

Abstract
Assessing mitochondrial dysfunction requires definition of the dysfunction to be investigated. Usually, it is the ability of the mitochondria to make ATP appropriately in response to energy demands. Where other functions are of interest, tailored solutions are required. Dysfunction can be assessed in isolated mitochondria, in cells or in vivo, with different balances between precise experimental control and physiological relevance. There are many methods to measure mitochondrial function and dysfunction in these systems. Generally, measurements of fluxes give more information about the ability to make ATP than do measurements of intermediates and potentials. For isolated mitochondria, the best assay is mitochondrial respiratory control: the increase in respiration rate in response to ADP. For intact cells, the best assay is the equivalent measurement of cell respiratory control, which reports the rate of ATP production, the proton leak rate, the coupling efficiency, the maximum respiratory rate, the respiratory control ratio and the spare respiratory capacity. Measurements of membrane potential provide useful additional information. Measurement of both respiration and potential during appropriate titrations enables the identification of the primary sites of effectors and the distribution of control, allowing deeper quantitative analyses. Many other measurements in current use can be more problematic, as discussed in the present review.
Keywords: membrane potential, mitochondrion, mitochondrial function, respiration, respiratory control
Abbreviations: ΔpH, pH gradient; Δψp, plasma membrane potential; Δψm, mitochondrial membrane potential; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; pmf, protonmotive force; PMPI, plasma membrane potential indicator; R123, rhodamine 123; RCR, respiratory control ratio (state 3/state 4); state 3u, uncoupled state; state 4o, state 4oligomycin; TMRM, tetramethylrhodamine methyl ester; TPMP, triphenylmethylphosphonium; TPP, tetraphenylphosphonium; UQ, ubiquinone
INTRODUCTION
The many genetically modified cell lines and organisms that have been generated for the study of physiological and pathological mechanisms often show features that suggest compromised energy metabolism or mitochondrial dysfunction. This is illustrated by the plethora of recent reviews associating mitochondrial dysfunction with specific diseases, e.g. [1–13]. In parallel, there is an increasing realization in the pharmaceutical industry that candidate drugs must be tested at an early stage for adverse effects on mitochondria [14]. In the present review, we address how to assay mitochondrial dysfunction in a way that is straightforward yet yields the most physiologically relevant, unambiguous and informative results in these genetic models, in tissues and cells from patients, and in response to bioactive compounds.
The terms mitochondrial ‘function’ and ‘dysfunction’ are widely employed in bioenergetics and cell biology, but precise definition is difficult because different studies have different goals. The predominant physiological function of mitochondria is the generation of ATP by oxidative phosphorylation, but additional functions include the generation and detoxification of reactive oxygen species, involvement in some forms of apoptosis, regulation of cytoplasmic and mitochondrial matrix calcium, synthesis and catabolism of metabolites and the transport of the organelles themselves to correct locations within the cell. Abnormality in any of these processes can be termed mitochondrial dysfunction.
The best approach to assessing dysfunction depends on whether function is to be determined with isolated mitochondria, intact cells or in vivo, whether the assessment needs to be qualitative or quantitative, which assays are theoretically the most appropriate and informative, and which assays are technically the most accessible. The major aim of the present review is to assess the different methods, their strengths and weaknesses and the information they provide.
We start by reviewing approaches to monitor the basic processes of oxidative phosphorylation by isolated mitochondria and then discuss how to extend investigation into intact cells. Techniques now exist to monitor mitochondrial bioenergetic function in cells with comparable precision to previous studies with isolated mitochondria. Of course there are problems (Table 1): the mitochondria are not directly accessible to the full range of substrates and inhibitors, and the complexity of cytoplasmic metabolism must be considered together with the presence of separate pools of adenine nucleotides, nicotinamide nucleotides and calcium in the cytoplasm and mitochondrial matrix. However, overriding all of these complexities is the greatly enhanced physiological relevance of working with intact cells. The mitochondria are present in a physiological environment, are exposed to a relevant mix of substrates and ions and interact with the cytoplasm, plasma membrane and other organelles and cell structures. However, even with isolated cells the true in vivo complexity of cell–cell interaction is lacking and can only be investigated fully in the intact animal or human subject. We therefore conclude by briefly reviewing techniques for investigating mitochondrial function in these intact systems.
Table 1. Advantages and disadvantages of working with isolated mitochondria and intact cells.
| Isolated mitochondria | Intact cells | ||
|---|---|---|---|
| Advantage | Disadvantage | Advantage | Disadvantage |
| Relatively simple and well understood. Better for studies of mechanism. No interference from cytosolic factors | Lack cellular context | Undisturbed cellular environment; greater physiological relevance. Interactions with the rest of the cell are preserved | More complex, more scope for errors of interpretation. Lack organismal context |
| Easy to isolate from many adult tissues of wild-type or genetically modified animals | Subject to damage and selection during isolation. Isolation from small or tough tissues can be problematic | No artefacts due to mitochondrial isolation. Cell lines are amenable to genetic manipulation and plate-based assays | Can be hard or impossible to isolate viable primary cells from adult tissues of transgenic animals |
| Reagents and substrates can be added directly; the experimenter has control over conditions | The experimenter has to choose appropriate experimental conditions | The cell sets the mitochondrial environment | Many reagents and substrates are cell-impermeant, restricting experimental options. The experimenter chooses extracellular substrates, hormones and conditions |
| Methods are generally very well established | Existing methods often need large amounts of sample; mitochondria from different cell types may be unavoidably aggregated | Plate-based assays allow measurements on tiny amounts of the sample or single cells | Many methods are not sufficiently specific or quantitative |
| Easy and usually meaningful to normalize to protein or cytochrome content | Effects due to mitochondrial proliferation, localization etc. lost during isolation | Effects due to mitochondrial proliferation and localization retained | The meaning of results changes with normalization (cell number, cell mass, DNA, cytochrome a etc.) |
THE PROTON CIRCUIT
The mitochondrial proton circuit [15] is central to the multiple physiological functions of mitochondria. Three electron transport complexes, I, III and IV, comprise the energy-conserving core of the electron transport chain and pump protons across the mitochondrial inner membrane (Figure 1). In each case, a drop in redox potential of the electrons passing through the complex is coupled to the extrusion of protons from the matrix. Several other important members of the electron transport chain are energetically and mechanistically incapable of pumping protons, specifically complex II (including succinate dehydrogenase), glycerol phosphate dehydrogenase and the electron-transferring flavoprotein quinone oxidoreductase of fatty acid β-oxidation. Ten protons are extruded for each electron pair passing from NADH to oxygen, or six protons for each electron pair passing from other quinone-linked dehydrogenases to oxygen [16,17]. The redox span across the entire electron transport chain is approximately 1100 mV, and the maximal pmf (protonmotive force) across the inner membrane is 180–220 mV [18–21].
Figure 1. The proton circuit across the mitochondrial inner membrane and an equivalent electrical circuit.

(A) The primary, ATP-generating, proton circuit is shown as bold lines/boxes and the pathway of electron flow as a dashed vertical line. Secondary pathways of proton re-entry include metabolite transport [represented in the present Figure by the phosphate carrier, the transhydrogenase for the reduction of NADP+ to NADPH, and the endogenous and UCP (uncoupling protein)-mediated proton leaks]. Ca2+ cycles between the uniport and the Na+/Ca2+ antiport, and a sodium circuit links the proton and calcium circuits. (B) The equivalent electrical circuit modelling the primary proton circuit. The resistances associated with each ‘complex’ reflect the observation that membrane potential drops as the proton current drawn increases.
The total outward (extrusion) and inward (re-entry) proton currents exactly balance under steady-state conditions. The dominant pathway of proton re-entry during active ATP synthesis is the ATP synthase. The proton pumps of the electron transport chain, together with the ATP synthase, thus create a proton circuit across the inner membrane, with terms of driving force or potential (the pmf in mV) and flux (the proton current in nmol of protons/min). This proton circuit is central to mitochondrial bioenergetics. Experimentally distinct, but parallel, experimental approaches are used to quantify the potential and flux components of the proton circuit, both with isolated mitochondria and intact cells. The pmf has two components, ΔpH (the pH gradient across the inner membrane) and Δψm (mitochondrial membrane potential, the difference in electrical potential between the cytoplasm and the matrix) (eqn 1):
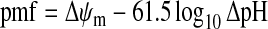 |
(1) |
The pmf is expressed in mV. By convention, electrophysiologists define membrane potentials with respect to the external medium, so both Δψp (plasma membrane potential) and Δψm have negative signs. However, bioenergeticists historically define potentials with respect to the mitochondrial matrix, so both potentials have positive signs. Context usually allows deciphering of the convention being used. In the present review, we use the bioenergetic convention for all potentials.
In addition to proton re-entry through the ATP synthase, all mitochondria possess a parallel endogenous proton leak [22], also present in mitochondria in situ within intact cells and thus not an artefact of isolation [23–26]. In the absence of ATP synthesis, the proton circuit is largely completed by the proton leak, which may serve an important purpose in limiting pmf, to prevent dielectric breakdown of the membrane and restrict leakage of single electrons from the electron transport chain to form superoxide [25,27]. Cycling of calcium across the mitochondrial inner membrane also uses the proton current [28], as do metabolite transport and other reactions such as NADH/NADP transhydrogenase and protein import.
Net forward flux through each electron transport complex requires a thermodynamic disequilibrium, i.e. the free energy available from electron transfer must be greater than that required to pump protons across the membrane against the pmf. The electron transport chain thus responds to the drop in pmf resulting from an increase in proton re-entry through the ATP synthase or proton leak with an increased flux. The increase in respiration is an approximately linear function of the fall in pmf until it plateaus at relatively low pmf (approximately 100 mV) when the maximal (‘uncontrolled’) respiration is attained [19,21], as shown by the ‘substrate oxidation’ curve in Figure 2(B).
Figure 2. Modular kinetic analysis and modular control analysis.

(A) Three modules of mitochondrial energy metabolism in isolated mitochondria or in intact cells connected by pmf. ‘Substrate oxidation’ consists of all reactions involved in substrate uptake, metabolism and electron transport, and generates pmf. ‘ATP turnover’ consists of all reactions involved in phosphorylation of ADP to ATP and the export and turnover of ATP in the extramitochondrial space, and consumes pmf. ‘Proton leak’ consists of all other reactions that consume pmf. The proton current through the system, measured as respiration rate, is generated by substrate oxidation, flows through pmf and is then divided between proton leak and ATP turnover. (B) Modular kinetic analysis of the three modules in mitochondria from A549.B2 human lung carcinoma cells. The three curves with filled symbols show the kinetic responses of the rates of each of the three modules in (A) to their common intermediate, pmf. Succinate (4 mM) was present as the substrate. The response of substrate oxidation rate to pmf was determined by titration of proton leak with FCCP; the response of proton leak to pmf was determined by titration of substrate oxidation with malonate, and the response of ATP turnover plus proton leak to pmf was determined by titration of state 3 respiration with malonate (the response of ATP turnover alone can be calculated by subtraction of the proton leak curve). Filled symbols, control; open symbols, plus 25 nM myxothiazol. In the presence of myxothiazol, the kinetics of substrate oxidation were significantly different at membrane potentials above 98 mV. Redrawn from [33] © the Biochemical Society. (C) Flux control coefficients of the three modules over respiration rate in rat liver mitochondria at different rates of oxidative phosphorylation (varied by titration with hexokinase in the presence of ATP), calculated from titrations similar to those in (B). The flux control coefficients describe quantitatively how strongly each of the modules in (A) controls the respiration rate under each defined condition. Redrawn from [107] © the Biochemical Society; data from [21].
A useful conceptual analogy of the proton circuit is a simple electrical circuit (Figure 1B). The electrical analogue can be quantified by voltage, current and (by Ohm's law) resistance or conductance. In the proton circuit, the corresponding variables are pmf (in mV), proton current (nmol of H+/min per unit of mitochondrial protein or cell number etc.) and proton conductance (nmol of H+/min per mV per unit).
Modular analysis of the proton circuit
A complementary conceptual model simplifies the proton circuit to three modules of reactions connected by their common intermediate, pmf [21,29–31] (Figure 2A). The ‘substrate oxidation’ module consists of all reactions involved in substrate uptake, metabolism and electron transport, and generates pmf. The ‘ATP turnover’ module consists of all reactions involved in phosphorylation of ADP to ATP and the export and turnover of ATP in the extramitochondrial space, and consumes pmf. The ‘proton leak’ module consists of all other reactions that consume pmf. The proton current through the system, measured as respiration rate, is generated by substrate oxidation, flows through pmf, and is then divided between proton leak and ATP turnover. The advantage of this conceptual simplification is that it encompasses and fully describes the whole of the function of oxidative phosphorylation in one simple scheme.
