum size of lipid droplets in each cell was quantified for 3–5 independent image fields for more than 20 cells. Error bars represent the standard error of the mean.
The finding that only four of the ten compounds showed activity in 3T3-L1 and S2 cells may mean that fat effects of some of the compounds are C. elegans specific. However, it is also likely that each cell-based assay only represents a relatively limited metabolic space compared to a whole organism system. For example, some compounds may modulate fat storage in C. elegans through cell non-autonomous processes that originate in the nervous system of the nematode. To begin to understand how these compounds modulate fat storage we returned to C. elegans to examine the genetic requirements for the compounds’ activities.
Genetic analyses of the mechanisms of fat modulation
We treated C. elegans with a variety of genetically altered backgrounds with each of the ten compounds. We first asked whether the Nile Red-increasing E8 and H6 could retain their effects in animals deficient in SREBP, a transcription factor that regulates expression of lipid synthesis genes in mammals and C. elegans [31, 32]. RNAi mediated depletion of sbp-1 dramatically reduced Nile Red staining [21] (Supplementary Fig. 3a, b) that was unaffected by concomitant E8 or H6 treatment suggesting that Nile Red increasing effects of these compounds cannot bypass sbp-1 function. We next queried the effects of compound treatment in animals deficient in fat metabolism due to loss of fatty acyl-CoA synthase genes, in nutrient sensing due to loss of either rictor/TORC2 or AMPK signaling pathways, in bbs-7 and tub-1, C. elegans orthologs of human obesity genes [33, 34], or in insulin signaling due to loss of daf-16, encoding a FOXO-like transcription factor that is required for many of the phenotypes caused by reduction of insulin signaling [35]. In all cases, animals were susceptible to fat reduction by each of the Nile Red reducing compounds (H17, F21, F17, I13, A13, A15, C5, and F14; Supplementary Fig. 3c, d, data not shown). The only exception was that animals deficient in aak-1, which encodes a catalytic subunit of AMPK, were selectively resistant to the Nile Red-reducing effects of F17 (Fig. 4a, b).
Figure 4. The fat and feeding phenotypes induced by F17 but not F21 are due to AMPK.

(a) Nile red staining of wild-type (top row), aak-1 mutant animals (middle row) and aak-2 mutant nematodes (bottom row) cultured with 0.1% DMSO (Control, left column), 10 µM F17 (middle column) and 10 µM F21 (right column). Scale bar = 20 µm (b) The integrated fluorescence intensity of the anterior intestine was quantified for wild-type, aak-1 and aak-2 mutant nematodes cultured with 0.1% DMSO (black), 10 µM F17 (light gray) and 10 µM F21 (dark gray). 7–10 animals were quantified for each condition. Error bars represent the standard error of the mean.
aak-1 selectively mediates the effects of F17
AMPK is an evolutionarily conserved hetero-trimeric serine/threonine protein kinase complex that negatively regulates energy consuming pathways such as fat synthesis and positively regulates pathways of energy production such as fatty acid β-oxidation and glycolysis [36]. Like humans, C. elegans has two different genes (aak-1, aak-2) that encode different catalytic α-subunits. Control treated aak-1 mutants exhibited Nile Red staining that was similar to wild-type animals (Fig. 4a top row); thus the resistance of aak-1 mutants to F17 induced fat reduction was not simply a consequence of excess basal staining (Fig. 4a middle row, b). Moreover, aak-1 mutants remained susceptible to the effects of F21, despite structural similarity of this compound to F17 (Fig. 2a), suggesting that distinct mechanisms underlie the observed effects of these newly discovered compounds. While the extended lifespan of insulin signaling deficient animals [37] and excess fat content of hibernating C. elegans [38] requires aak-2, the reduced fat phenotype of F17 or F21 (Fig. 4a, b, Supplementary Fig. S4a) did not depend on the presence of this AMPK catalytic subunit.
