

Abstract
Dimerization or oligomerization of many G protein-coupled receptors, including the CB1 receptor, is now widely accepted and may have significant implications towards medications development targeting these receptor complexes. A library of bivalent ligands composed of two identical CB1 antagonist pharmacophores derived from SR141716 linked by spacers of various lengths were developed. The affinities of these bivalent ligands at CB1 and CB2 receptors were determined using radiolabeled binding assays. Their functional activities were measured using GTP-γ-S accumulation and intracellular calcium mobilization assays. The results suggest that the nature of the linker and its length are crucial factors for optimum interactions of these ligands at CB1 receptor binding sites. Finally, selected bivalent ligands (5d and 7b) were able to attenuate the antinociceptive effects of the cannabinoid agonist CP55,940 in a rodent tail-flick assay. These novel compounds as probes will enable further evaluation of CB1 receptor dimerization and oligomerization, its functional significance, and may prove useful in the development of new therapeutic approaches to G protein-coupled receptor mediated disorders.
Introduction
The endocannabinoid system (ECS) is comprised of the CB1 and CB2 receptors, their endogenous ligands (endocannabinoids), and the proteins involved in endocannabinoid synthesis and inactivation, as well as the intracellular signaling pathways affected by endocannabinoids.1 Increasing evidence suggest that the endocannabinoid system is critically involved in a variety of physiological and pathological conditions. More importantly, modulation of the endocannabinoid system may hold therapeutic promise in a wide range of disparate diseases such as pain, inflammatory diseases, peripheral vascular disease, appetite enhancement or suppression, and locomotor disorders.2 Most of the actions exerted by exogenous cannabinoids or endocannabinoid in the brain are mediated by the CB1 receptor, which belongs to the G-protein-coupled receptors (GPCRs) super family, the largest class of cell surface receptors. GPCRs, including the class A family of which the CB1 receptor is a member, are attractive targets for medication development.
While GPCRs were traditionally considered monomeric, it is now well accepted that many GPCRs, including the CB1 receptor,3, 4 exist on the cell membrane as homo- and hetero-dimers or higher-order oligomers.5 Moreover, receptor oligomerization is often essential for receptor function (e.g., the GABAB receptor)6, and can also modulate ligand interaction, activation, signal transduction, and internalization.7-11 For example, it has been proposed that a μ-δ opioid receptor heterodimer is the fundamental signaling unit that mediates opioid tolerance and dependence through specific signal transducer(s) that recognize and couple to the heterodimer but not to μ-receptor monomers/homomers.12 In an analogous fashion, modulation of the CB1 receptor dimers or oligomers may offer novel opportunities to uniquely target and manipulate function of the endocannabinoid system.
The importance of GPCR dimerization and oligomerization in vivo remains to be elucidated and exploited, largely due to a lack of selective pharmacological tools and immunological reagents. Among various efforts to modulate the GPCR oligomers, bivalent ligands, which are defined as two pharmacophores linked by spacers, represent a unique and promising approach and may provide such a tool.13, 14 Bivalent ligands, provided they have suitable functional affinity at the monomeric receptor, are expected to selectively bind with greatly enhanced affinity to ligand recognition sites on heterodimers and oligomers due to the small containment volume for the second pharmacophore after the binding of the first one and the formation of thermodynamically more stable complexes. At the same time, bivalent ligands may display unique properties since they interact with more than one receptor simultaneously. Indeed, bivalent ligands have been developed for variety of G protein-coupled receptor targets, including opioids,13, 15 adrenergic,16, 17 dopamine,18 serotonin19, 20 and muscarinic receptors.21, 22 These bivalent ligands have been shown to be able to selectively target homo- or heterodimers and display unique pharmacological properties as compared to their monomeric subunits. However, to the best of our knowledge, there are no bivalent ligands developed for the CB1 receptor to date.
Here we present our efforts in the design and synthesis of symmetrical bivalent ligands targeting CB1 receptor dimers. The bivalent ligands confer two identical core structure of 1,5-diarylpyrazole derived from 1 (SR141716, or rimonabant, Figure 1) joined by a variety of linkers. Compound 1 was initially reported by Sanofi-Recherche as a highly potent and selective CB1 receptor antagonist/inverse agonist. It was the first drug to selectively block both the in vitro and in vivo effects of cannabinoids that are mediated by the CB1 receptor. Compound 1 was approved for the treatment of obesity in Europe before its recent withdrawal from the market due to undesirable psychological effects. This compound also shows great promise in many potential therapeutic applications including smoking addiction, drug and alcohol dependence, cognitive disorders, inflammation and arthritis.23, 24 By developing bivalent ligands with 1 as the pharmacophore, we aim to affect the binding affinities of these ligands to cannabinoid receptor monomers/dimers and perhaps alter their efficacies or signal transduction pathways as antagonists/inverse agonists. We hereby describe the synthesis and preliminary pharmacological examination of a series of bivalent ligands that possess linkers of various lengths, and describe the results with respect to the optimal linker length for affinity, and their related pharmacological activity in two signal transduction pathways. For comparative purposes, corresponding monovalent ligands were synthesized to evaluate the contribution of the presence of the linkers to activity. In doing so, these bivalent ligands enhance our understanding of the structure and function of CB1 receptor dimers.