The kinetic response of each module to pmf is easy to measure: rate and pmf are simply titrated with suitable inhibitors or activators of another module [31]. In this way, the response of substrate oxidation rate to pmf can be determined by titration of proton leak with uncoupler; the kinetic response of proton leak to pmf can be determined by titration of substrate oxidation using respiratory inhibitors, and the kinetic response of ATP turnover to pmf can be determined by titration of respiration during ATP synthesis with respiratory inhibitors, and subtraction of the relevant proton leak rate at each point (Figure 2B).
The kinetics of the three modules describe fully the overall properties of oxidative phosphorylation in isolated mitochondria or in intact cells. Any change that affects overall function must be reflected by a primary change in the kinetics of one or more of the modules [32]. The consequent changes in pmf will then have secondary effects on the rates of the other modules. If required, the module that is directly affected can be broken into two or three smaller modules connected by further intermediates, and the analysis repeated to home in on the primary molecular event [29]. The abilities to reveal any functionally relevant change in kinetics, to distinguish primary changes from secondary changes and to bore down to molecular events are powerful advantages of modular kinetic analysis. For example, Figure 2(B) shows that a sub-maximal concentration of myxothiazol (an inhibitor of complex III) slows respiration in mitochondria exclusively by its primary effect on substrate oxidation (because there is a statistically significant change in the kinetic curve describing this module), whereas its effects on the rates of proton leak and ATP synthesis are secondary to the change in pmf, since there is no significant primary change in their kinetics [33].
Once the modular kinetics are known, the distribution of control by each module over rates and over the value of pmf can be calculated (modular control analysis) [31] (Figure 2C). If the modular kinetics are remeasured after some functional change, one can quantify how that change propagates through the system (modular regulation analysis) [34]. We shall exploit these powerful concepts below.
ISOLATED MITOCHONDRIA
Fluxes: mitochondrial proton current (respiration rate)
Measurement of proton current is generally more informative than measurement of pmf if only one is to be measured (but it is best to know both). In the same way, it is more informative to know the current drawn from the electricity grid by your domestic appliances than to know the voltage drop across your electricity supply box.
The tight coupling between electron transport and proton extrusion in both mitochondria and cells [35–37] means that for a given substrate the rate of mitochondrial oxygen utilization is an accurate measure of the total proton current. This rate measures the activity of a single process: the transfer within complex IV of four electrons to a molecule of oxygen to generate two molecules of water. Despite this apparent limitation, experiments can be designed to obtain information on a wide variety of processes, including substrate transport into the cell, cytoplasmic metabolism, transport into the mitochondrion, mitochondrial metabolism, electron delivery to the respiratory chain, the activities of complex I or II, complex III, complex IV, ATP synthesis, proton leak, ATP export to the cytoplasm and cell ATP utilization. The art of using respiration to uncover dysfunction in a wide variety of modules and reactions lies in designing experiments that move control on to that reaction so that the dysfunction is most easily recognized and quantified. The Clark oxygen electrode has been used for 50 years to measure mitochondrial respiration [38], but other methods, such as plate-based fluorescence assays [39,40], can provide the same information.
Mitochondrial respiratory control: the best general measure of mitochondrial function in isolated mitochondria
The classic oxygen electrode experiments to determine mitochondrial bioenergetic function were devised by Chance and Williams [38]. Substrate is added to a mitochondrial incubation, followed by a small amount of ADP, allowing the ATP synthase to function, pmf to drop and electron transport to accelerate (‘state 3ADP’). When the ATP/ADP ratio approaches equilibrium, pmf rises, proton re-entry through the synthase stops and respiration slows (‘state 4’). In practice, the presence of ATP in the incubation may have additional effects in state 4. Any contaminating ATPase activity (e.g. broken mitochondria with uncoupled ATPases or residual muscle fibres in a muscle mitochondrial preparation) will prevent the restoration of a low respiration. For this reason, state 3ADP respiration can be terminated by adding the ATP synthase inhibitor oligomycin to achieve a ‘state 4o’ (‘state 4oligomycin’) rate, where ATP recycling cannot contribute. Oligomycin addition can be followed by a carefully titrated concentration of a protonophore, such as FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone), to give uncoupled respiration (‘state 3FCCP’ or ‘state 3u’). In some mitochondria, such as those from brown fat where the ATP synthase activity is very limited, state 3u may be much higher than the maximal state 3ADP [41].
Two ‘pre-chemiosmotic’ parameters are still used to quantify the behaviour of mitochondria in this classic experiment: the mitochondrial RCR [respiratory control ratio (state 3/state 4)], defined as the respiration in state 3ADP divided by that in state 4, and the P/O ratio, mol of ATP synthesized per mol of O (1/2O2) used. With rare exceptions, such as brown fat mitochondria whose uncoupling protein is activated by fatty acid [42], ‘healthy’ mitochondria under correctly designed incubation conditions show high respiratory control: a large increase in respiration rate with ADP followed by a return to state 4. Mitochondrial respiratory control encapsulates the main function of mitochondria: their ability to idle at a low rate yet respond to ADP by making ATP at a high rate. A high RCR implies that the mitochondria have a high capacity for substrate oxidation and ATP turnover and a low proton leak. However, there is no absolute RCR value that is diagnostic of dysfunctional mitochondria, because values are substrate- and tissue-dependent. Mitochondrial respiratory control is a complex function whose value depends on numerous factors, and this complexity is its main strength: a change in almost any aspect of oxidative phosphorylation will change RCR.
State 3ADP is controlled approximately equally (depending on the tissue and conditions) by the activity of ATP turnover (primarily the adenine nucleotide translocase, phosphate transporter and ATP synthase) and substrate oxidation (including substrate uptake, processing enzymes, relevant electron-transport-chain complexes, pool sizes of UQ (ubiquinone) and cytochrome c, and [O2]) (Figure 2C). Inhibition of any of these processes will decrease the state 3 rate.
State 3u is controlled exclusively by substrate oxidation and will detect dysfunction in respiratory chain components, substrate translocases or dehydrogenases. Different substrates allow multiple metabolic pathways or respiratory complexes to be probed.
State 4 is controlled predominantly by the proton leak and any contaminating ATPases that recycle synthesized ATP as ADP (and to a small extent by the activity of substrate oxidation) (Figure 2C).
State 4o is controlled by the activity of the proton leak (and approximately 10% by substrate oxidation). A low state 4o rate indicates that the mitochondria maintain a sufficiently high pmf to restrict electron transport (see the ‘substrate oxidation’ curve in Figure 2B, where pmf must be >100–120 mV to slow respiration rate). A higher state 4o rate indicates altered proton leakiness in vivo or inappropriate isolation techniques that induce an artefactual proton leak.
Thus the RCRs, either state 3ADP/state 4 or, to a lesser extent, state 3u/state 4o, are strongly influenced by almost every functional aspect of oxidative phosphorylation, making them good indicators of dysfunction. However, the rates are very sensitive to errors in measurement; if the electrode system being used has significant back-diffusion of oxygen from the Teflon stirrer bar (which can hold half of the oxygen in a suspension) or from the surrounding vessel or atmosphere, state 4 rates can be significantly underestimated, resulting in some of the very high apparent RCR values found in the literature. The test for this is to repeat the state 3/state 4 cycle to ensure that the RCR remains the same at lower oxygen tension, or to fully inhibit respiration and check for an upward drift.
Substrates that feed electrons to the UQ pool, such as succinate (and glycerol phosphate in appropriate tissues), translocate fewer protons per electron pair than NADH-linked substrates, so the same proton cycling rate requires a higher respiration rate. This does not affect RCR, as both state 3 and state 4 change by the same factor. However, RCR tends to be lower with these substrates because they generate a higher pmf, for kinetic and thermodynamic reasons. Since the proton leak rate is voltage-dependent [22] (see the ‘proton leak’ curve in Figure 2B), this increases the state 4 rate with no change in the proton leakiness. Furthermore, under identical incubation conditions, very different RCRs may be seen with mitochondria from different tissues, reflecting differing substrate oxidation kinetics and different physiological roles of the endogenous proton leak [43].
In conclusion, the respiratory control ratio is the single most useful general measure of function in isolated mitochondria. High RCR indicates good function, and low RCR usually indicates dysfunction. The utility of RCR rests on its complexity: virtually any change in oxidative phosphorylation will change RCR. If absolute rates and RCR are unaffected by a treatment, it is safe to assert that there is no overall bioenergetic dysfunction manifest in isolated mitochondria. Once a dysfunction in RCR has been identified and its immediate causes determined (was the change in state 3 rate or state 4 rate?), further experiments will be required to pinpoint the primary causes, using modular kinetic analyses or candidate activity assays.
Absolute respiration rate can give useful insights into the mechanism of dysfunction
A straightforward indicator of the function of isolated mitochondria is their absolute respiration rate, normalized to protein or cytochrome a, in state 3ADP or state 3u. A damaged electron transport chain may be unable to support high rates of respiration if, for example, substantial cytochrome c has been lost from the intermembrane space due to damage to the outer membrane. Small changes in absolute respiration rate can be caused by changes in purity that may not be directly related to mitochondrial dysfunction. Once RCR has been determined, absolute respiration rates are a useful way to gain more insight into the site(s) of the dysfunction: is respiratory control decreased because of lower state 3u rate (dysfunction localized to substrate oxidation) or only state 3 rate (dysfunction in ATP synthesis), or because of higher state 4o rate (dysfunction in proton conductance) or only state 4 rate (dysfunction by ATPase contamination)? If state 3u is low, is it low for all substrates (complex IV defect) or only for particular substrates (e.g. complex II if only succinate oxidation is compromised)?
P/O ratio: a poor reporter of mitochondrial dysfunction
The P/O ratio is the maximum number of ATP molecules made as an electron pair passes down the respiratory chain from substrate to oxygen. It is the H+/O stoichiometry of respiration divided by the H+/ATP stoichiometry of ATP synthesis, and is thus a mechanistic constant for any particular substrate. Its value will not change in dysfunction unless the coupling mechanism itself is altered (e.g. the respiratory complexes slip and pump fewer protons than normal). Literature reports of changed P/O ratios almost always reflect alterations in the correction for proton leak as state 3 and state 4 rates change, so it is better to report those rates and RCR directly.
The effective P/O ratio (or coupling efficiency) is an empirical value that makes no correction for proton leak, but reports the relative fluxes through the ATP synthase and proton leak pathways [44]. The coupling efficiency near state 4 is sensitive to changes in proton leakiness and ATP turnover, and is a useful measure in intact cells (see below).
Forces: mitochondrial pmf
In the electrical analogue of the proton circuit, the oxygen electrode corresponds to the electricity meter monitoring the total current consumption by the appliances in a house: switch on the television and the current increases. We do not normally monitor the mains voltage separately because the drop in voltage when you switch on the television is small. The equivalent variable in the proton circuit, pmf, is less constant, falling slightly (by 10–20%) during a state 4–3 transition or when Ca2+ is accumulated or rising slightly when substrate supply is enhanced [45]. Monitoring pmf, ideally in parallel with respiration, can help to remove ambiguity. For example, a treatment could increase state 4 respiration by stimulation of uncoupling or ATP synthesis, or by increased substrate availability; pmf, monitored in parallel, can distinguish between the former (depolarizing) and the latter (hyperpolarizing).
The pmf has two components: membrane potential (Δψm) and pH gradient (ΔpH) (eqn 1). Although Δψm is always dominant, under extreme conditions (phosphate-depleted mitochondria accumulating Ca2+), ΔpH can contribute 50% of the pmf. Alternatively, ΔpH is zero in a KCl-based medium in the presence of the K+/H+ ionophore nigericin. The careful use of nigericin simplifies experiments with isolated mitochondria by allowing pmf to be equated with Δψm, although for qualitative studies in the presence of phosphate, changes in Δψm tend to be paralleled by similar, but smaller, changes in ΔpH [45].
Δψm
In contrast with cellular electrophysiology, where individual cells can be patched or penetrated by electrodes for the direct determination of membrane potential, indirect techniques must be employed for the much smaller mitochondria. Δψm is estimated by monitoring the distribution of membrane permanent monovalent cations (C+) between the incubation medium and the mitochondrial matrix following the Nernst equilibrium (eqn 2):
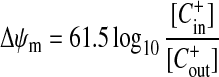 |
(2) |
Typical Δψm values of 150–180 mV correspond to 300–1000-fold accumulation. The logarithmic relationship in eqn (2) means that signals are sensitive to small changes in potential, but that errors in the estimation of low potentials can be large.