Mice deficient in SCD-1 exhibit increased fatty acid oxidation in the liver that is correlated with increased activation of AMPK [39]. Consistent with this finding, RNAi mediated inactivation of fat-7 in C. elegans results in reduced fat storage [23]. Given our earlier observation that treatment of C. elegans with F17 decreased fat-7 expression (Fig. 2e, f), we investigated whether the F17-aak-1 mediated fat reduction required fat-7. We subjected both wild-type and aak-1 mutants to either fat-7 RNAi or vector control and then treated them with F17 or vehicle control. Wild-type animals treated with fat-7 RNAi exhibited decreased Nile Red staining that was decreased even further with F17 treatment (Supplementary Fig. S4b). While mutants in aak-1 partially abrogated the effects of F17, exposure to fat-7 RNAi induced a similar percentage change in fat reduction whether animals were treated with vehicle control or with F17 (Supplementary Fig. S4b). The additive, independent effects of these interactions indicate that the loss of FAT-7:GFP expression upon treatment with F17 is likely a consequence of the fat reduction rather than the mechanism by which F17 reduces fat storage.
The existence of a discreet function for aak-1 has until now remained largely uncharacterized. With several known direct and indirect agonists of AMPK complexes already known, we examined the effects of these molecules on wild-type and aak-1 animals to discern whether any of these known agents exhibit F17’s selective dependence on aak-1. The riboside AICAR is converted in cells to an AMP mimetic (ZMP) and is thought to activate AMPK directly by binding to the AMP-binding allosteric regulatory site [40]. C. elegans treated with 1 mM AICAR have reduced Nile Red staining (Fig. 1). However, aak-1 mutants fail to suppress the fat-reducing effects of AICAR indicating that, unlike F17, AICAR reduces fat in the nematode through a mechanism independent of aak-1 (Supplementary Fig. S4c). Another class of AMPK activating agents, the biguanides (i.e., metformin, phenformin, buformin) are thought to act indirectly on AMPK by elevating the AMP:ATP ratio through interference with mitochondrial electron transport [41]. Treating animals with 7.5 mM phenformin (13) resulted in a developmental delay that was not suppressed in aak-1 mutants (Supplementary Fig. S4d). In addition, we also treated C. elegans with the direct AMPK activator A-766962 [42]. However this β1-complex selective agonist did not affect Nile Red staining at concentrations up to 300 µM (data not shown). Thus, F17 possesses the unique ability to modulate C. elegans fat levels potently through a pathway that utilizes an AMPK complex whose function until now has been undefined.
Structural requirements for fat reduction through aak-1
To understand the structure-activity relationship for the 4-hydroxy 2-oxo-quinoline scaffold found in F17 and F21 better, we examined analogs with different 4-hydroxy quinoline substituents (R1, R2: Table 1). Compounds bearing 6-membered and larger ring systems such as pyridyl and pyrimidyl in place of the thiazolyl substituent at R2 did not exhibit low fat phenotypes (Table 1, compounds F17-3115 (14),-9110 (15),-8895 (16),-4816 (17),-4038 (18), -0796 (19)). However, other 5-membered thiazol substituted compounds reduced levels of fat storage relative to control treated animals (Table 1, compounds F17-1564 (20), -4059 (21), -8860 (22), -5735 (23), -8949 (24)) Compound F17-5735 that was substituted with a 4-phenyl group on the thiazol ring (R2) and an N-allyl group on the quinoline ring (R1) had the most potent fat reducing effect and, similar to F17, required presence of aak-1. The presence of the large-branched alkyl side chain at the R1 position of the quinoline ring was not absolutely required for aak-1 mediated fat reduction since F17-1564 had an activity profile comparable to F17. While most conservative substitutions around the thiazol ring system were tolerated in terms of targeting the aak-1 pathway, the fat-reducing effects of compound F17-8949 that has a 5-ethyl substituted thiadiazol ring system at R2 were unaffected by loss of aak-1. These findings echo the preceding epistasis data (Fig. 4) demonstrating that the low fat phenotypes exhibited by F17 and F21-treated animals are due to the interaction of these structurally similar compounds with distinct pathways. Additionally, we found that more potent modulators of the AMPK pathway can be identified through modification of the core scaffold of F17.
Table 1.
Nile Red phenotypes of different 4-hydroxy-2-oxo-quinoline carboxamides.
| Compound | 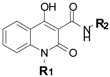 |
Nile Red (% of Control)1 |
aak-1 suppressed | |
|---|---|---|---|---|
| R1 | R2 | |||
| F17 | 14 | yes | ||
| F17-1564 | methyl | 28 | yes | |
| F17-4059 | 40 | yes | ||
| F17-8860 | 16 | yes | ||
| F21 | 7 | no | ||
| F17-5735 | 1 | yes | ||
| F17-8949 | 22 | no | ||
| F17-3115 | 100 | nd2 | ||
| F17-9110 | 100 | nd | ||
| F17-8895 | 100 | nd | ||
| F17-4816 | 100 | nd | ||
| F17-4038 | 100 | nd | ||
| F17-0796 | 237 | nd | ||
Integrated intensity of wild-type C. elegans treated with compound expressed as a percentage of DMSO-treated control.