Figure 1.

SR141716 (1) and 3- substituted analogs with alkyl or polar linkers
Results and Discussions
Bivalent ligand design
In order to focus our efforts on the efficient development of bivalent ligands, we selected a prototypical CB1 receptor inverse agonist, SR141716 (1). In addition to the high affinity and potency at the CB1 receptor in vitro and in vivo, the structure-activity relationships on this class of compounds have been well studied and documented. This allowed for an informed selection of appropriate positions to attach the linkers to the molecules without likely altering their activity significantly. It also permitted efficient synthesis following known procedures with minimal modifications. In particular, SAR results on this structure class indicate that the 3-carboxyamide position generally tolerates the replacement of the 1-aminopiperidyl group with a variety of substituents including alkyl groups and aromatic groups (2, Figure 1).25-27 Therefore, bivalent ligands linked through the 3-position were initially developed.
A series of bivalent ligands with linkers of varying lengths were synthesized and evaluated in efforts to optimize the linker for bridging of the receptors dimers. The optimal linker lengths, or the distances between the binding sites on neighboring receptors in receptor dimers or oligomers, have been reported on a number of GPCRs. Molecular modeling studies based on the crystal structure of rhodopsin suggested a distance between the individual receptors to be in the vicinity of ∼35Å, although the receptor dimer was in a head-to-tail orientation.28 Similarly, molecular modeling on the opioid receptor suggested that the distance between the recognition sites of either the interlocking or contact dimers with a TM5,6-interface is ∼27Å, while it's greater (∼32Å) in dimers with TM4,5-interface.13 However, during their studies on opioid bivalent ligands, Portoghese and co-workers discovered that optimal activity was obtained when spacers are in the range of 22Å (∼19 atoms).29 Neumeyer and coworkers found that bivalent ligands for the opioid receptors having spacers containing 10 methylene units or less displayed the highest affinities.30, 31 More recently, a series of adenosine A2A antagonist/dopamine D2 agonist bivalent ligands were developed where linkers ranged between 26 and 66 atoms.32 Interestingly, affinities of the bivalent ligands to both receptors stayed almost identical with the elongation of the linkers. The authors indicated that linkers with 26 atoms were of sufficient length to allow the bivalent ligands to bind to receptor dimers according to receptor docking experiments, and suggested that the lack of correlation between binding affinity and linker length might be due to the high flexibility of the mixed peptide/polyethylene glycol linkers. Based on these findings and others, linkers between 5 and 23 atoms were initially examined in our laboratory to determine optimal linker length.
Three types of linkers have been considered in the design of the bivalent ligands. The first class investigated were polyethylene glycol linkers. The second category is composed of small peptides (Figure 1). These two classes of linkers are readily available and have been employed in bivalent ligand development by a number of groups.13, 22, 33 These linkers are not only readily available but also offer the advantage of gradually increasing the linker length. However, our preliminary results with these two types of linkers failed to show promise. Both compounds 3 and 4 had low affinity in radiolabeled binding and were inactive in GTP-γ-S and calcium assays (data not shown). This is consistent with literature results that suggest hydrophobic groups are generally preferred at this 3-carboxamide position.25, 26, 34 Linkers composed of alkylamines were also examined (Figure 2). The selection of these hydrophobic molecules was based on the SAR studies in our laboratory and also by Wiley and co-workers that indicate substitution of the 3-carboxamide with hydrocarbons usually retains or sometimes even improves the affinity or antagonist activity of 1.25, 26, 34 Accordingly, a series of alkyl triamines were selected to construct the bivalent ligands (5a-f, Figure 2). A protonatable nitrogen atom was introduced in the middle of the chain in order to reduce the incremental increases in hydrophobicity upon elongation of the alkyl chains. This nitrogen not only provides symmetry of the bivalent ligands, it also facilitates the construction of long alkyl linkers. Additionally, the N-methyl series of analogs (6a-d) were prepared to examine the possible hydrogen bonding effects of the alkyl amine linker.
Figure 2.

Bivalent and monovalent ligands with triamine linkers
Chemistry
Compound 3 was obtained by coupling between the pyrazole carboxylic acid (9), which was readily prepared from commercially available 4-chloropropiophenone in three steps following the procedure developed in our laboratory,35, 36 and 1,11-diamino-3,6,9-trioxaundecane using BOP as the coupling agent (Scheme 1). In the preparation of 4, acid 9 was coupled to glycine methyl ester hydrochloride under standard coupling conditions that employed HOBt, EDCI and triethylamine in THF to give the methyl ester (10) in almost quantitative yield. Hydrolysis of 10 in methanolic sodium hydroxide at room temperature furnished 11 in quantitative yield. Coupling of 11 with glycine methyl ester hydrochloride under identical conditions as that of 9, followed by mild hydrolysis (LiOH, MeOH/THF/H2O), provided 12 in excellent yield. Finally, reaction of 12 with excess ethylenediamine furnished 4.