Reasonably accurate absolute values of Δψm may be obtained by determining cation accumulation and matrix volume with correction for the apparent activity coefficient of the cation. Matrix volume can be estimated by suspending mitochondria in medium containing 3H2O and [14C]sucrose, centrifuging and determining the water-permeable and sucrose-impermeable volume of the pellet [46]. A typical volume is 0.5–1 μl per mg of mitochondrial protein. More conveniently, the apparent binding coefficient of the probe often depends on matrix volume, so Δψm can be calculated using a pre-measured volume-dependent binding correction that allows routine measurement of Δψm without the need to measure the matrix volume or to assume that it remains constant during an experiment [47,48].
A common technique to monitor Δψm is to equilibrate mitochondrial incubations with low concentrations of lipophilic membrane-permeant cations, such as TPP+ (tetraphenylphosphonium ion) or TPMP+ (triphenylmethylphosphonium ion), and to quantify uptake of the cations from the decrease in the medium concentration monitored by an external macro-electrode [48,49]. Membrane-permanent cations that possess distinctive absorbance or fluorescence are also employed. The most common method is to suspend mitochondria in a temperature-regulated stirred cuvette or multiwell plate and to determine the change in total transmission or emission of the incubation in response to effectors of the membrane potential. The cations are employed in a concentration range sufficient to result in aggregation and fluorescent quenching within the matrix, and as a result the total signal from the cuvette decreases as a function of the matrix accumulation, and hence Δψm. The fluorescent response is usually calibrated in parallel by varying external [K+] in the presence of valinomycin and applying the Nernst equation assuming constant internal [K+] [50]. There are steep relationships between pmf and the fluxes through the three modules of oxidative phosphorylation at physiological values of pmf (Figure 2B), so large changes in respiration are associated with rather small changes in pmf. Although pmf does report bioenergetic status, it is less sensitive to changes in function and less useful generally than measurements of respiratory rates. However, the combination of pmf and respiration rates is much more powerful and informative than measurement of either alone.
Modular kinetic analysis
Quantitative measurement of both proton current and pmf enables a modular kinetic analysis, allowing a full and quantitative description of any change in function and its primary and secondary sites of action. For example, an important role of quantitative Δψm determination, in combination with respiration, is to detect and measure subtle changes in proton leak and efficiency of coupling between mitochondrial preparations. In the equivalent electrical circuit (Figure 1), the conductance of a component is defined using Ohm's law as the current per unit of potential difference. A similar approach has been used to quantify the proton conductance of the mitochondrial inner membrane. Δψm (or better, pmf) is determined in parallel with respiration, usually with oxygen and TPMP+ electrodes inserted into the same incubation. In the absence of net ATP synthesis, this allows the basal proton conductance of the membrane to be calculated [21,45]; this is non-ohmic, increasing disproportionately at high pmf [19,51]. To obtain most information about the proton leak it is therefore necessary to generate a current/voltage plot for the conductance over a range of potentials, by starting in state 4 and progressively limiting electron transport, for example by titrating malonate into an isolated mitochondrial incubation oxidizing succinate and simultaneously determining respiration and pmf [19,22] or Δψm with a TPMP electrode in the presence of nigericin [33,52] (the ‘proton leak’ curve in Figure 2B). This modular kinetic analysis can then be repeated for the other two modules of oxidative phosphorylation (substrate oxidation and ATP turnover), giving the full picture of the functional state of the mitochondria (Figure 2B).
Modular kinetic analysis describes fully (at the system level) any functional change in oxidative phosphorylation. Once a dysfunction is discovered by measuring RCR and absolute respiration rates, modular kinetic analysis is the method of choice for uncovering the primary and secondary causes of that dysfunction.
Concentrations and activities of candidate complexes and enzymes
A very common approach to address mitochondrial bioenergetic dysfunction is to measure the expression, concentration or maximum activity of a few candidate electron transport complexes or metabolic enzymes, such as complex I, complex IV or tricarboxylic acid-cycle enzymes. One reason for the popularity of the candidate approach is the ease with which specific transcripts can be assayed in microarrays, specific proteins can be assayed using antibody technology, and specific enzyme activities can be assayed using commercial kits. The implicit assumption is that these proteins control respiratory rate and bioenergetic function, so if they are altered, function must be altered, and if they are not altered, function must be intact. This is incorrect for two reasons. First, control over the rate of ATP turnover or substrate oxidation in isolated mitochondria is widely shared, with no one enzyme being rate-limiting, and the distribution of control alters as conditions change (e.g. Figure 2C). It may be that the candidate has significant control under the conditions chosen and a change in its activity correctly reports a bioenergetic dysfunction. However, it is just as likely that moderate changes in activity of the candidate complex have little effect on the overall system behaviour, leading to false positives. Secondly, function depends on the integrity of many processes, so even steps that have no control under normal conditions may become strongly rate-controlling if they are compromised. Omitting these steps from the candidate list will lead to false negatives.
Other specific functions and dysfunctions
The choice of which dysfunction to measure depends on the particular question being asked. Specialized functions of mitochondria include the urea cycle, γ-aminobutyric acid cycle, amino acid metabolism, one-carbon metabolism, FeS protein synthesis, haem synthesis, fatty acid metabolism, calcium transport and homoeostasis, reactive oxygen species metabolism, the mitochondrial permeability transition pore in the control of cytochrome c release and apoptosis, and fission and fusion. Assays of dysfunctions in these specific pathways will require specialized approaches rather than the general bioenergetic approaches outlined in the present paper.
Summary: assaying dysfunction in isolated mitochondria
The simplest and most revealing test for energetic dysfunction in isolated mitochondria is to measure mitochondrial respiratory control. Absolute values of respiration rates in different states and conditions, and qualitative measurements of pmf, can clarify mechanisms, but the best way to gain mechanistic insight is to simultaneously titrate both respiration rate and pmf and plot the kinetics of the different modules of oxidative phosphorylation (Figure 2B). Candidate assays of the amounts or activities of specific complexes and enzymes, mitochondrial morphology and responses to specific stressors can be useful to test specific hypotheses, but should generally be held in reserve and not used as the primary assay for mitochondrial dysfunction.
PERMEABILIZED CELLS
Permeabilized cells or skinned muscle fibre preparations can be considered as a subset of isolated mitochondria, with similar strengths and weaknesses. The advantage is that the use of permeabilized cells can decrease problems associated with the preparation time required, the general or selective loss of some of the mitochondria, and damage caused during mitochondrial isolation. Permeabilization may also leave intact many of the intracellular interactions with proteins, the cytoskeleton and other membranes, particularly the mitochondrial-associated membranes that are involved in intracellular calcium signalling, and so can be considered to have many of the advantages of intact cells without the significant disadvantage of the permeability barrier imposed by the plasma membrane [53]. Compared with isolated mitochondria, mitochondria in permeabilized preparations have poor purity, and there is the additional risk of outer membrane damage and cytochrome c release caused by the permeabilization protocol. It is also critical that the supporting medium is ‘mitochondria friendly’ at the instant of permeabilization, particularly in relation to Ca2+.
INTACT CELLS
A major goal of current bioenergetic research is the development and application of techniques to quantify mitochondrial function and cellular bioenergetics in intact cells, avoiding many of the artefacts associated with mitochondrial isolation or cell permeabilization while offering the overwhelming advantage of greater physiological relevance (Table 1). However, isolated cells still lack their in vivo context, and the experimenter has to choose what substrates to add, how much serum to use (with its uncharacterized cargo of hormones, growth factors, cytokines and other effectors), and the pH and bicarbonate and oxygen concentrations. These choices may determine the outcome of the investigation. Glucose is often included in cell incubation media, but many cells do not metabolize it (particularly if they have insulin-dependent glucose uptake and insulin is not present) and prefer to oxidize either endogenous substrates or other components of the medium, such as fatty acids or amino acids, particularly glutamine and alanine. Compared with isolated mitochondria, cells are more complex and have the major restriction that the cell membrane is effectively impermeable to adenine nucleotides, most mitochondrial substrates and some useful inhibitors.
A bioenergetic analysis of the proton circuit similar to that for isolated mitochondria can be performed with intact cells. Mitochondria are responsible for the majority of cellular respiration, except in cells such as neutrophils that can activate non-mitochondrial oxygenases, and in any cell the contribution of non-mitochondrial respiration can be determined at the termination of an experiment by the residual respiration in the presence of electron transport inhibitors. Fluorescence methods for measuring pmf (or at least Δψm) in cells are improving all the time, making the voltage term qualitatively (and, increasingly, quantitatively) accessible. Although measurement of the proton current is generally more informative than measurement of pmf, fluorescence microscopy possesses the considerable advantage that Δψm can be monitored at single-cell, or even single-mitochondrial, resolution in parallel with indicators of Ca2+, reactive oxygen species, Δψp etc., allowing complex stochastic events to be analysed. As with isolated mitochondria, the combination of respiration and potential is much more informative than either alone.
Modular analysis can be applied to cells and is even more valuable than with isolated mitochondria because it simplifies modules that are more complex: substrate oxidation now contains the whole of cellular catabolism, and ATP turnover includes all cellular ATP demand. It is also helpful to follow the rate of glycolysis (monitored by measurements of lactate production or extracellular acidification) for a full understanding of mitochondrial function.
One important difference between isolated mitochondria and cells is often overlooked. The respiration rates of mitochondrial preparations are usually normalized to mitochondrial concentration, whereas the respiration rates of cells are normalized to cell number, cell protein etc. Thus a change in mitochondrial density in the cell may profoundly affect the cellular respiration and fluorescence signals while being invisible in experiments on mitochondria isolated from the same cells. It is therefore crucial to take the method of normalization into account when interpreting the mechanistic basis for observed changes (or lack of changes) in the proton current. Finally, with both isolated mitochondria and intact cells, care should be taken when inferring that an observed dysfunction is the primary cause of a cellular or whole-organism phenotype. Mitochondrial ATP generation is a crucial cellular function, so mitochondria have evolved powerful feedback loops to maintain it in the face of dysfunction. Retrograde signalling from dysfunctional mitochondria to the nucleus can therefore change other mitochondrial properties in an attempt to maintain function, making cause-and-effect difficult to disentangle.
Fluxes: mitochondrial proton current (respiration rate) in cells
When using intact cells, the experimenter sets the extracellular conditions, but the cell determines what mitochondria are exposed to, making analysis more complicated, but also more physiologically relevant. The basal respiration of most cells corresponds neither to the state 3ADP rate (unlimited availability of substrate and ADP) nor to the state 4o rate (unlimited availability of substrate, but zero ATP synthesis), but is generally substrate-limited while satisfying an intermediate ATP demand. As with isolated mitochondria, the proton current generated by basal respiration supplies ATP synthesis and the proton leak, but in cells substrate supply and ATP demand are both strongly influenced by extramitochondrial processes not necessarily under the control of the experimenter.
Until recently, the only apparatus commonly used to monitor cell respiration was the Clark-type oxygen electrode and chamber. However this is limited to cells that can be obtained in high yield and in suspension. It has been used extensively and successfully with nerve terminals (synaptosomes) [54,55], primary hepatocytes [23] and cells trypsinized from plates (e.g. [56]), but there must always be reservations about the metabolic status of cells stripped from their normal growth niche and subjected to rapid stirring in suspension. As most cell preparations are available in limited amounts growing in two dimensions on a coverslip or multiwell plate, a different technology is required. One solution, the cell respirometer, superfuses incubation medium over a cell monolayer within a closed perfusion chamber [57]. Micro flow-through oxygen electrodes placed upstream and downstream allow continuous monitoring of the cellular respiration rate, and the chamber is mounted on a fluorescence microscope, allowing fluorescence signals to be monitored in parallel. The equipment has been used to monitor the respiration of cultured neurons, with a focus on the energy demands of pathological glutamate receptor activation [57–62].
Several techniques have been described for monitoring cell respiration in a multiwell format using oxygen-dependent fluorescence quenching to monitor the oxygen concentration in the medium. These techniques either operate at steady-state, where oxygen consumption is balanced by inward diffusion of oxygen [63], or measure oxygen depletion in a sealed chamber [64,65]. Although these can be used to monitor or compare basal cell respiration rates, their low time resolution and the limited ability to make additions during the run preclude them from being used for sophisticated respiration assays. The Seahorse XF24 and XF96 extracellular flux analysers refine the technology by using a piston to reversibly enclose a small volume (7 μl in the XF-24) above the cells, monitoring oxygen uptake in that volume for 2–5 min, then raising the piston, allowing the bulk incubation medium (~1 ml) to re-equilibrate [39,40]. The ability to make up to four additions during the experiment allows cell respiratory control to be measured, with the possibility of a further initial addition of a compound to screen its effects. The Seahorse instrument can be used to monitor the respiration of cells and organelles that can be attached to the multiwell plate, either spontaneously in the case of cultured cells [39] or by centrifugation of isolated mitochondria [40] or synaptosomes [66].