Not Determined (nd)
F17’s fat phenotype also depends on a transcription factor
Although loss of aak-1 abrogated the fat reducing effects of F17, the requirement for aak-1 was only partial. To gain further insights into the fat reducing effects of F17, we screened through 975 clones representing approximately 70% of the genes that are predicted to encode kinases and transcription factors in the C. elegans genome. Among these, we found that RNAi-mediated inactivation of K08F8.2, which encodes a transcription factor that contains a basic leucine-zipper (bZIP) DNA binding domain [43], blocked approximately one third of F17’s Nile Red reducing phenotype (Fig. 5. Supplementary Fig. S5). While aak-1 suppressed 60–65% of the fat reducing effects of F17, RNAi inactivation of K08F8.2 in the aak-1 mutant background resulted in virtually complete abrogation of F17’s fat reducing effects (Fig. 5). Although not definitive proof, this additive suppressive interaction suggests that K08F8.2 acts in parallel to aak-1 to mediate F17’s reduced fat phenotype, rather than as a pathway component of AAK-1 signaling. Like aak-1 mutants, K08F8.2 RNAi failed to suppress the fat reducing effects of F21 (Fig. 5), further indicating that F17 and F21 modulate fat metabolism through distinct mechanisms.
Figure 5. Loss of K08F8.2 partially suppresses the F17 low fat phenotype.

First larval stage wild-type and aak-1 mutant C. elegans were cultured on HT115 E .coli expressing either a control double stranded RNA or double stranded RNA targeting K08F8.2 along with Nile Red. After 2 days of culture at 20 °C the medium was supplemented with either 10 µM F17, 10 µM F21 or 0.1% DMSO as a vehicle control. After an additional 24 hr of culture, the integrated fluorescence intensity of the first two intestinal cells of gravid adults was quantified. For each genetic background, fluorescence intensities from compound-treated animals were normalized to DMSO-treated animals of the same genetic background to correct for modest fat increasing effects of K08F8.2 RNAi. Error bars represent the standard error of the mean of 5–7 individual measurements. * p < 0.02: two-tailed t-test.
F17 activates AMPK in human liver-derived cells
To assess whether F17 affects the AMPK pathway in organisms other than C. elegans, we treated HepG2 human hepatocarcinoma cells with this compound. We used this cell line because it has been used frequently to study AMPK and its activating compounds and because liver cells are important sites of fat metabolism. Fixation and staining of this cell line with a neutral lipid specific stain revealed the presence of numerous lipid droplets contained within the cytoplasm of each cell (Fig. 6a). Treatment with F17 (25 µM) markedly reduced the number of lipid droplets in each cell (Fig. 6a, Supplementary Fig. S6a). To assess the effects of F17 on AMPK signaling directly, we treated cells with either F17, AICAR as a positive control, F17 with AMPK inhibitor compound C [44], F21 or DMSO as a vehicle control. AMPK inhibits fat synthesis by phosphorylation and inactivation of acetyl coenzyme A carboxylase (ACC) [45], which catalyzes the rate-limiting step in de novo fatty acid synthesis. Treatment with either AICAR or F17, but not F21, induced phosphorylation of ACC. F17-induced ACC phosphorylation was inhibited by simultaneous treatment with compound C (Fig. 6b, Supplementary Fig. S6b). We also observed that phosphorylation of AMPK-α, which is characteristic of its activation, was induced by AICAR and F17 treatment, but not by F21 ((Fig. 6b, Supplementary Fig. S6b). These results indicate that F17 acts as an agonist of AMPK signaling in human cells at much lower concentrations than AICAR, the widely used direct activator of AMPK. The concentration at which F17 activates the AMPK pathway in mammalian cells was also similar to that of a recently reported agonist A-592107 (25), a compound discovered through screening ~700,000 small molecules in an in vitro assay directly targeting purified AMPK [42]. Thus, these findings indicate that screening in C. elegans can yield compounds that act on pathways of therapeutic interest across phyla.