Scheme 1.

Synthesis of compounds 3 and 4.
Reagents and conditions: a). Benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP), 1,11-diamino-3,6,9-trioxaundecane, THF; b). Glycine methyl ester hydrochloride, HOBt, EDCI, Et3N, CH2Cl2;c). NaOH, MeOH/H2O; d). LiOH, THF/MeOH/H2O; e). Ethylenediamine, HOBt, EDCI, Et3N, CH2Cl2.
The route to bivalent ligands 5a-f and monovalent ligands 7a-f required the use of the N-H triamine linkers 18a-f. While 18a-b were commercially available, 18c-f were prepared in our laboratory as shown in Scheme 2. For 18c-d, the starting bromoalkyl nitriles (15c-d) were commercially available. Intermediates 15e (m = 8) and 15f (m = 10) in the preparation of 18e-f needed to be synthesized from dibromides 13 and 14, respectively. This was readily accomplished by displacing one of the bromides in these dibromoalkanes with a cyano group using sodium cyanide in DMSO. Thereafter, bis-alkylation of benzylamine with bromides 15c-f in the presence of potassium carbonate in 1-butanol or dimethylformamide (DMF) furnished amines 16c-f in excellent yields. Reduction of these nitriles was readily accomplished by hydrogenation catalyzed with Raney Nickel to give 17c-f. Another hydrogenation using palladium on carbon removed the benzyl groups to afford triamines 18c-f, which were generally of sufficient purity and were used in the following step without further purification. It is worth noting that the sequence of the hydrogenations was important and debenzylation followed by reduction of nitriles failed to give the desired products in satisfactory yields.
Scheme 2.

Synthesis of N-H triamine linkers 18c-f
Reagents and conditions: (a). NaCN, K2CO3, DMSO; (b). benzylamine, K2CO3, 1-butanol or DMF, 100° C; (c). H2, Raney Nickel, ethanol, 2N. NaOH; (d). H2, Pd/C, ethanol.
The coupling of acid 9 and triamines 18a-f was then attempted using several methods in order to furnish the bivalent ligands. Initial trials on the coupling employing the acid chloride or activation of acid 9 with agents such as chloroformates or BOP all failed to display selectivity and products with acylation at all three amino sites were obtained as the primary products. Eventually carbonyldiimidazole (CDI) appeared to provide satisfactory selectivity, and the desired products 5a-f, where acylation occurred at the two primary amino sites, were obtained in reasonable yields (Scheme 3). Under similar conditions, acylation at only one of the primary amino groups could be readily accomplished with the employment of excess triamines 18a-f to provide 7a-f.
Scheme 3.

Synthesis of bivalent ligands 5a-f and monovalent ligands 7a-f
Reagents and Conditions: CDI (0.5 equiv. for 5a-f, 3 equiv. for 7a-f), CH2Cl2.
Following a procedure analogous to that of scheme 2, the N-methyl triamine linkers were prepared as depicted in Scheme 4. Bis-alkylation of methylamine in methanol or methylamine hydrochloride with bromides 15c-f in ethanol under microwave conditions or heated in sealed pressure tubes provided 19a-d in almost quantitative yields. Hydrogenation catalyzed by palladium on carbon provided the triamines (20a-d) in excellent yields. Similar to Scheme 3, the N-methyl bivalent ligands 6a-d and the monovalent controls 8a-d were obtained by coupling reactions between acid 9 and amines 20a-d using CDI.
Scheme 4.

Synthesis of N-Me bivalent ligands 6a-d and monovalent ligands 8a-d
Reagents and Conditions: (a). Methylamine in methanol or methylamine hydrochloride, K2CO3, ethanol, microwave or heated in pressure tube; (b). H2, Pd/C, ethanol; (c). CDI (0.5 equiv. for 6a-d, 3 equiv. for 8a-d), CH2Cl2.
Pharmacology
Affinity of bivalent ligands
All the target compounds were evaluated in competition binding assays using both rat whole brain membrane preparations and cells stably transfected with either the human CB1 or CB2 receptors. The receptor binding affinities were determined in competitive displacement assays using radioligands [3H]-1 and [3H]-CP55,940. The results are summarized in Tables 1 and 2.
Table 1.