Synaptosomes can be considered as miniature cells for the purpose of bioenergetic analysis [67]. A representative cell respiratory control experiment [66] is shown in Figure 3(A). Predominant control over the oxygen consumption rate shifts in this experiment from proton re-entry through ATP synthase and proton leak (during basal and oligomycin-resistant respiration) to substrate oxidation (in the presence of protonophore) and to non-mitochondrial respiration (after addition of electron transport inhibitors). In the illustrative experiment in Figure 3(B), the cell respiratory control protocol is combined with the effect of a partial inhibitor of pyruvate transport, α-cyano-4-hydroxycinnamate [68]. Basal and oligomycin-resistant respiration are unaffected, but FCCP-stimulated respiration is inhibited. The lack of inhibition when proton re-entry exerts predominant control, prior to the addition of protonophore, together with the inhibition of uncontrolled respiration, indicates that the inhibitor is acting on the substrate oxidation module at some stage upstream of the proton circuit. Conversely, as shown in Figure 3(C), addition of the sodium channel activator veratridine, to enhance extramitochondrial ATP turnover, increases basal respiration. That the increase is largely oligomycin-sensitive indicates an action on ATP turnover rather than proton leak.
Figure 3. Cell respiratory control.

(A) Representative cell respiratory control experiment. Rat cortical neurones in the presence of glucose were exposed sequentially to oligomycin (oligo), FCCP and rotenone/myxothiazol. Non-mitochondrial respiration after the final addition (e) was subtracted from the other values. a, basal respiration; b, oligomycin-insensitive (leak) respiration; c, oligomycin-sensitive (ATP turnover) respiration; d, maximal respiration in the presence of FCCP. Derived parameters: coupling efficiency (c/a); respiratory control ratio (d/b); spare respiratory capacity (d−a). (B) Mouse cortical synaptosomes respiring on glucose in the presence of varying concentrations of the pyruvate transport inhibitor α-cyanocinnamate (CCIN). Note that inhibition is only seen when respiratory control is relieved by the protonophore, indicating an effect upstream of the proton circuit. (C) Synaptosomes respiring on glucose plus pyruvate; effect of the Na+ channel activator veratridine (Vt). Note the oligomycin-sensitive increase in basal respiration, indicating increased ATP utilization, and the unexpected decrease in maximal respiration. (D) Effect of supplementing glucose in the medium with pyruvate. Note that respiration is increased at all stages, showing that enhanced substrate supply exerts some control over basal and oligomycin-insensitive respiration (mediated by a slight mitochondrial hyperpolarization), although the major enhancement is seen in the presence of FCCP. Rates in (B–D) are expressed as a percentage of basal respiration in the presence of glucose. Adapted from [66].
With isolated mitochondria, substrate supply is usually in excess and exerts little control over respiration. In intact cells, substrate delivery is under complex metabolic regulation. Figure 3(D) shows the effect of augmenting glycolytic delivery of pyruvate to the synaptosomal mitochondria by adding exogenous pyruvate. The most dramatic effect is a doubling of the rate of FCCP-stimulated respiration, indicating that glycolysis was exerting considerable control over maximum respiration rate. However, both basal and oligomycin-resistant respiration are also enhanced, and parallel experiments monitoring Δψm show that the addition of exogenous pyruvate activates substrate oxidation at lower rates as well, leading to significant mitochondrial hyperpolarization and driving an increased proton current [66]. This demonstrates one of the limitations inherent in simply measuring the current component of the circuit without the potential. Ignoring potential can lead to significant errors. For example, pmf and the distribution of control over respiration rate may be very different between control and mutated cells. Erroneous conclusions may be drawn if the difference in respiration rates alone is plotted to show the effect of the mutation, ignoring the differences in pmf. The effect of ignoring pmf on the measurement of coupling efficiency in cells is significant but relatively small (see below).
Cell respiratory control: the best general measure of mitochondrial function in cell populations
In cells that have sufficiently active glycolysis to support metabolism while mitochondrial function is manipulated, all of the major aspects of mitochondrial coupling and respiratory control can be measured in a single experiment. Basal respiration, ATP turnover, proton leak, coupling efficiency, maximum respiration rate, apparent respiratory control ratio, spare respiratory capacity and non-mitochondrial respiration can all be determined by the sequential additions of the ATP synthase inhibitor oligomycin, a protonophoric uncoupler such as FCCP, and electron transport inhibitors such as rotenone and antimycin A. If glycolysis is too restricted and cell bioenergetics becomes unstable after manipulation of mitochondrial function, separate parallel oligomycin and FCCP experiments may be performed instead.
The measurement of cell respiratory control is the single most useful general test of mitochondrial function in cell populations. It is so rich in information about mitochondrial function and dysfunction that the individual steps in Figure 3(A) are discussed separately below.
Non-mitochondrial respiration
In cells such as macrophages, the activity of non-mitochondrial NADPH oxidases may dominate cellular oxygen uptake. Most other cells have low, but significant, non-mitochondrial oxygen consumption (e in Figure 3A), approximately 10% of the total [23,39,56,57,69,70], caused by various desaturase and detoxification enzymes. At the end of each cell respiratory control experiment, it is therefore usual to inhibit the mitochondrial electron transport chain completely using specific high-affinity inhibitors (Figure 3A) such as rotenone plus antimycin A (or better, myxothiazol or stigmatellin, which inhibit more completely, but can be hard to source). Cyanide, to inhibit complex IV, should be avoided because it also inhibits many haem-containing enzymes, including some of those responsible for non-mitochondrial oxygen consumption. The non-mitochondrial rate is assumed to be constant and is subtracted from all other rates.
Basal respiration
Basal respiration (a in Figure 3A) is usually controlled strongly by ATP turnover and partly by substrate oxidation and proton leak [30,71]. Therefore it alters in response to ATP demand, but is rather insensitive to small changes in maximum respiratory capacity or in proton leakiness. Basal respiration can alter dramatically, depending on the presence or absence of different substrates and hormones in the incubation medium, or triggering of ATP demand by stimulation of ion pumps or metabolic cycles. For example, hepatocyte basal respiration can be greatly increased by addition of fatty acids, pyruvate, lactate or adrenaline [72,73], insulinoma cell basal respiration depends on glucose concentration [56], and partial permeabilization of the plasma membrane in any cell type can lead to a high ATP demand for ion pumps [74]. A change in basal rate in a given cell population indicates some change in the cell, and its nature can be elucidated by the remainder of the cell respiratory control experiment. However, it can be difficult to interpret a difference in absolute rate between two cell populations. For example, if cells double in size but otherwise stay the same, the respiration rate per cell will double, but respiration rate per unit of cell protein or per nucleus or amount of DNA may not. Because of inherent noise, many replicates may be needed for statistical significance of small effects. For these reasons, ratios of rates can be more informative (see below).
ATP turnover
The rate of mitochondrial ATP synthesis in a defined basal state can be estimated from the decrease in respiration on inhibiting the ATP synthase with oligomycin (c in Figure 3A). Since ATP synthase inhibition results in a slight mitochondrial hyperpolarization and the proton leak is voltage-dependent, this approach underestimates ATP synthesis and exaggerates the proton leak prior to inhibition. In many cells the error is fairly small, underestimating ATP synthesis by less than 10% [75] and overestimating proton leak rate by 15–20%. It can be ignored where semi-quantitative answers are adequate, or overcome by measuring the modular kinetics of proton leak and correcting for the hyperpolarization. Since oligomycin shifts the entire cellular ATP production to glycolysis, this experiment requires that glycolysis accelerates perhaps 10-fold to maintain ATP production. Although most cells have sufficient glycolytic capacity to allow this, in some cells oligomycin induces an ATP crisis, causing a failure of glycolysis with its two ATP-requiring reactions (and also failure of fatty acid activation and catabolism), leading to failure of substrate supply and a progressive decrease in respiration [23,30]. If this is observed, the cell respiratory control experiment can be redesigned by making separate additions of inhibitors and uncouplers. A change in basal rate caused by a change in ATP turnover is most likely to be simply the response of the mitochondria to altered ATP demand elsewhere in the cell. However, if the response to an imposed ATP demand (see state 3ADP below) is altered, but state 3u is not, a dysfunction in the mitochondrial ATP synthetic machinery is indicated.
Proton leak
The respiration rate in the presence of oligomycin (b in Figure 3A) is a direct measure of the proton leak rate across the mitochondrial membrane in situ. After correction for hyperpolarization, it reports the proton leak rate in the preceding basal state. Under these state 4o conditions, respiration is controlled very strongly by proton leak kinetics and partially by substrate oxidation, and is therefore responsive to dysfunction caused by uncoupling, relatively insensitive to changes in substrate oxidation and completely insensitive to changes in ATP turnover. A large increase may mean that the mitochondria are severely damaged (uncoupled). Alternatively, a modest change in leak rate may indicate a change in proton leakiness or a change in Δψm caused by altered substrate oxidation. This can be tested by monitoring Δψm in parallel (uncoupling decreases potential; increased substrate oxidation increases it), or from changes in maximum respiration rate (see below).
Coupling efficiency
This can be determined from the change in basal respiration rate on addition of oligomycin. It is the fraction of basal mitochondrial oxygen consumption used for ATP synthesis (c/a in Figure 3A) and is subject to the hyperpolarization error discussed above. The coupling efficiency varies with ATP demand (it drops to zero in state 4o), but is usually fairly high: approximately 70% in hepatocytes and up to 90% in other cells [23,44,57,70]. It can be much lower in specialized cells, such as pancreatic β-cells (insulinoma cells) (30% [56,75]) or activated brown adipose tissue (almost zero). Although coupling efficiency is most sensitive to changes in proton conductance, a modular kinetic analysis is needed to resolve the mechanism of an observed change unambiguously. As an index of dysfunction, coupling efficiency has two main advantages compared with its component fluxes. First, it is sensitive to changes in all bioenergetic modules, so is likely to change in any dysfunction. Secondly, as a ratio it is internally normalized.
Maximum respiration rate
The maximum respiration rate caused by addition of an uncoupler such as FCCP (d in Figure 3A) reflects a more complicated and unstable state in cells than it does in isolated mitochondria, where substrate is added in excess and state 3u is limited by the Vmax of substrate uptake and metabolism and the activity of the respiratory chain. In cells, addition of uncoupler tends to stimulate respiration as in isolated mitochondria, but the resulting rate may be considerably less than the mitochondrial Vmax. It is critical to titrate the uncoupler in each new cell line and condition, adding just enough to give fully uncontrolled respiration yet limiting the drop in Δψm. This artificial energy demand may not evoke a physiological matching of substrate supply controlled by factors such as cytoplasmic Ca2+, and may cause a lack of cytoplasmic ATP to sustain activation of catabolic pathways. These factors are apparent in synaptosomes, where addition of pyruvate to circumvent limited glycolytic flux in the presence of uncoupler strongly increases ‘maximum’ respiratory rate [66] (Figure 3D). In addition, uncouplers act as protonophores across all membranes, acidifying the cytosolic compartment and disrupting the function of endosomes and other compartments. Together, these effects can inhibit substrate supply and cause the uncoupled rate to be lower than the true maximum respiratory rate. In cells where secondary effects of uncoupling have no significant consequences, carefully titrated uncoupled rates can report the maximum activity of electron transport and substrate oxidation that is achievable by the cells under the assay conditions. A decrease in maximum respiratory capacity is then a strong indicator of potential mitochondrial dysfunction. However, because of these complexities, caution should be exercised, as an apparent dysfunction in substrate oxidation might have more complex causes than expected.