Figure 6. F17 activates the AMPK pathway and reduces the number of lipid droplets in HepG2 hepatocarcinoma cells.

(a) HepG2 cells were treated with F17 (25 µM) or 0.25% DMSO as a control. The cells were fixed and stained with LipidTOX neutral lipid stain (red) and counterstained with DAPI (blue). Scale bars represent 10 µm. (b) HepG2 cells were treated with either 2 mM AICAR or AICAR vehicle control (0.2% DMSO) 30 µM F17, F17 vehicle control (0.3% DMSO), simultaneously with 30 µM F17 and 20 µM Compound C, or with 30 µM F21. Western blots of extracts for anti-phospho ACC (pACC), anti-ACC total protein (ACC), anti-phospho AMPKα (pAMPKα) or anti-AMPKα (AMPKα) are shown. Full length blots are shown in Supplementary Fig. S7.
DISCUSSION
Discovery of fat regulatory compounds in whole organisms
Just as small molecules have helped drive understanding of many biological pathways at the cellular level, there is a need for pharmacological tools to interrogate complex processes of whole organism physiology. Fish, amphibians and nematodes [5–8] have proven to be compatible with the demands of systematic forward chemical screening in intact, multicellular animals. Although our C. elegans fat screen is amenable to very large scale screening, our findings indicate that even a small screen is sufficient to identify diverse, novel compounds that modulate fat content, a physiologically relevant phenotype that is the outcome of an array of integrated pathways.
Here, we have reported on numerous novel small molecules that modulate intestinal labeling of C. elegans with vital dyes that stain lipid-containing compartments, modulate total triglyceride content and affect the expression of a key lipogenesis gene, whose expression is correlated with triglyceride storage in nematodes and mammals. In addition, we found that several of these molecules also modulate fat accumulation in both mammalian and Drosophila cell culture. These findings indicate that C. elegans vital Nile Red staining provides an effective strategy for identifying small molecules that perturb fat storage in C. elegans and mammalian systems. Our results stand in contrast to several recent publications that have implied that vital dye staining is non-predictive of changes in global fat metabolism in C. elegans [46–48]. One explanation for this discrepancy is that biochemical measurements of bulk triglycerides extracted from populations of whole animals do not exhibit the same sensitivity and/or compartment specificity afforded by vital dye-based imaging.
Use of small molecules to uncover physiological functions
One of the compounds identified by the primary Nile Red screen, F17, reduced fat content in C. elegans through pathways involving the AMPK-α1 catalytic subunit. Similar to the human and mouse genomes, the C. elegans genome encodes two α, two β, and five γ subunits that may interact to form different AMPK complexes with discrete activities [36]. In mammals, AMPK complexes exhibit varying tissue expression patterns and even different subcellular localizations [49]. In addition, individual complexes respond to different stimuli to mediate varied functions from distinct peripheral energy consumption pathways to feeding behaviors in the central nervous system [36]. In C. elegans, the function of the catalytic subunit encoded by aak-1 is dispensable for both the insulin-dependent lifespan extension phenotype mediated by aak-2 [37] and in the maintenance of an alternative hibernative state that also requires aak-2 [38]. As aak-1 mutants exhibit no fat storage phenotype of their own, it was only through F17-treatment that we identified a function for aak-1 in modulating fat storage in C. elegans. Similarly, we found that F17’s fat reducing effects depend on K08F8.2 a transcription factor containing a bZIP DNA binding motif [43]. As in aak-1, inactivation of K08F8.2 has not been previously implicated in fat regulation, highlighting the utility of small molecules in uncovering physiological roles for various pathways. While the direct binding target(s) of F17 remains to be elucidated, these studies highlight the power of combining pharmacology with genetics to identify in vivo pathways through which a compound exerts its effects. Finally, in addition to causing fat reduction, F17 treatment caused feeding increase, however, the molecular mechanisms through which F17 elicits the observed feeding effects remain to be elucidated.