Binding affinities of N-H bivalent ligands 5a-f and 7a-f against [3H]-CP55,940 and [3H]-1
| Cmpd | n | Linker (atoms) | Displacement Assay vs. Tritiated Ligands: Ki (nM) in hCB1 (n=2) | Displacement vs. 3H-CP55,940: Ki (nM) in hCB2 | CB1/CB2 | |||
|---|---|---|---|---|---|---|---|---|
| 3H-CP55,940 | SEM | 3H-1 | SEM | |||||
| 1 | 6.18 | 1.2 | 1.18 | 0.1 | 313 | 50.6 | ||
| 5a | 2 | 5 | 229 | 75.0 | 94.0 | 8.00 | 1285 | 5.6 |
| 5b | 3 | 7 | 174 | 1.0 | 41.9 | 5.40 | 496 | 2.9 |
| 5c | 5 | 11 | 68.1 | 12.6 | 30.4 | 4.40 | 451 | 6.6 |
| 5d | 7 | 15 | 12.3 | 1.10 | 4.41 | 0.34 | 553 | 45.0 |
| 5e | 9 | 19 | 54.1 | 16.3 | 57.4 | 44.7 | a | >46 |
| 5f | 11 | 23 | 99.3 | 35.8 | 37.0 | 4.55 | a | >25 |
| 7a | 2 | 5 | 1225 | 359 | 506 | 56.5 | a | >2 |
| 7b | 3 | 7 | a | - | a | - | a | - |
| 7c | 5 | 11 | 349 | 36.5 | 230 | 4.50 | a | >7 |
| 7d | 7 | 15 | 46.7 | 1.85 | 19.5 | 1.35 | 622 | 13.3 |
| 7e | 9 | 19 | 14.0 | 2.10 | 5.44 | 0.62 | 419 | 29.9 |
| 7f | 11 | 23 | 4.56 | 0.83 | 2.30 | 0.20 | 305 | 66.9 |
Ki >highest standard of 2500 nM
Table 2.
Binding affinities of N-Me bivalent ligands 6a-d and monovalent ligands 8a-d against [3H-CP55,940] and [3H-1]
| Compd | n | Linker (atoms) | Displacement Assay vs. Tritiated Ligands: Ki (nM) in hCB1 (n=2) | Displacement vs. 3H-CP55,940: Ki (nM) in hCB2 | CB1/CB2 | |||
|---|---|---|---|---|---|---|---|---|
| 3H-CP55,940 | SEM | 3H-1 | SEM | |||||
| 6a | 5 | 11 | 38.7 | 4.0 | 6.35 | 1.07 | 1037 | 26.8 |
| 6b | 7 | 15 | 17.3 | 0.45 | 27.5 | 1.90 | 683 | 39.5 |
| 6c | 9 | 19 | 247 | 29.0 | 94.1 | 25.9 | a | >10 |
| 6d | 11 | 23 | 1885 | 450 | 1292 | 524 | a | >1.3 |
| 8a | 5 | 11 | 162 | 22.5 | 88.8 | 0.45 | a | >15 |
| 8b | 7 | 15 | 37.5 | 4.45 | 15.5 | 0.40 | 1934 | 51.6 |
| 8c | 9 | 19 | 10.0 | 1.27 | 6.12 | 1.06 | 265 | 26.5 |
| 8d | 11 | 23 | 14.7 | 5.27 | 5.51 | 1.81 | 426 | 29.0 |
Ki >highest standard of 2500 nM
Most of the bivalent ligands displayed nM affinity at the CB1 receptor, albeit somewhat lower than the parent compound 1. Similarly to 1, all bivalent ligands and monovalent controls also showed reasonable selectivity for the CB1 receptor over the CB2 receptor, displaying little or no affinity at the CB2 receptor. Noticeably, all compounds exhibited higher affinity (2-3 fold) for the CB1 receptor in the displacement of [3H]1 than the structurally different [3H]CP55,940. This is in agreement with observations previously reported by Wiley and coworkers where CP55,940 derivatives were usually better ligands in displacement of radiolabeled CP55,940 than 1.37
Interestingly, the binding affinity of the N-H bivalent ligand series at the CB1 receptor appeared to be sensitive to the length of the linkers (Table 1). Specifically, the affinity initially increased with increasing linker length, and then decreased as the linker was extended. The peak affinity was obtained with 5d (n = 7), where the linker is composed of 15 atoms, against both radioligands (12.3 nM vs. [3H]CP55,940 and 4.41 nM vs. [3H]1). Such an initial increase, followed by a subsequent decrease in affinity, is consistent with observations in the bivalent opioid ligands made by the Portoghese and Neumeyer groups.13, 30, 38, 39 Thus, it is hypothesized that linker length is critical for the ability of the bivalent ligands to bind two CB1 receptors simultaneously, as insufficient length would not permit bridging and spacers of excessive length would reduce bridging due to increased confinement volume. Such a transition suggests that bridging of vicinal receptors by bivalent ligands occurs most efficiently with optimal linker length. This hypothesis is also supported by the different pattern that was observed for the corresponding monovalent controls in this series (7a-f, Table 1), where the affinity increased as the spacer became longer, with the most potent compound determined to be 7f (n = 11, 23 atoms). The observation that monovalent ligands display comparable or even higher affinity than the corresponding bivalent ligands (5e vs. 7e and 5f vs. 7f) has also been previously reported in bivalent ligands for other GPCRs including opioids, 5-HT4 and GnRHR. 19, 30, 33, 40 One possibility is that the linker may represent an additional recognition site and only one pharmacophore is needed when the spacer is of sufficient length. Most significantly, the highest affinity bivalent ligand (5d), where the linker is composed of 15 atoms, displays higher affinity (∼4 fold for both radioligands) than the corresponding monovalent control (7d), indicating that the presence of the second pharmacophore increases the ability for the compounds to bind to the receptor, possibly by simultaneous occupying vicinal recognition sites of neighboring receptors in the receptor dimer. Again, this affinity enhancement of bivalent ligands over their corresponding monovalent ligands has been widely observed in previous studies on the opioid receptors, although it is often a modest (∼ 2 fold) difference.30 The optimal linker length of 15 atoms in the present study is consistent with the range reported for bivalent ligands developed for other GPCRs.29, 30, 41, 42
A similar trend in affinity was also observed in the bivalent ligands of the N-Me series (6a-d, Table 2) with respect to their ability to compete for [3H]CP55,940 and [3H]1 binding. Affinity initially increased with linker length, with 6a (n = 5) and 6b (n = 7) displaying the greatest ability in displacing either [3H]1 or [3H]CP55,940. Slightly different than the N-H monovalent ligand series, the binding affinity of 8a-d at the CB1 receptor initially increased and then remained relatively constant with the elongation of the linker, with 8c and 8d displaying almost identical Ki's. Interestingly, when the linkers are of the same length, the affinities of the bivalent ligands from both the N-H and N-Me series are relatively similar; indicating that the presence of the N-methyl group did not appear to interfere with the interaction of the bivalent ligands with the receptors.