Cell respiratory control ratio
The ratio of the uncoupled rate (state 3u) to the rate with oligomycin present (state 4o) (d/b in Figure 3A) is analogous to the (uncoupled) respiratory control ratio of isolated mitochondria. It is sensitive to changes in substrate oxidation and proton leak, but not to ATP turnover. Ideally, it would be better to measure the ‘true’ respiratory control ratio (state 3ADP/state 4o) in cells, as this would be sensitive to changes in all bioenergetic modules. State 3ADP can be approached by activating extramitochondrial ATPases. To some extent this is possible in neurons and synaptosomes, where the plasma membrane Na+/K+-ATPase has a high maximal activity, normally kept in check by the low Na+ conductance of the plasma membrane limiting ion cycling and hence ATP utilization. Increasing the sodium conductance by locking open voltage-activated sodium channels, for example with veratridine [55] (Figure 3C), or activating ionotropic glutamate receptors [57] can dramatically increase extramitochondrial ATP utilization and hence respiration. A similar approach can be taken in other cells by using limiting concentrations of the sodium ionophore gramicidin, with careful controls to check that the increased respiration rates are fully sensitive to ouabain (an inhibitor of the Na+/K+-ATPase) and not caused by the known protonophoric activity of gramicidin [74,76]. This general approach is not without problems, however. First, the imposed ATPase activity may be insufficient to maximally activate oxidative phosphorylation. If it is more than sufficient, it may deplete ATP and compromise substrate oxidation. Secondly, a genuine physiological energy demand may orchestrate matching increases in substrate oxidation using signalling cascades that are not recruited by drugs and ionophores, so the physiological maximum mitochondrial ATP synthesis rate may still exceed the artificial one. Thirdly, the activation of plasma membrane channels and receptors affects ion balances across the plasma membrane, increasing cytoplasmic sodium, depleting potassium and acidifying the cytoplasm. Because it is a ratio of two rates, the cell respiratory control ratio has the same two main advantages as coupling efficiency: sensitivity to several potential sites of dysfunction, and internal normalization. In principle, it is an excellent marker of mitochondrial dysfunction in cells, but the requirement for careful titration of FCCP and the ambiguity in what limits the maximum respiration rate, even at the peak of the titration curve or with an imposed ATP demand, makes it an uncertain index.
Spare respiratory capacity
A potentially important diagnostic of the bioenergetics of a cell that can experience a variable ATP demand, such as a neuron or muscle cell, is the spare respiratory capacity: the ability of substrate supply and electron transport to respond to an increase in energy demand [62,66]. This is measured by the difference between state 3u (or better, state 3ADP) and the basal rate (d−a in Figure 3A). Subject to the same cautions about the determination of maximum rates as above, the lack of spare respiratory capacity indicates a mitochondrial dysfunction that may not be particularly apparent under basal conditions, when respiration rate is strongly controlled by ATP turnover, but becomes manifest only under load when ATP demand increases and substrate oxidation more strongly limits respiration (e.g. Figure 3B). Conceptually, spare respiratory capacity has the advantage of indicating how close to its bioenergetic limit a cell is operating.
Use of ionophores in cells
A general caution is required about the use of ionophores in cell experiments. By their nature, ionophores show little or no membrane selectivity, since they mediate ion transport across lipid bilayers. As detailed above, a protonophore that can be used with confidence with isolated mitochondria when applied to cells will in addition acidify the cytoplasm, release transmitters from synaptic vesicles and release calcium from mitochondria. The electroneutral calcium/proton antiport ionophores ionomycin and A23187 have been used in more than 20000 publications to increase cytoplasmic free calcium and investigate its messenger roles. Under normal conditions, calcium cycling across the mitochondrial inner membrane uses only 1–2% of the maximal proton current, limited by the high calcium concentration dependence of the mitochondrial calcium uniporter and the low activity of the calcium efflux pathway [77]. Both pathways are activated dramatically by ionomycin, which induces an artefactual calcium efflux pathway in the mitochondria, while the increased cytoplasmic calcium generated by the ionophore at the plasma membrane increases calcium entry into the mitochondria through the uniporter. The overall effect is to increase the effective proton conductance of the mitochondrial membrane, i.e. to induce a protonophore-like proton leak. There is in practice a very narrow range of ionophore concentrations that allows cytoplasmic calcium to be stably enhanced without damaging mitochondrial function [78]. Unless Δψm or respiration are monitored in parallel, effects ascribed to a signalling function of calcium may instead be artefacts resulting from the induced mitochondrial dysfunction.
Forces: mitochondrial membrane potential
Because of its apparent simplicity and the availability of imaging microscopes, fluorescent monitoring of Δψm with ‘mitochondrial membrane potential indicators’ is the most common technique for monitoring mitochondrial function in intact cells at the single-cell or even single-mitochondrial level {ΔpHm (the difference in pH across the mitochondrial inner membrane) is rarely measured, although monitoring matrix pH with targeted green fluorescent protein variants has provided important information [79]}. However, the simplicity is deceptive, since the correct design and interpretation of experiments is not trivial. A well-designed experiment will give much the same information as with isolated mitochondria, with the proviso that although accurate relative values of potential can be obtained [78,80–82], absolute calibration has only recently been achieved ([83,84] and A.A. Gerencser, C. Chinopoulos, M.J. Birket, M. Jastroch, C. Vitelli, D.G. Nicholls, and M.D. Brand, unpublished work). Instead, studies are overwhelmingly qualitative (mitochondria are either ‘polarized’ or ‘depolarized’) or semi-quantitative, monitoring changes in potential from an initial arbitrary starting value. The practical details of monitoring changes in Δψm in cell culture have been reviewed extensively [85,86] and only the key factors will be summarized below.
Does the cell possess a multidrug resistance transporter capable of pumping the probe out of the cytoplasm? If so, it must be inhibited. Note that R123 (rhodamine 123) is not only a probe of potential, but is also frequently employed to test for the presence of such transporters [87].
Choice of probe
No probe is perfect, but TMRM (tetramethylrhodamine methyl ester), TMRE (tetramethylrhodamine ethyl ester) and R123 are the indicators of choice. JC-1 and DiOC(6)3 are subject to artefacts and should be avoided [86].
Loading
Low nanomolar concentrations of TMRM usually equilibrate within 60 min. No washing is performed. The more hydrophilic R123 is normally loaded into cells by 10–20 min exposure to a high (1–20 μM) concentration and the external probe is washed off before the experiment [88].
Single-mitochondrial resolution
If individual mitochondria can be resolved in a wide field-or confocal fluorescence microscope, changes in Δψm can be monitored at single-mitochondrial resolution by determining the concentration gradient of the cation between the cytoplasm and matrix [89]. The experiment must be performed in non-quench mode (see below). Light microscopy has insufficient resolution to determine the matrix volume, so relative changes in Δψm are obtained unless this value is assumed. This technique requires a very high dynamic detection range, since a Δψm of 150 mV implies a 300-fold concentration gradient between matrix and cytoplasm (eqn 2). To compensate for movement in and out of the focal plane, a mitochondrial-targeted fluorescent protein may be used to provide a reference signal [90]. Additional variables such as motility, fission and fusion, and heterogeneity can be determined in parallel [91].
Single-cell resolution
If individual mitochondria within a cell behave relatively homogeneously, quite detailed information can be obtained at single-cell resolution using the total fluorescence signal from a region of interest encompassing the cell. Although the probes do not discriminate between the plasma and mitochondrial membranes, the cell and the mitochondrial matrix possess very different surface-to-volume ratios, so the probe equilibrates across the latter hundreds of times more rapidly than between the medium and the cytoplasm. This kinetic distinction facilitates discrimination between changes in the two potentials.
Analyses performed at single-cell resolution have to take into account Δψp. Relatively permanent cations such as TMRM equilibrate across both membranes within ~60 min, and are employed in this equilibrium loading condition (without washing). The matrix accumulation is dependent on the sum of Δψp and Δψm and is equally sensitive to changes in either potential (eqn 3). The equilibrium concentration of TMRM+ in the mitochondrial matrix (m) relative to the external medium (e) is given at 37 °C by eqn (3):
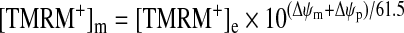 |
(3) |
where both potentials are defined as positive. On the basis of three principles, a, eqn (3), b, differential rates of equilibration across the two membranes and c, probe aggregation at a critical concentration within the matrix, a simple Excel [80,86] (or more sophisticated MATLAB [81]) program predicts with considerable accuracy the single-cell fluorescence responses to changes in Δψm and Δψp. These programs can be used to interpret complex fluorescence responses when both potentials change, for example in neurons during ionotropic receptor activation [80].
Quench or non-quench mode?
Unlike single-mitochondrial resolution (and flow cytometry), which must be performed with sufficiently low probe concentrations to avoid aggregation and quenching in the matrix, either mode may be used at single-cell resolution. In quench mode, a fall in Δψm results in a transient increase in whole-cell fluorescence as probe redistributes from the quenched matrix to the cytoplasm. The excess probe now present in the cytoplasm slowly re-equilibrates across the plasma membrane and the fluorescence returns to its initial value (Figure 4). Quench mode provides a sensitive means to detect small rapid changes in Δψm occurring during the experiment, but cannot be used to compare populations of cells with a pre-existing difference in Δψm. The differential kinetics of equilibration across plasma (slow) and mitochondrial membranes (fast) often allow changes in Δψm and Δψp to be distinguished even when they occur simultaneously [80]. R123 may be the probe of choice for many quench mode experiments, since its re-equilibration across the plasma membrane is particularly slow and may be ignored in very-short-term experiments [80,92]. Note that quench mode must never be used in flow cytometry to compare Δψm between cells; in fact, any persistent differences under these conditions are most probably due to differences in Δψp or mitochondrial amount [93]. The threshold to quench mode is the concentration at which addition of FCCP starts to cause a transient spike in whole-cell fluorescence [78].
Figure 4. Mitochondrial membrane potential changes in situ.

(A) TMRM fluorescence (in quench mode) from a single cerebellar granule neuron exposed to glutamate plus glycine (Glut/gly) to activate NMDA (N-methyl-D-aspartate) receptors. Where indicated, oligomycin and FCCP were added. (B) Values for Δψp and Δψm input to a spreadsheet simulating Nernst equilibria across both membranes, differential equilibration times across plasma (slow) and mitochondrial (fast) membranes and matrix quenching at a critical concentration. (C) Simulated trace generated from (B). Note that the small initial increase indicates a slight (5 mV) mitochondrial depolarization; the plasma membrane depolarization resulting from receptor activation causes a slow decrease in fluorescence; the 10 mV hyperpolarization during the oligomycin ‘null-point test’ indicates that the mitochondria were still generating ATP, and the spike with FCCP confirms that the experiment was performed in quench mode. Adapted from [80]. min/div, min per time division shown.
Equilibration of a typical cell with 1–20 nM TMRM will result in the probe retaining full fluorescence within the mitochondrial matrix (non-quench mode). The single-cell response to a partial mitochondrial depolarization is now totally different. Since there is no change in quantum yield between matrix and cytoplasm, the total fluorescence from a single cell will not change as a result of the probe translocation. All that will be seen is the slow re-equilibration of probe across the plasma membrane to restore the Nernst equilibrium. Equilibration can be speeded up by the presence of tetraphenylboron [55]. Non-quench mode does not distinguish at single-cell resolution between mitochondrial and plasma membrane depolarization.
Simultaneous monitoring of plasma and mitochondrial membrane potential changes
The membrane ambiguity inherent in non-quench mode is serious if Δψp changes during the experiment. In such cases, experiments may be performed in the simultaneous presence of a fluorescent cation, such as TMRM, and a fluorescence anion that enters the cytoplasm as the plasma membrane is depolarized. One such study [78] has been performed with cultured neurons using an anionic probe, termed PMPI (plasma membrane potential indicator), from a commercial kit. Since the intracellular concentration of PMPI is lower than that in the medium, interference from extracellular fluorescence is prevented by a hydrophilic quencher. The two independent signals from the cationic and anionic indicators allow changes in Δψp and Δψm to be estimated independently [78]. More sophisticated models allow fully quantitative calculation of both potentials using this approach ([83,84] and A.A. Gerencser, C. Chinopoulos, M.J. Birket, M. Jastroch, C. Vitelli, D.G. Nicholls and M.D. Brand, unpublished work).
What does measurement of Δψm tell us?
If only a single technique is to be used, then respiration, i.e. proton current, is generally the method of choice for subtle changes, since the steep relationships between pmf and the fluxes through oxidative phosphorylation (Figure 2B) mean that small changes in pmf are associated with large changes in respiration. However, for quantitative analysis, the combination of potential and respiration is more informative than measurement of either alone. If respiration increases, the direction of the change in potential indicates whether the primary effect is upstream of Δψm (potential rises) or downstream (potential drops). Modular kinetic analysis in cells quantitatively extends this approach to identify the primary causes of any bioenergetic change and to rank their importance. Modular kinetic analysis and control analysis have been applied to cells in bulk using the Clark electrode to measure respiration and radioactive tracers to measure membrane potentials [32,94–96], but the combination of plate-based respirometry and fluorescence microscopy should allow the approach to be used more easily in cell monolayers and has great promise. As with the simpler system of isolated mitochondria, modular kinetic analysis fully describes (at the system level) any functional change in cellular bioenergetics. Once a dysfunction is discovered, modular kinetic analysis will uncover the primary and secondary causes of that dysfunction.