Perspectives
An advantage of whole animal phenotypic screening is that it does not depend on any preconceived notions as to which pathways should be targeted for generating desired outcomes. Counter to the prevailing paradigm for generation of therapeutics through target-based screening, many of the staples of modern medicine emerged serendipitously when unanticipated effects of particular compounds were noted in whole animals [50]. It remains to be seen whether any newly identified C. elegans fat regulatory compounds will exert similar effects in other intact species. It is, however, noteworthy that the chemical structures of most enzymatic cofactors, metabolic intermediates, neurotransmitters, and lipid-based signaling intermediates are absolutely conserved from nematodes to humans. Given that known mammalian fat regulatory compounds exert similar effects in C. elegans and at least one of the newly discovered compounds, F17, activates AMPK signaling in C. elegans and mammalian cell lines, it is possible that many of the newly identified compounds will be portable to other systems.
METHODS
Materials
The diverse compound screening library was purchased from SPECS via the Small Molecule Development Center at UCSF. Compounds H17, F21, F17, A13, I13, A15, C5, F14, H6 and E8 for follow-up studies were repurchased from SPECS. Compounds F17, F21, F17-1564, F17-4059, F17-8860, F17-8949, F17-3115, F17-9110, F17-8895, F17-4816, F17-4038, and F17-0796 were purchased from Chembridge. BODIPY labeled fatty acid (BODIPY-FA: 4,4-difluoro-5-octyl-4-bora-3a,4a-diaza-s-indacene-3-pentanoic acid) and lipidTOX neutral lipid stain were from Invitrogen. AMPK agonist AICAR was from Toronto Research Chemicals, Inc. AMPK agonist A-766962 was from Tocris Bioscience. The AMPK antagonist compound C was from EMD4Biosciences. Except where noted, all other chemicals were from Sigma-Aldrich. All antibodies were from Cell Signaling Technologies. All cell culture media were purchased from Invitrogen. Cell lines and heat inactivated fetal bovine serum (FBS) were obtained from the UCSF cell culture facility.
Nematode strains
Wild-type: N2 (Bristol), aak-1 (tm1944) III, aak-2 (ok524) X, tub-1 (nr2044) II, osm-12 (n1606) III, lpo-6 (mg360) II, daf-16 (MgDf47) I, mod-1(ok103) V, fat-7::gfp: lin-15 (n765) X; waEx15[fat-7::gfp].
Compound screening
(See Supplemental Methods for a detailed description) Compounds from a SPECS diversity library assembled by the Small Molecule Discovery Center (QB3/UCSF) were screened by culturing synchronized L1 animals with E. coli OP-50, 100 nM Nile Red and the compound library at an average concentration of 5 µM in 384-well plates. After 2 days of culture at 20 °C, brightfield and red fluorescence images were acquired using an automated microscope. Phenotypes for growth, and red-fluorescence staining patterns were scored if more than 75% of animals in a well exhibited the phenotype.
Quantitative imaging
All image data intended for quantitative comparison were acquired at the same sub-saturating exposure time. Fluorescence images of 7–10 nematodes were used to quantify lipid storage using ImageJ (http://rsbweb.nih.gov/ij/). The total integrated fluorescence intensity from the two most anterior intestinal cells in 100–200 × images was quantified. Alternatively, to measure the integrated intensity of intestinal lipid droplets, independent of intestinal haze, a 2-dimensional spot-enhancing Gaussian filter was used to create a binary mask that identified the fluorescent lipid droplets in the first two anterior pairs of intestinal cells from 160–200 × images of nematodes. The mask was applied to the original image and the sum of the unmasked pixel intensities was evaluated.
Pharyngeal pumping assay
Developmentally synchronized L1 animals were added to 24 well plates (approximately 25 nematodes per well) containing NGM-agar seeded with E.coli OP-50 and either experimental compound (2.5–10 µM), 5-hydroxytryptamine (5 mM) or DMSO. Plates were incubated for 46–48 hr at 20 °C. The number of pharyngeal contractions in a 10 s period was recorded using an electronic counter. The pumping rate reported is the average rate of 10–15 young adult animals per condition.
C. elegans growth and egg-laying assays, cell culture assays, western blotting and RNAi treatments, are described in Supplementary Methods.