Inverse agonist/antagonist activity of bivalent ligands
All compounds were examined in vitro using both [35S]GTP-γ-S accumulation and intracellular calcium mobilization assays to characterize their efficacy, inverse agonist activity, and apparent affinity (pA2). The results are shown in table 3 and 4.
Table 3.
Functional assessment of the alkyl N-H series of bivalent ligands 5a-f and monovalent ligands 6a-f at the CB1 receptor
| Cmpd | Linker | GTP-γ-S Assay in Rat Brain | GTP-γ-S Assay in hCB1 | GTP-γ-S Assay in hCB1 | Calcium Assay | ||||
|---|---|---|---|---|---|---|---|---|---|
| EC50 (nM) | Emax | EC50 (nM) | Emax | PA2 in hCB1 | +/- 95% confidence limits | Ke (nM) | SEM | ||
| 1 | 56305 | -37.8 | ND | - | 8.59 | 0.08 | 1.1 | 0.12 | |
| 5a | 5 | b | -35.9 | 237 | -25.5 | 7.08 | 0.52 | 2702 | 411 |
| 5b | 7 | 718 | -37.2 | 84.2 | -31.9 | 7.41 | 0.27 | 1304 | 279 |
| 5c | 11 | b | -29.6 | 33.0 | 8.5 | 7.53 | 0.28 | 476 | 69 |
| 5d | 15 | 1193 | -25.0 | 179 | -38.8 | 8.08 | 0.24 | 567 | 64 |
| 5e | 19 | 1.34 | 10.4 | 27.9 | 6.5 | 7.76 | 0.60 | 4165 | 1142 |
| 5f | 23 | b | -12.2 | a | - | 7.56 | 0.35 | d | - |
| 7a | 5 | 7243 | -22.9 | ND | - | c | c | d | - |
| 7b | 7 | b | -34.8 | ND | - | c | c | d | - |
| 7c | 11 | 2222 | -40.7 | ND | - | 6.12 | 0.32 | 5399 | 1105 |
| 7d | 15 | 3279 | -52.8 | ND | - | 7.69 | 0.46 | 502.6 | 225 |
| 7e | 19 | 2393 | -65.2 | ND | - | 8.25 | 0.28 | 31.6 | 3.1 |
| 7f | 23 | 3616 | -29.2 | ND | - | 8.50 | 0.26 | 146.5 | 19.1 |
EC50 >highest standard of 25,000 nM;
EC50 >highest standard of 10,000 nM;
Does not converge, unable to calculate value;
no shift observed at 10,000 nM;
ND: not done.
Table 4.