Whereas respiration can currently be determined only in cell populations, fluorescence microscopy can analyse stochastic changes at single-cell or single-mitochondrial resolution. Monitoring Δψm in a stressed cell can establish the time at which its mitochondria fail (i.e. depolarize). If this changes, or if the concentration of stressor that triggers failure differs between control and experimental cells, one possibility is that the mitochondria in the experimental cells are in some way dysfunctional. The ability to combine multiple probes for Ca2+, superoxide etc. means that temporal relationships can be established. For example, in an individual neuron exposed to excitotoxic glutamate, irreversible mitochondrial depolarization is synchronous with a massive rise in cytoplasmic Ca2+ and superoxide levels. Experiments can then be devised to establish cause-and-effect [97].
It is frequently important to establish whether the mitochondria in a stressed cell still generate ATP or are damaged and maintain their membrane potential by hydrolysing glycolytic ATP. If mitochondria show a slight hyperpolarization on adding oligomycin to the cells they were still (partially) functional. If oligomycin causes a depolarization they were relying on glycolytic ATP. This ‘oligomycin null-point test’ has established at what point mitochondria fail during staurosporine-induced apoptosis [98], neuronal excitotoxicity [80] and glutathione depletion [59].
Adenine nucleotides
Several bioenergetic intermediates other than Δψm can be measured in cell monolayers, particularly ATP (e.g. with luciferase) and NADH (using autofluorescence). In general, they give much the same information as Δψm, since they are closely linked by near-equilibrium reactions. Measurement of the amount of ATP in a cell is conceptually and practically appealing. However, the amount of cellular ATP does not safely report mitochondrial function.
Generally, most of the adenine nucleotide in cells is present as ATP. For example, in rat hepatocytes, the adenine nucleotide pool is approximately 71% ATP, 19% ADP and 10% AMP [99,100]. There is essentially no scope for an increase in the total amount of ATP in a cell due solely to ‘improved’ bioenergetic function (with the exception of highly specialized cells, such as the insulin-secreting β-cell [101]). Instead, reports of increases of 100% or more in cellular ATP content following various manipulations must reflect large increases in total adenine nucleotide pool size, and do not report bioenergetic status. Nonetheless, an increase in pool size may sometimes be related to bioenergetics because of a slow cycle catalysed by AMP deaminase, which converts AMP into IMP and depletes the adenine nucleotide pool, and adenine nucleotide resynthesis, which refills it. If cellular ATP/ADP decreases, adenylate kinase activity consumes ADP and raises AMP. If the depression is sustained, AMP deaminase degrades AMP to IMP, decreasing the adenine nucleotide pool size. Conversely, when ATP/ADP is high, AMP degradation by AMP deaminase slows and the pool size may rise [102]. Whether an observed change in ATP content in cells reflects a change in mitochondrial function or an independent change in the metabolism of adenine nucleotides is impossible to determine without further experiments, so total cellular ATP is a very poor reporter of mitochondrial dysfunction.
Other criteria for mitochondrial function and dysfunction in cells
Classical assays of mitochondrial dysfunction
Several classical indicators retain some value as first indicators of possible mitochondrial dysfunction. The reduction of tetrazolium salts such as MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide] or water-soluble tetrazolium derivatives (in the presence of membrane-permeant redox mediators, such as phenazine methosulfate) to purple formazan products is a quick and dirty indicator of the availability of reducing power in a preparation [103]. It has been used to flag redox derangements that might be related to mitochondrial dysfunction, but clearly needs to be followed up by more specific approaches before any conclusions can be drawn. The ability of cells to respire on galactose is sometimes used as an index of mitochondrial function on the grounds that mitochondrial ATP production is more prominent with this substrate. An increase in lactate production by cells might indicate a mitochondrial dysfunction that leads to a compensatory increase in glycolytic rate. In each case, the classical experiments may indicate a dysfunction, but measurement of cell respiratory control is much more informative.
In passing, we note that inappropriate knockout experiments are sometimes used to infer the involvement of mitochondria in cellular processes. The loss of some phenotype in rho-zero cells (which lack mitochondrial DNA and therefore lack oxidative phosphorylation) is sometimes taken to demonstrate mitochondrial involvement in generating the phenotype. Similarly, the knockout of a specific respiratory complex is sometimes used to argue that this complex is directly involved in causing a phenotype. In both cases, however, the gross derangement of cellular oxidative metabolism and energetics that will follow the knockout means that virtually any process in the cell will be affected, and simple interpretations are very dangerous.
Assays of mitochondrial proliferation or appearance
Mitochondrial proliferation might indicate the engagement of feedback loops to compensate for compromised mitochondrial function. A popular indicator is gene expression, where over-representation (or under-representation) of genes coding for the components of oxidative metabolism, or transcription factors involved in mitochondrial biogenesis, such as PGC-1α (peroxisome-proliferator-activated receptor γ co-activator 1α), may flag an underlying energetic dysfunction. The same is true for decreases or increases in the concentrations and activities of candidate respiratory complexes and enzymes in cells. Similarly, gross changes in mitochondrial shape, distribution or appearance (e.g. orthodox or condensed cristae [104]) might indicate altered function. All of these markers might (or might not) flag a mitochondrial dysfunction, and indicate that cell respiratory control should be measured.
Reactive oxygen species, oxidative stress and other reactions with a mitochondrial connection
Mitochondrial dysfunction may lead to increased radical production and consequent ‘oxidative stress’. Observations of altered glutathione concentrations, protein thiol status, lipid peroxidation markers, protein carbonyls, sensitivity to radicals such as paraquat or sensitivity to mitochondrial poisons may suggest that direct assay of mitochondrial function is required to see if it has changed as a cause or consequence of these phenotypes. The same inference can be drawn from alterations in other reactions where mitochondria have a role, such as cytosolic calcium homoeostasis, cytochrome c release, caspase activation, apoptosis, necrosis or metabolism. In short, mitochondrial dysfunction can affect almost every cellular function, so the observation of almost any cellular dysfunction can suggest a mitochondrial dysfunction that should be tested by measuring cell respiratory control.
Summary: assaying mitochondrial dysfunction in intact cells
The measurement of cell respiratory control is the single most useful general test of mitochondrial function in cells. The experiment is easy to perform using the appropriate apparatus, and yields information that allows a quick, simple and full assessment of the bioenergetic status of the cells: how fast they are turning over ATP, how well-coupled their mitochondria are, and how much spare respiratory capacity they have available to deal with energetic demands. Measurements of Δψm are subject to several pitfalls that need to be understood, but can also be very useful. The best way to gain mechanistic insight is to monitor both respiration and potential, to assay the mitochondrial proton circuit in situ. Other indirect assays of mitochondrial function in cells, such as measurements of dye reduction or total cell ATP, can be problematic.
IN VIVO BIOENERGETIC MONITORING
In principle, measurement of mitochondrial function in vivo is better than measurement in vitro. However, the gain in physiological relevance is offset by the loss in specificity and the greater difficulties of interpretation. Many symptoms may suggest an underlying mitochondrial dysfunction. Prominent among these are failure to thrive or muscular or neurological clinical symptoms in patients with mitochondrial diseases. In animal models, there may be exercise intolerance, poor grip strength, altered metabolic efficiency or failure to put on body weight. Often, a more indirect indication of mitochondrial dysfunction is raised by the pattern of changes in gene arrays, proteomics or metabolomics, where GO (Gene Ontology) categories may suggest altered energy metabolism. Sometimes a specific gene that might affect mitochondria is known to be mutated, raising the hypothesis that its phenotype is caused by mitochondrial dysfunction.
Sometimes it is most appropriate to study muscle biopsies or tissue slices. These have the advantage of maintained physical structure, but many disadvantages, particularly those caused by lack of rapid diffusion of oxygen into the preparation. With tissue slices, many attempts have been made to maintain good aeration by working with very thin slices, but these contain a greater proportion of broken cells, because of their increased surface-to-volume ratio, and may still have a hypoxic core. Often the next step in assaying mitochondrial dysfunction is to isolate mitochondria or revert to a cellular system to simplify the study; the advantages and disadvantages of these are discussed above.
Whole-body respirometry to study mitochondrial function in vivo pre-dates studies of cells and isolated mitochondria. Most of the oxygen we breathe is consumed by cytochrome oxidase in the tissues, so whole-body respiration (or tissue respiration inferred from flow rates and arterio-venous differences in oxygen tension) is used by respiratory physiologists to measure the overall rate of mitochondrial electron transport. However, the complexity of whole organisms and tissues makes it hard to infer mitochondrial dysfunction from changes in oxygen consumption or aerobic scope, because it is so difficult to distinguish a dysfunction in the mitochondria from a dysfunction in the supply of substrates or the consumption of ATP in the tissues in the absence of other information. In a few cases, the whole-body problem can be simplified. An excellent example is the hummingbird in flight, where nearly all oxygen consumption is used to power muscle contraction and ATP consumption can be calculated from the dynamics of wing movements. In this extreme system, it is possible to measure the effective P/O ratio of oxidative phosphorylation in vivo, and to distinguish fatty acid oxidation after overnight fasting from glucose oxidation following the first morning sugar feed [105].
Techniques such as positron emission tomography now allow direct measurement of glucose uptake by cells in vivo and provide a rich three-dimensional and dynamic picture of energy use in different locations. Much of this will involve mitochondrial oxidative phosphorylation, but again, unpicking changes in energy demand from dysfunctions in energy supply is not straightforward. There have been major improvements in 31P NMR, allowing a great expansion of in vivo bioenergetics. Using 31P NMR it is possible to measure ATP in intact organs. More usefully, the creatine phosphate signal can be used to infer the creatine phosphate/creatine ratio and, from the equilibrium of creatine kinase, the ATP/ADP ratio. By occluding blood flow and then releasing it, the rate of ATP resynthesis can be measured, giving an estimate of the maximum capacity of oxidative phosphorylation to generate ATP, the rate of ATP turnover in steady-state and (using related measures of oxygen consumption) the effective P/O ratio [106]. Such approaches hold great promise for the direct study of mitochondrial function and dysfunction in vivo.
FUNDING
Work in the authors' laboratories is supported by the National Institutes of Health [grant numbers P01 AG025901, PL1 AG032118, P30 AG025708 and R01 AG033542], the W.M. Keck Foundation (to M.B. and D.N.) and the Ellison Medical Foundation [grant number AG-SS-2288-09 (to M.B.)].