Supplementary Material
Acknowledgements
We are grateful to Dr. David Lum for his help and advice with the initial C. elegans experiments. We are thankful to Dr. Brendan Mullaney and Dr. Katherine Cunningham for helpful discussions and comments on the manuscript. We thank Dr. Kurt Thorn and the Nikon Imaging Center for use of the Nikon 6D automated epifluorescence microscope and help with imaging as well as the Small Molecule Discovery Center at the California Institute for Quantitative Biomedical Research at UCSF for providing the small molecule library. This work was supported by grants from the National Cancer Institute and National Institute of Environmental Health Sciences (ES012801, ES019458 and CA056721) to Z.W., the National Institute of Diabetes and Digestive and Kidney Diseases (DK070149) to K.A., the National Institute of General Medicine (GM081863) to R.B. and a Byers Award to K.A. and R.B.
Footnotes
Author Contributions. G.A.L., K.A. and Z.W. conceived the study design. G.A.L., J.L., and N.M. performed the experiments. G.A.L., K.A., R.B. and Z.W. analyzed the data. G.A.L., K.A. and Z.W. wrote the paper. All the authors read, revised and approved the manuscript.
Competing Interests. The authors declare no competing interests
References
- 1.Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 2.Kopelman PG. Obesity as a medical problem. Nature. 2000;404:635–643. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 3.Knight ZA, Shokat KM. Chemical genetics: where genetics and pharmacology meet. Cell. 2007;128:425–430. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 4.Stelling J, et al. Robustness of cellular functions. Cell. 2004;118:675–685. doi: 10.1016/j.cell.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Wheeler GN, Brandli AW. Simple vertebrate models for chemical genetics and drug discovery screens: Lessons from zebrafish and Xenopus. Dev Dyn. 2009;238:1287–1308. doi: 10.1002/dvdy.21967. [DOI] [PubMed] [Google Scholar]
- 6.Moy TI, et al. High-throughput screen for novel antimicrobials using a whole animal infection model. ACS Chem Biol. 2009;4:527–533. doi: 10.1021/cb900084v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrascheck M, Ye X, Buck LB. An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature. 2007;450:553–556. doi: 10.1038/nature05991. [DOI] [PubMed] [Google Scholar]
- 8.Kwok TC, et al. A small-molecule screen in C. elegans yields a new calcium channel antagonist. Nature. 2006;441:91–95. doi: 10.1038/nature04657. [DOI] [PubMed] [Google Scholar]
- 9.Jones AK, Buckingham SD, Sattelle DB. Chemistry-to-gene screens in Caenorhabditis elegans. Nat Rev Drug Discov. 2005;4:321–330. doi: 10.1038/nrd1692. [DOI] [PubMed] [Google Scholar]
- 10.Schafer WR, Sanchez BM, Kenyon CJ. Genes affecting sensitivity to serotonin in Caenorhabditis elegans. Genetics. 1996;143:1219–1230. doi: 10.1093/genetics/143.3.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinshenker D, Garriga G, Thomas JH. Genetic and pharmacological analysis of neurotransmitters controlling egg laying in C. elegans. J Neurosci. 1995 Oct;15(10):6975–6985. doi: 10.1523/JNEUROSCI.15-10-06975.1995. 15, 6975–6985 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watts JL. Fat synthesis and adiposity regulation in Caenorhabditis elegans. Trends Endocrinol Metab. 2009;20:58–65. doi: 10.1016/j.tem.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez CL, Van Gilst MR. A 13C isotope labeling strategy reveals the influence of insulin signaling on lipogenesis in C. elegans. Cell Metab. 2008;8:266–274. doi: 10.1016/j.cmet.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Jones KT, Ashrafi K. Caenorhabditis elegans as an emerging model for studying the basic biology of obesity. Dis Model Mech. 2009;2:224–229. doi: 10.1242/dmm.001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKay RM, et al. C elegans: a model for exploring the genetics of fat storage. Dev Cell. 2003;4:131–142. doi: 10.1016/s1534-5807(02)00411-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- 17.Kimura KD, et al. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan S, et al. Serotonin regulates C. elegans fat and feeding through independent molecular mechanisms. Cell Metab. 2008;7:533–544. doi: 10.1016/j.cmet.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Bono M, Bargmann CI. Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell. 1998;94:679–689. doi: 10.1016/s0092-8674(00)81609-8. [DOI] [PubMed] [Google Scholar]
- 20.Suo S, Culotti JG, Van Tol HH. Dopamine counteracts octopamine signalling in a neural circuit mediating food response in C. elegans. EMBO J. 2009;28:2437–2448. doi: 10.1038/emboj.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashrafi K, et al. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature. 2003;421:268–272. doi: 10.1038/nature01279. [DOI] [PubMed] [Google Scholar]