Functional assessment of the N-Me series of bivalent ligands 6a-d and monovalent ligands 8a-d at the CB1 receptor
| Cmpd | Linker | GTP-γ-S Assay in Rat Brain | GTP-γ-S Assay in hCB1 | GTP-γ-S Assay in hCB1 | Calcium Assay | ||||
|---|---|---|---|---|---|---|---|---|---|
| EC50 (nM) | Emax | EC50 (nM) | Emax | PA2 in hCB1 | +/- 95% confidence limits | Ke (nM) | SEM | ||
| 6a | 11 | 1.78 | -3.8 | 59.8 | -13.1 | 7.96 | 0.22 | 107.7 | 20.8 |
| 6b | 15 | 1460 | -40.8 | 2238 | -32.7 | 8.37 | 0.24 | 478.3 | 14.3 |
| 6c | 19 | b | - | 161 | -23.0 | 7.53 | 0.25 | c | - |
| 6d | 23 | 29.3 | 17.3 | a | - | 6.30 | 0.32 | c | - |
| 8a | 11 | 3037 | -53.3 | ND | - | 6.51 | 0.77 | 2873 | 418 |
| 8b | 15 | 4367 | -62.0 | ND | - | 7.66 | 0.55 | 219.8 | 2.8 |
| 8c | 19 | 3404 | -67.9 | ND | - | 8.34 | 0.25 | 140.9 | 29.1 |
| 8d | 23 | 8768 | -49.3 | ND | - | 7.63 | 0.65 | 205.5 | 2.9 |
EC50 >highest standard of 25,000 nM;
EC50 >highest standard of 10,000 nM;
no shift observed at 10,000 nM
In the [35S]GTP-γ-S assay using whole rat brain, most dimers and monomers appeared to act as weak inverse agonists. Similar to 1, most compounds required μM concentrations to show inverse agonist activity in hCB1 transfectants, and the change from basal activity was relatively modest (<25% decrease in basal binding under the conditions used). However, most compounds potently shifted the concentration-response curve of the agonist CP55,940, indicating that they were high affinity antagonists. Significantly, the pA2 values against CP55,940 stimulated [35S]-GTP-γ-S binding in hCB1 cells were correlated with their Ki values in both the N-H and N-Me series. Specifically, the pA2 values first increased and then decreased for the bivalent ligands and always increased for the monovalent ligands. The bivalent ligand with the highest apparent affinity in the N-H series was 5d whereas 6b showed the highest pA2 value in the N-Me series. The higher potencies of the bivalent ligand 5d and 6b than the monovalent ligands 7d and 8b, respectively, is consistent with the binding of these bivalent ligands to CB1 receptor dimers.
All the compounds were also tested using a calcium mobilization assay as a measure of CB1 receptor function and again showed nM potency. The same trend of an initial increase followed by a subsequent decrease in potency with increasing linker length was observed for bivalent ligands in both series (5a-f and 6a-d), with the exception of 6a, whereas the potency generally increased and stayed consistent for the monovalent ligands (7a-f and 8a-d). 5c and 5d showed the greatest potency in N-H bivalent ligands and 6a was the most potent N-Me bivalent ligand. However, no potency enhancement at the optimal linker (15 atoms) was observed in this assay between the bivalent and monovalent ligands (5d vs. 7d and 6b vs. 8b). While data from the three in vitro assays trended well, differences were noted between Ki, pA2 and Ke values. This is not surprising as different endpoints and biological systems were experimentally employed for ligand characterization.
Tail-flick Studies
The bivalent ligands with the highest affinity and potency, 5d and 6b, and their monovalent controls, 7d and 8b, were evaluated for their ability to block the antinociceptive effects of the cannabinoid agonist CP55,940 in a rodent tail-flick assay. In the experiment, the tail was exposed to 55°C warm water and the amount of time taken for the animal to move (flick) its tail away from the heat was recorded. Test compounds or vehicle were administered i.p. at 10 mg/kg i.p. to male mice 30 minutes prior to the administration of vehicle or 1.5 mg/kg CP55,940. Tail-flick times were measured 30 minutes after treatment with CP55,940. Antinociceptive response was calculated as percentage of maximum possible effect.
As shown in Figure 3, a single 10 mg/kg i.p. dose of the bivalent ligand 6b and its monomeric control 8b could significantly attenuate the antinociceptive response to CP55,940. However, the N-H analogs (5d and 7d) were considerably less active. It remains to be determined if the differences in potencies in vivo can be attributed to differences in biodistribution or metabolism of the various ligands. Nevertheless, these results suggest that these bivalent compounds, despite their high molecular weight, are able to attenuate nociceptive responses by central and or peripheral mechanisms by antagonizing the CB1 receptor complexes.
Figure 3.
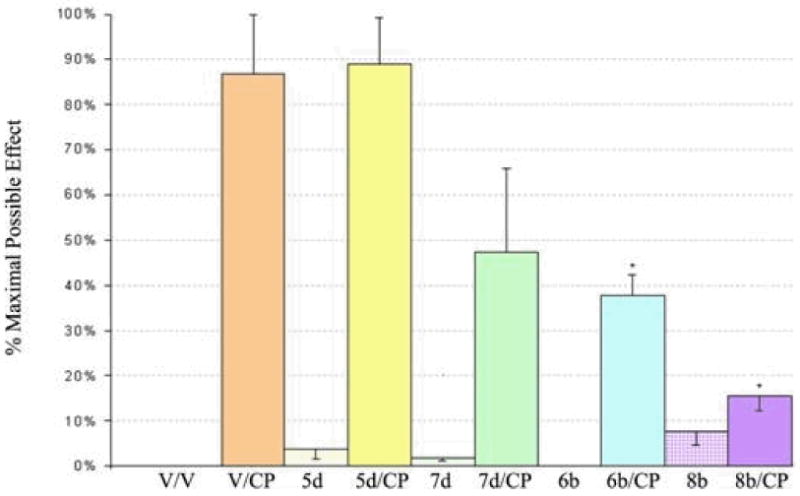
Blockade of the antinociceptive effect of CP55,940 by bivalent and monovalent ligands. Results are expressed as the percent maximal possible effect (% MPE, where % MPE = [(test _ control)/(maximum latency _control) · 100]) as defined by a 10 sec cut-off for the noxious stimulus. Significant differences (p<0.05) from vehicle-CP55,940 treated controls denoted with an asterisk (*).