References
- 1.De Kock I., Van Daele C., Poelaert J. Sepsis and septic shock: pathophysiological and cardiovascular background as basis for therapy. Acta Clin. Belg. 2010;65:323–329. doi: 10.1179/acb.2010.070. [DOI] [PubMed] [Google Scholar]
- 2.Fernyhough P., Roy Chowdhury S. K., Schmidt R. E. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev. Endocrinol. Metab. 2010;5:39–49. doi: 10.1586/eem.09.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferreira I. L., Resende R., Ferreiro E., Rego A. C., Pereira C. F. Multiple defects in energy metabolism in Alzheimer's disease. Curr. Drug Targets. 2010;11:1193–1206. doi: 10.2174/1389450111007011193. [DOI] [PubMed] [Google Scholar]
- 4.Haas R. H. Autism and mitochondrial disease. Dev. Disabil. Res. Rev. 2010;16:144–153. doi: 10.1002/ddrr.112. [DOI] [PubMed] [Google Scholar]
- 5.Jarrett S. G., Lewin A. S., Boulton M. E. The importance of mitochondria in age-related and inherited eye disorders. Ophthalmic Res. 2010;44:179–190. doi: 10.1159/000316480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawamata H., Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 2010;131:517–526. doi: 10.1016/j.mad.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kones R. Parkinson's disease: mitochondrial molecular pathology, inflammation, statins, and therapeutic neuroprotective nutrition. Nutr. Clin. Pract. 2010;25:371–389. doi: 10.1177/0884533610373932. [DOI] [PubMed] [Google Scholar]
- 8.Ren J., Pulakat L., Whaley-Connell A., Sowers J. R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010;88:993–1001. doi: 10.1007/s00109-010-0663-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rocha M., Apostolova N., Hernandez-Mijares A., Herance R., Victor V. M. Oxidative stress and endothelial dysfunction in cardiovascular disease: mitochondria-targeted therapeutics. Curr. Med. Chem. 2010;17:3827–3841. doi: 10.2174/092986710793205444. [DOI] [PubMed] [Google Scholar]
- 10.Rosenstock T. R., Duarte A. I., Rego A. C. Mitochondrial-associated metabolic changes and neurodegeneration in Huntington's disease – from clinical features to the bench. Curr. Drug Targets. 2010;11:1218–1236. doi: 10.2174/1389450111007011218. [DOI] [PubMed] [Google Scholar]
- 11.Sas K., Pardutz A., Toldi J., Vecsei L. Dementia, stroke and migraine – some common pathological mechanisms. J. Neurol. Sci. 2010;299:55–65. doi: 10.1016/j.jns.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Scaglia F. The role of mitochondrial dysfunction in psychiatric disease. Dev. Disabil. Res. Rev. 2010;16:136–143. doi: 10.1002/ddrr.115. [DOI] [PubMed] [Google Scholar]
- 13.Waldbaum S., Patel M. Mitochondrial dysfunction and oxidative stress: a contributing link to acquired epilepsy? J. Bioenerg. Biomembr. 2010;42:449–455. doi: 10.1007/s10863-010-9320-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Will Y., Dykens J. A., Nadanaciva S., Hirakawa B., Jamieson J., Marroquin L. D., Hynes J., Patyna S., Jessen B. A. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol. Sci. 2008;106:153–161. doi: 10.1093/toxsci/kfn157. [DOI] [PubMed] [Google Scholar]
- 15.Nicholls D. G., Ferguson S. J. Bioenergetics. 3rd edn. London: Academic Press; 2002. [Google Scholar]
- 16.Brand M. D. The efficiency and plasticity of mitochondrial energy transduction. Biochem. Soc. Trans. 2005;33:897–904. doi: 10.1042/BST0330897. [DOI] [PubMed] [Google Scholar]
- 17.Hinkle P. C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta. 2005;1706:1–11. doi: 10.1016/j.bbabio.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell P., Moyle J. Estimation of membrane potential and pH difference across the cristae membrane of rat liver mitochondria. Eur. J. Biochem. 1969;7:471–484. doi: 10.1111/j.1432-1033.1969.tb19633.x. [DOI] [PubMed] [Google Scholar]
- 19.Nicholls D. G. Hamster brown-adipose-tissue mitochondria. The control of respiration and the proton electrochemical potential gradient by possible physiological effectors of the proton conductance of the inner membrane. Eur. J. Biochem. 1974;49:573–583. doi: 10.1111/j.1432-1033.1974.tb03861.x. [DOI] [PubMed] [Google Scholar]
- 20.Stumpf D. A., Haas R., Eguren L. A., Parks J. K., Eilert R. E. Protonmotive force in muscle mitochondria. Muscle Nerve. 1982;5:14–19. doi: 10.1002/mus.880050104. [DOI] [PubMed] [Google Scholar]
- 21.Hafner R. P., Brown G. C., Brand M. D. Analysis of the control of respiration rate, phosphorylation rate, proton leak rate and protonmotive force in isolated mitochondria using the ‘top-down’ approach of metabolic control theory. Eur. J. Biochem. 1990;188:313–319. doi: 10.1111/j.1432-1033.1990.tb15405.x. [DOI] [PubMed] [Google Scholar]
- 22.Nicholls D. G. The effective proton conductance of the inner membrane of mitochondria from brown adipose tissue. Dependency on proton electrochemical potential gradient. Eur. J. Biochem. 1977;77:349–356. doi: 10.1111/j.1432-1033.1977.tb11674.x. [DOI] [PubMed] [Google Scholar]
- 23.Nobes C. D., Brown G. C., Olive P. N., Brand M. D. Non-ohmic proton conductance of the mitochondrial inner membrane in hepatocytes. J. Biol. Chem. 1990;265:12903–12909. [PubMed] [Google Scholar]
- 24.Brand M. D. The proton leak across the mitochondrial inner membrane. Biochim. Biophys. Acta. 1990;1018:128–133. doi: 10.1016/0005-2728(90)90232-s. [DOI] [PubMed] [Google Scholar]
- 25.Brand M. D., Chien L. F., Ainscow E. K., Rolfe D. F. S., Porter R. K. The causes and functions of mitochondrial proton leak. Biochim. Biophys. Acta. 1994;1187:132–139. doi: 10.1016/0005-2728(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 26.Rolfe D. F. S., Newman J. M., Buckingham J. A., Clark M. G., Brand M. D. Contribution of mitochondrial proton leak to respiration rate in working skeletal muscle and liver and to SMR. Am. J. Physiol. 1999;276:C692–C699. doi: 10.1152/ajpcell.1999.276.3.C692. [DOI] [PubMed] [Google Scholar]
- 27.Rolfe D. F. S., Brand M. D. The physiological significance of mitochondrial proton leak in animal cells and tissues. Biosci. Rep. 1997;17:9–16. doi: 10.1023/a:1027327015957. [DOI] [PubMed] [Google Scholar]
- 28.Nicholls D. G. Mitochondria and calcium signaling. Cell Calcium. 2005;38:311–317. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Brown G. C., Hafner R. P., Brand M. D. A ‘top-down’ approach to the determination of control coefficients in metabolic control theory. Eur. J. Biochem. 1990;188:321–325. doi: 10.1111/j.1432-1033.1990.tb15406.x. [DOI] [PubMed] [Google Scholar]
- 30.Brown G. C., Lakin-Thomas P. L., Brand M. D. Control of respiration and oxidative phosphorylation in isolated rat liver cells. Eur. J. Biochem. 1990;192:355–362. doi: 10.1111/j.1432-1033.1990.tb19234.x. [DOI] [PubMed] [Google Scholar]
- 31.Brand M. D. Top down metabolic control analysis. J. Theor. Biol. 1996;182:351–360. doi: 10.1006/jtbi.1996.0174. [DOI] [PubMed] [Google Scholar]
- 32.Brand M. D. Top-down elasticity analysis and its application to energy metabolism in isolated mitochondria and intact cells. Mol. Cell. Biochem. 1998;184:13–20. [PubMed] [Google Scholar]
- 33.Amo T., Brand M. D. Were inefficient mitochondrial haplogroups selected during migrations of modern humans? A test using modular kinetic analysis of coupling in mitochondria from cybrid cell lines. Biochem. J. 2007;404:345–351. doi: 10.1042/BJ20061609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brand M. D. Regulation analysis of energy metabolism. J. Exp. Biol. 1997;200:193–202. doi: 10.1242/jeb.200.2.193. [DOI] [PubMed] [Google Scholar]
- 35.Hafner R. P., Brand M. D. Effect of protonmotive force on the relative proton stoichiometries of the mitochondrial proton pumps. Biochem. J. 1991;275:75–80. doi: 10.1042/bj2750075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brand M. D., Chien L. F., Diolez P. Experimental discrimination between proton leak and redox slip during mitochondrial electron transport. Biochem. J. 1994;297:27–29. doi: 10.1042/bj2970027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porter R. K., Brand M. D. Mitochondrial proton conductance and H+/O ratio are independent of electron transport rate in isolated hepatocytes. Biochem. J. 1995;310:379–382. doi: 10.1042/bj3100379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chance B., Williams G. R. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem. 1955;217:383–393. [PubMed] [Google Scholar]
- 39.Wu M., Neilson A., Swift A. L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 40.Gerencser A. A., Neilson A., Choi S. W., Edman U., Yadava N., Oh R. J., Ferrick D. A., Nicholls D. G., Brand M. D. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal. Chem. 2009;81:6868–6878. doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicholls D. G., Bernson V. S. Inter-relationships between proton electrochemical gradient, adenine-nucleotide phosphorylation potential and respiration, during substrate-level and oxidative phosphorylation by mitochondria from brown adipose tissue of cold-adapted guinea-pigs. Eur. J. Biochem. 1977;75:601–612. doi: 10.1111/j.1432-1033.1977.tb11560.x. [DOI] [PubMed] [Google Scholar]
- 42.Nicholls D. G. The physiological regulation of uncoupling proteins. Biochim. Biophys. Acta. 2006;1757:459–466. doi: 10.1016/j.bbabio.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Affourtit C., Brand M. D. Stronger control of ATP/ADP by proton leak in pancreatic β-cells than skeletal muscle mitochondria. Biochem. J. 2006;393:151–159. doi: 10.1042/BJ20051280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brand M. D., Harper M. E., Taylor H. C. Control of the effective P/O ratio of oxidative phosphorylation in liver mitochondria and hepatocytes. Biochem. J. 1993;291:739–748. doi: 10.1042/bj2910739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicholls D. G. The influence of respiration and ATP hydrolysis on the proton-electrochemical gradient across the inner membrane of rat-liver mitochondria as determined by ion distribution. Eur. J. Biochem. 1974;50:305–315. doi: 10.1111/j.1432-1033.1974.tb03899.x. [DOI] [PubMed] [Google Scholar]
- 46.Nicholls D. G., Grav H. J., Lindberg O. Mitochondrial from hamster brown-adipose tissue. Regulation of respiration in vitro by variations in volume of the matrix compartment. Eur. J. Biochem. 1972;31:526–533. doi: 10.1111/j.1432-1033.1972.tb02561.x. [DOI] [PubMed] [Google Scholar]
- 47.Brown G. C., Brand M. D. Proton/electron stoichiometry of mitochondrial complex I estimated from the equilibrium thermodynamic force ratio. Biochem. J. 1988;252:473–479. doi: 10.1042/bj2520473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brand M. D. Measurement of mitochondrial protonmotive force. In: Brown G. C., Cooper C. E., editors. Bioenergetics, a practical approach. Oxford: IRL Press; 1995. pp. 39–62. [Google Scholar]
- 49.Kamo N., Muratsugu M., Hongoh R., Kobatake Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J. Membr. Biol. 1979;49:105–121. doi: 10.1007/BF01868720. [DOI] [PubMed] [Google Scholar]
- 50.Akerman K. E. O., Wikstrom M. K. Safranine as a probe of the mitochondrial membrane potential. FEBS Lett. 1976;68:191–197. doi: 10.1016/0014-5793(76)80434-6. [DOI] [PubMed] [Google Scholar]
- 51.Nicholls D. G. The non-ohmic proton leak – 25 years on. Biosci. Rep. 1997;17:251–257. doi: 10.1023/a:1027376426860. [DOI] [PubMed] [Google Scholar]
- 52.Cadenas S., Echtay K. S., Harper J. A., Jekabsons M. B., Buckingham J. A., Grau E., Abuin A., Chapman H., Clapham J. C., Brand M. D. The basal proton conductance of skeletal muscle mitochondria from transgenic mice overexpressing or lacking uncoupling protein-3. J. Biol. Chem. 2002;277:2773–2778. doi: 10.1074/jbc.M109736200. [DOI] [PubMed] [Google Scholar]
- 53.Appaix F., Kuznetsov A. V., Usson Y., Kay L., Andrienko T., Olivares J., Kaambre T., Sikk P., Margreiter R., Saks V. Possible role of cytoskeleton in intracellular arrangement and regulation of mitochondria. Exp. Physiol. 2003;88:175–190. doi: 10.1113/eph8802511. [DOI] [PubMed] [Google Scholar]
- 54.Bradford H. F. Respiration in vitro of synaptosomes from mammalian cerebral cortex. J. Neurochem. 1969;16:675–684. doi: 10.1111/j.1471-4159.1969.tb06444.x. [DOI] [PubMed] [Google Scholar]
- 55.Scott I. D., Nicholls D. G. Energy transduction in intact synaptosomes. Influence of plasma-membrane depolarization on the respiration and membrane potential of internal mitochondria determined in situ. Biochem. J. 1980;186:21–33. doi: 10.1042/bj1860021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Affourtit C., Brand M. D. Uncoupling protein-2 contributes significantly to high mitochondrial proton leak in INS-1E insulinoma cells and attenuates glucose-stimulated insulin secretion. Biochem. J. 2008;409:199–204. doi: 10.1042/BJ20070954. [DOI] [PubMed] [Google Scholar]