- 22.Greenspan P, Mayer EP, Fowler SD. Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol. 1985;100:965–973. doi: 10.1083/jcb.100.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Gilst MR, et al. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol. 2005;3:e53. doi: 10.1371/journal.pbio.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones KS, et al. A high throughput live transparent animal bioassay to identify non-toxic small molecules or genes that regulate vertebrate fat metabolism for obesity drug development. Nutr Metab (Lond) 2008;5:23. doi: 10.1186/1743-7075-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan JE, et al. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353:33–36. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- 26.Watts JL, Browse J. A palmitoyl-CoA-specific delta9 fatty acid desaturase from Caenorhabditis elegans. Biochem Biophys Res Commun. 2000;272:263–269. doi: 10.1006/bbrc.2000.2772. [DOI] [PubMed] [Google Scholar]
- 27.Van Gilst MR, Hadjivassiliou H, Yamamoto KR. A Caenorhabditis elegans nutrient response system partially dependent on nuclear receptor NHR-49. Proc Natl Acad Sci U S A. 2005;102:13496–13501. doi: 10.1073/pnas.0506234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang F, et al. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature. 2006;442:700–704. doi: 10.1038/nature04942. [DOI] [PubMed] [Google Scholar]
- 29.MacDougald OA, Lane MD. Transcriptional regulation of gene expression during adipocyte differentiation. Annu Rev Biochem. 1995;64:345–373. doi: 10.1146/annurev.bi.64.070195.002021. [DOI] [PubMed] [Google Scholar]
- 30.Guo Y, et al. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453:657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horton JD, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kniazeva M, et al. Monomethyl branched-chain fatty acids play an essential role in Caenorhabditis elegans development. PLoS Biol. 2004;2:E257. doi: 10.1371/journal.pbio.0020257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shiri-Sverdlov R, et al. Identification of TUB as a novel candidate gene influencing body weight in humans. Diabetes. 2006;55:385–389. doi: 10.2337/diabetes.55.02.06.db05-0997. [DOI] [PubMed] [Google Scholar]
- 34.Goldstone AP, Beales PL. Genetic obesity syndromes. Front Horm Res. 2008;36:37–60. doi: 10.1159/000115336. [DOI] [PubMed] [Google Scholar]
- 35.Ogg S, et al. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 36.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 37.Apfeld J, et al. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narbonne P, Roy R. Caenorhabditis elegans dauers need LKB1/AMPK to ration lipid reserves and ensure long-term survival. Nature. 2009;457:210–214. doi: 10.1038/nature07536. [DOI] [PubMed] [Google Scholar]
- 39.Dobrzyn P, et al. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc Natl Acad Sci U S A. 2004 Apr 27;101(17):6409–6414. doi: 10.1073/pnas.0401627101. Epub 2004 Apr 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corton JM, et al. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 41.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 42.Cool B, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Jia H, Chamberlin HM. The bZip proteins CES-2 and ATF-2 alter the timing of transcription for a cell-specific target gene in C. elegans. Dev Biol. 2006;289:456–465. doi: 10.1016/j.ydbio.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 44.Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davies SP, Sim AT, Hardie DG. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur J Biochem. 1990;187:183–190. doi: 10.1111/j.1432-1033.1990.tb15293.x. [DOI] [PubMed] [Google Scholar]
- 46.O'Rourke EJ, et al. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 2009;10:430–435. doi: 10.1016/j.cmet.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brooks KK, Liang B, Watts JL. The influence of bacterial diet on fat storage in C. elegans. PLoS One. 2009;4:e7545. doi: 10.1371/journal.pone.0007545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morck C, et al. Statins inhibit protein lipidation and induce the unfolded protein response in the non-sterol producing nematode Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2009;106:18285–18290. doi: 10.1073/pnas.0907117106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kodiha M, et al. Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK-->ERK1/2 pathway. Am J Physiol Cell Physiol. 2007;293:C1427–C1436. doi: 10.1152/ajpcell.00176.2007. [DOI] [PubMed] [Google Scholar]
- 50.MacRae CA, Peterson RT. Zebrafish-based small molecule discovery. Chem Biol. 2003;10:901–908. doi: 10.1016/j.chembiol.2003.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.