Conclusions
The concept of homo- and heterodimerization has opened new potential avenues for the development of drugs targeted at GPCRs. One emerging approach is to employ bivalent ligands that specifically bind to these receptor dimers. Ideally, bivalent ligands with linkers of optimal length will bind to receptor dimers with greatly enhanced affinity due to the formation of thermodynamically stable complexes. Indeed, significant progress has been made in a number of GPCRs including opioids,13, 15 adrenergic,16, 17 dopamine,18 serotonin19, 20 and muscarinic receptors.21, 22 Most significantly, much success has been recently achieved by Portoghese and co-workers with bivalent opioid ligands in vivo.42, 43 In particular, μ-opioid (MOP) agonist/δ-opioid (DOP) antagonist bivalent ligands were shown to be potent analgesics after systemic administration, but did not produce the tolerance or dependence seen with traditional monovalent opioid analgesics.12 However, to the best of our knowledge, there are no bivalent ligands developed for the CB1 receptor to date. It is now well established that this receptor is a viable target to treat various indications including smoking addiction, drug and alcohol dependence, metabolic syndrome, cancer, fibrosis and inflammation. The consequences of altered cellular function as a result of dimerization and oligomerization of CB1 receptors are being explored. Availability of specific probes to understand the physiological importance of such interactions hold the key to better understand the role of cannabinoid signaling in the context of health and disease.
In the present study, we synthesized a library of symmetrical bivalent ligands containing two SR141716 moieties joined by aminoalkyl linkers. All the target compounds were evaluated in radiolabeled binding assays at the CB1 and CB2 receptors, functional [35S]GTP-γ-S accumulation assay and functional calcium mobilization assay. In all three assays, a clear trend could be detected where the bivalent ligands showed initially increased and then decreased affinity/activity with elongation of the linkers, whereas the monovalent ligands generally continued increasing or stayed consistent once the linker length was sufficiently long. The fact that the bivalent ligands showing higher affinity and activity than their respective monovalent controls (5d vs. 7d and 6b vs. 8b) when the spacer was comprised of 15 atoms in both the radiolabeled binding and [35S]GTP-γ-S assay, respectively, strongly suggests bridging of neighboring receptors. It is worth noting that, although only moderate affinity or potency enhancement was observed for the bivalent over the monovalent ligands in our binding or functional assays, additional evidence in support of this hypothesis has been reported using techniques such as FRET as demonstrated by Russo and coworker in 5-HT4 receptors.19 Finally, selected bivalent ligands (5d and 7b) and the corresponding monovalent controls (6d and 8b) were able to attenuate the antinociceptive effects of the cannabinoid agonist CP55,940 in the tail-flick assay. Taken together, further evaluation of this bivalent ligand approach of the CB1 receptor dimerization or oligomerization is clearly needed and may serve as the basis for the ultimate development of new medications.
Experimental
Chemistry
Reactions were conducted under N2 atmospheres using oven-dried glassware. All solvents and chemicals used were reagent grade. Anhydrous tetrahydrofuran, dichloromethane, and N,N-dimethylformamide (DMF) were purchased from Aldrich and used as such. Unless otherwise mentioned, all reagents and chemicals were purchased from commercial vendors and used as received. Flash column chromatography was carried out on a Teledyne ISCO CombiFlash Companion system using RediSep Rf prepacked columns. Purity and characterization of compounds were established by a combination of HPLC, TLC, gas chromatography mass spectrometry (GC-MS), and NMR analytical techniques described below. 1H and 13CNMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in CHCl3-d or MeOH-d4 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference unless otherwise noted. Chemical shifts are reported in ppm relative to the solvent signal, and coupling constant (J) values are reported in hertz (Hz). Thin-layer chromatography (TLC) was performed on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or I2 detection. Low-resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). High-resolution mass spectra were obtained in the Mass Spectrometry Laboratory, Department of Chemistry, University of Michigan. All test compounds were greater than 95% pure as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1x150 mm, 5 μm column with gradient elution using the mobile phases (A) H2O containing 0.1% CF3COOH and (B) MeCN. A flow rate of 0.5 mL/min was used for 5a-f and 7a-d and 1.0 mL/min for 6a-f and 8a-d.
15c-d and 18a-b were purchased from Aldrich and were used as such.