- 57.Jekabsons M. B., Nicholls D. G. In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J. Biol. Chem. 2004;279:32989–33000. doi: 10.1074/jbc.M401540200. [DOI] [PubMed] [Google Scholar]
- 58.Jekabsons M. B., Nicholls D. G. Bioenergetic analysis of cerebellar granule neurons undergoing apoptosis by potassium/serum deprivation. Cell Death Differ. 2006;13:1595–1610. doi: 10.1038/sj.cdd.4401851. [DOI] [PubMed] [Google Scholar]
- 59.Vesce S., Jekabsons M. B., Johnson-Cadwell L. I., Nicholls D. G. Acute glutathione depletion restricts mitochondrial ATP export in cerebellar granule neurons. J. Biol. Chem. 2005;280:38720–38728. doi: 10.1074/jbc.M506575200. [DOI] [PubMed] [Google Scholar]
- 60.Oliveira J. M., Jekabsons M. B., Chen S., Lin A., Rego A. C., Goncalves J., Ellerby L. M., Nicholls D. G. Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem. 2007;101:241–249. doi: 10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- 61.Johnson-Cadwell L. I., Jekabsons M. B., Wang A., Polster B. M., Nicholls D. G. ‘Mild uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J. Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- 62.Yadava N., Nicholls D. G. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J. Neurosci. 2007;27:7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deshpande R. R., Heinzle E. On-line oxygen uptake rate and culture viability measurement of animal cell culture using microplates with integrated oxygen sensors. Biotechnol. Lett. 2004;26:763–767. doi: 10.1023/b:bile.0000024101.57683.6d. [DOI] [PubMed] [Google Scholar]
- 64.Hynes J., Floyd S., Soini A. E., O'Connor R., Papkovsky D. B. Fluorescence-based cell viability screening assays using water-soluble oxygen probes. J. Biomol. Screen. 2003;8:264–272. doi: 10.1177/1087057103008003004. [DOI] [PubMed] [Google Scholar]
- 65.Zwicker K., Galkin A., Drose S., Grgic L., Kerscher S., Brandt U. The Redox-Bohr group associated with iron–sulfur cluster N2 of complex I. J. Biol. Chem. 2006;281:23013–23017. doi: 10.1074/jbc.M603442200. [DOI] [PubMed] [Google Scholar]
- 66.Choi S. W., Gerencser A. A., Nicholls D. G. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J. Neurochem. 2009;109:1179–1191. doi: 10.1111/j.1471-4159.2009.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nicholls D. G. Bioenergetics and transmitter release in the isolated nerve terminal. Neurochem. Res. 2003;28:1433–1441. doi: 10.1023/a:1025653805029. [DOI] [PubMed] [Google Scholar]
- 68.Halestrap A. P., Denton R. M. Specific inhibition of pyruvate transport in rat liver mitochondria and human erythrocytes by α-cyano-4-hydroxycinnamate. Biochem. J. 1974;138:313–316. doi: 10.1042/bj1380313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Porter R. K., Brand M. D. Causes of differences in respiration rate of hepatocytes from mammals of different body mass. Am. J. Physiol. 1995;269:R1213–R1224. doi: 10.1152/ajpregu.1995.269.5.R1213. [DOI] [PubMed] [Google Scholar]
- 70.Amo T., Yadava N., Oh R., Nicholls D. G., Brand M. D. Experimental assessment of bioenergetic differences caused by the common European mitochondrial DNA haplogroups H and T. Gene. 2008;411:69–76. doi: 10.1016/j.gene.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ainscow E. K., Brand M. D. Top-down control analysis of ATP turnover, glycolysis and oxidative phosphorylation in rat hepatocytes. Eur. J. Biochem. 1999;263:671–685. doi: 10.1046/j.1432-1327.1999.00534.x. [DOI] [PubMed] [Google Scholar]
- 72.Nobes C. D., Hay W. W., Jr, Brand M. D. The mechanism of stimulation of respiration by fatty acids in isolated hepatocytes. J. Biol. Chem. 1990;265:12910–12915. [PubMed] [Google Scholar]
- 73.Korzeniewski B., Harper M. E., Brand M. D. Proportional activation coefficients during stimulation of oxidative phosphorylation by lactate and pyruvate or by vasopressin. Biochim. Biophys. Acta. 1995;1229:315–322. doi: 10.1016/0005-2728(95)00008-7. [DOI] [PubMed] [Google Scholar]
- 74.Nobes C. D., Lakin-Thomas P. L., Brand M. D. The contribution of ATP turnover by the Na+/K+-ATPase to the rate of respiration of hepatocytes. Effects of thyroid status and fatty acids. Biochim. Biophys. Acta. 1989;976:241–245. doi: 10.1016/s0005-2728(89)80236-1. [DOI] [PubMed] [Google Scholar]
- 75.Affourtit C., Brand M. D. Measuring mitochondrial bioenergetics in INS-1E insulinoma cells. Methods Enzymol. 2009;457:405–424. doi: 10.1016/S0076-6879(09)05023-X. [DOI] [PubMed] [Google Scholar]
- 76.Lou P. H., Hansen B. S., Olsen P. H., Tullin S., Murphy M. P., Brand M. D. Mitochondrial uncouplers with an extraordinary dynamic range. Biochem. J. 2007;407:129–140. doi: 10.1042/BJ20070606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nicholls D. G., Chalmers S. The integration of mitochondrial calcium transport and storage. J. Bioenerg. Biomembr. 2004;36:277–281. doi: 10.1023/B:JOBB.0000041753.52832.f3. [DOI] [PubMed] [Google Scholar]
- 78.Nicholls D. G. Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J. Biol. Chem. 2006;281:14864–14874. doi: 10.1074/jbc.M510916200. [DOI] [PubMed] [Google Scholar]
- 79.Wiederkehr A., Park K. S., Dupont O., Demaurex N., Pozzan T., Cline G. W., Wollheim C. B. Matrix alkalinization: a novel mitochondrial signal for sustained pancreatic β-cell activation. EMBO J. 2009;28:417–428. doi: 10.1038/emboj.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ward M. W., Rego A. C., Frenguelli B. G., Nicholls D. G. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ward M. W., Huber H. J., Weisova P., Dussmann H., Nicholls D. G., Prehn J. H. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J. Neurosci. 2007;27:8238–8249. doi: 10.1523/JNEUROSCI.1984-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gerencser A. A., Mark K. A., Hubbard A. E., Divakaruni A. S., Mehrabian Z., Nicholls D. G., Polster B. M. Real-time visualization of cytoplasmic calpain activation and calcium deregulation in acute glutamate excitotoxicity. J. Neurochem. 2009;110:990–1004. doi: 10.1111/j.1471-4159.2009.06194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Birket M. J., Orr A. L., Gerencser A. A., Madden D. T., Vitelli C., Swistowski A., Brand M. D., Zeng X. A reduction in ATP demand and mitochondrial activity with neural differentiation of human embryonic stem cells. J. Cell Sci. 2011;124:348–358. doi: 10.1242/jcs.072272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chinopoulos C., Gerencser A. A., Mandi M., Mathe K., Torocsik B., Doczi J., Turiak L., Kiss G., Konrad C., Vajda S., et al. Forward operation of adenine nucleotide translocase during F0F1-ATPase reversal: critical role of matrix substrate-level phosphorylation. FASEB J. 2010;24:2405–2416. doi: 10.1096/fj.09-149898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davidson S. M., Yellon D., Duchen M. R. Assessing mitochondrial potential, calcium, and redox state in isolated mammalian cells using confocal microscopy. Methods Mol. Biol. 2007;372:421–430. doi: 10.1007/978-1-59745-365-3_30. [DOI] [PubMed] [Google Scholar]
- 86.Nicholls D. G. Fluorescence measurement of mitochondrial membrane potential changes in cultured cells. In: Palmeira C. M., Moreno A. J., editors. Mitochondrial Bioenergetics : Methods and Protocols. New York: Humana Press; 2011. in the press. [Google Scholar]
- 87.Bartosiewicz D., Krasowska A. Inhibitors of ABC transporters and biophysical methods to study their activity. Z. Naturforsch. C. 2009;64:454–458. doi: 10.1515/znc-2009-5-625. [DOI] [PubMed] [Google Scholar]
- 88.Duchen M. R. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem. J. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chacon E., Reece J. M., Nieminen A. L., Zahrebelski G., Herman B., Lemasters J. J. Distribution of electrical potential, pH, free Ca2+, and volume inside cultured adult rabbit cardiac myocytes during chemical hypoxia: a multiparameter digitized confocal microscopic study. Biophys. J. 1994;66:942–952. doi: 10.1016/S0006-3495(94)80904-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G., et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wikstrom J. D., Twig G., Shirihai O. S. What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int. J. Biochem. Cell Biol. 2009;41:1914–1927. doi: 10.1016/j.biocel.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 92.Duchen M. R., Biscoe T. J. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. J. Physiol. 1992;450:33–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zamzami N., Marchetti P., Castedo M., Zanin C., Vayssiere J. L., Petit P. X., Kroemer G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med. 1995;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Harper M. E., Brand M. D. The quantitative contributions of mitochondrial proton leak and ATP turnover reactions to the changed respiration rates of hepatocytes from rats of different thyroid status. J. Biol. Chem. 1993;268:14850–14860. [PubMed] [Google Scholar]
- 95.Buttgereit F., Grant A., Muller M., Brand M. D. The effects of methylprednisolone on oxidative phosphorylation in concanavalin-A-stimulated thymocytes. Top-down elasticity analysis and control analysis. Eur. J. Biochem. 1994;223:513–519. doi: 10.1111/j.1432-1033.1994.tb19020.x. [DOI] [PubMed] [Google Scholar]
- 96.Krauss S., Buttgereit F., Brand M. D. Effects of the mitogen concanavalin A on pathways of thymocyte energy metabolism. Biochim. Biophys. Acta. 1999;1412:129–138. doi: 10.1016/s0005-2728(99)00058-4. [DOI] [PubMed] [Google Scholar]
- 97.Vesce S., Kirk L., Nicholls D. G. Relationships between superoxide levels and delayed calcium deregulation in cultured cerebellar granule cells exposed continuously to glutamate. J. Neurochem. 2004;90:683–693. doi: 10.1111/j.1471-4159.2004.02516.x. [DOI] [PubMed] [Google Scholar]
- 98.Rego A. C., Vesce S., Nicholls D. G. The mechanism of mitochondrial membrane potential retention following release of cytochrome c in apoptotic GT1–7 neural cells. Cell Death Differ. 2001;8:995–1003. doi: 10.1038/sj.cdd.4400916. [DOI] [PubMed] [Google Scholar]
- 99.Akerboom T. P., Bookelman H., Zuurendonk P. F., Van Der Meer R., Tager J. M. Intramitochondrial and extramitochondrial concentrations of adenine nucleotides and inorganic phosphate in isolated hepatocytes from fasted rats. Eur. J. Biochem. 1978;84:413–420. doi: 10.1111/j.1432-1033.1978.tb12182.x. [DOI] [PubMed] [Google Scholar]
- 100.Schwenke W. D., Soboll S., Seitz H. J., Sies H. Mitochondrial and cytosolic ATP/ADP ratios in rat liver in vivo. Biochem. J. 1981;200:405–408. doi: 10.1042/bj2000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nilsson T., Schultz V., Berggren P. O., Corkey B. E., Tornheim K. Temporal patterns of changes in ATP/ADP ratio, glucose 6-phosphate and cytoplasmic free Ca2+ in glucose-stimulated pancreatic β-cells. Biochem. J. 1996;314:91–94. doi: 10.1042/bj3140091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chapman A. G., Miller A. L., Atkinson D. E. Role of the adenylate deaminase reaction in regulation of adenine nucleotide metabolism in Ehrlich ascites tumor cells. Cancer Res. 1976;36:1144–1150. [PubMed] [Google Scholar]
- 103.Ishiyama M., Tominaga H., Shiga M., Sasamoto K., Ohkura Y., Ueno K. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol. Pharm. Bull. 1996;19:1518–1520. doi: 10.1248/bpb.19.1518. [DOI] [PubMed] [Google Scholar]
- 104.Hackenbrock C. R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. II. Electron transport-linked ultrastructural transformations in mitochondria. J. Cell Biol. 1968;37:345–369. doi: 10.1083/jcb.37.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Welch K. C., Jr, Altshuler D. L., Suarez R. K. Oxygen consumption rates in hovering hummingbirds reflect substrate-dependent differences in P/O ratios: carbohydrate as a ‘premium fuel’. J. Exp. Biol. 2007;210:2146–2153. doi: 10.1242/jeb.005389. [DOI] [PubMed] [Google Scholar]
- 106.Conley K. E., Amara C. E., Jubrias S. A., Marcinek D. J. Mitochondrial function, fibre types and ageing: new insights from human muscle in vivo. Exp. Physiol. 2007;92:333–339. doi: 10.1113/expphysiol.2006.034330. [DOI] [PubMed] [Google Scholar]
- 107.Brand M. D., Chien L. F., Rolfe D. F. S. Control of oxidative phosphorylation in liver mitochondria and hepatocytes. Biochem. Soc. Trans. 1993;21:757–762. doi: 10.1042/bst0210757. [DOI] [PubMed] [Google Scholar]