5-(4-chlorophenyl)-N-{13-[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]-13-oxo-3,6,9-trioxa-12-azatridec-1-yl}-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (3)
Benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP) (116mg, 0.262 mmol) was added to a solution of acid 9 (100 mg, 0.262 mmol) in 15 mL of THF. After 5 min, 1,11-diamino-3,6,9-trioxaundecane (30 mg, 0.157 mmol) was added. The reaction was stirred at room temperature for 1h. The solvent was removed and the resulting slurry was diluted with saturated aqueous NaHCO3 and extracted with ethyl acetate (2 × 30mL). The combined organic layers were washed with brine and dried. The residue was purified on silica using MeOH-CHCl3-NH4OH and EtOAc to give 3 (95mg, 78.9%) as a solid. 1H NMR (CDCl3) δ 2.38 (s, 6H), 3.58 (m, 16H), 7.05 (d, J = 9.0, 4H), 7.28 (m, 8H), 7.41 (s, 2H). MS: C25H29Cl3N4O4, [M+H]+ 555.2.
Methyl N-{[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]carbonyl}glycinate (10)
To a solution of acid 9 (2g, 5.24 mmol) in 60 mL of CH2Cl2 was added sequentially HOBt (0.78g, 5.76 mmol), EDCI (1.1g, 5.76 mmol) and glycine methyl ester hydrochloride (0.66g, 5.24 mmol). The reaction was stirred at room temperature for 15 min before Et3N was added. The reaction was stirred for 12h. The reaction was diluted with CH2Cl2 (100 mL) and washed with 1N HCl, NaHCO3 and then brine. The organic layer was dried with Na2SO4 and concentrate to give 10 as a white solid. 1H NMR (CDCl3) δ 2.36 (s, 3H), 3.78 (s, 3H), 4.22 (d, J = 5.7, 2H), 7.05 (d, J = 6.6, 2H), 7.30 (m, 4H), 7.40 (t, J = 3.0, 1H), 7.43 (s, 1H).
The product was of sufficient purity and was used in the next step without further purification.
N-{[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]carbonyl}glycine (11)
A solution of 10 in 30 mL of MeOH and 30 mL of 2N NaOH was stirred at room temperature for 16h. The solvent was concentrated in vacuo and the resulting solution was washed with ether. The aqueous solution was acidified with 6N HCl and then extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with water, brine and dried with Na2SO4. The solvent was removed in vacuo to give 11 (2.09g, 90.9% over both steps). 1H NMR (CDCl3) δ 2.35 (s, 3H), 4.26 (d, J = 6.0, 2H), 7.06 (d, J = 9.0, 2H), 7.31 (m, 4H), 7.42 (s, 1H), 7.58 (t, 3.0, 1), 10.78 (bs, 1H).
N-{[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]carbonyl}glycylglycine (12)
Following the procedure for the preparation of 10, 11 was coupled to glycine methyl ester hydrochloride to provide the methyl ester in 69.5% yield. 1H NMR (CDCl3) δ 2.36 (s, 3H), 3.75 (s, 3H), 4.07 (d, J = 6.0, 2H), 4.16 (d, J = 6.0, 2H), 6.77 (t, J = 3.0, 1H), 7.07 (d, J = 6.0, 2H), 7.29 (m, 4H), 7.43 (s, 1H), 7.53 (t, J = 3.0, 1H).
A solution of the above methyl ester (200mg, 0.40 mmol) and LiOH (25mg, 1.2 mmol) in 10 mL of THF-MeOH (3:1) and 2 mL of water was stirred at room temperature for 12h. The reaction was acidified with 3N HCl and extracted with EtOAc (2 × 25 mL). The combined organic layers were washed with water, brine and dried with Na2SO4. The solvent was evaporated to give 12 (175mg, 88.4%) as a white solid. 1H NMR (CDCl3) δ 2.31 (s, 3H), 3.78 (s, 2H), 4.09 (s, 2H), 7.19 (d, J = 8.4, 2H), 7.37 (d, J = 8.4, 2H), 7.46 – 7.56 (m, 3H).
N-{[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]carbonyl}glycyl-N-(2-aminoethyl)glycinamide (4)
To a solution of 12 (140mg, 0.28 mmol) in THF at room temperature was added BOP (125.0mg, 0.28 mmol) and ethylenediamine (9.0 μL, 1.41 mmol). The mixture was stirred for 15 min before Et3N was added. The reaction was stirred for 12h. The reaction was quenched with water and extracted with EtOAc (2 × 40 mL). The combined organic layers were washed with 1N HCl, saturated NaHCO3 and brine and then dried with Na2SO4. The solvent was removed and the residue was purified on silica gel using MeOH-CHCl3-NH4OH and EtOAc to give 4 (35mg, 34.5%) as a solid. 1H NMR (CDCl3) δ 2.31 (s, 3H), 2.87 (d, J = 6.0, 2H), 3.34 (d, J = 6.0, 2H), 3.96 (d, J = 6.0, 2H), 4.08 (d, J = 6.0, 2H), 7.06 (m, 3H), 7.20 – 7.40 (m, 5H), 7.42 (s, 1H), 7.80 (t, J = 3.0, 1H). MS: C23H23Cl3N6O3, [M+H]+ 537.4.
9-Bromononanenitrile (15e)
Sodium cyanide (3.6g, 73.5 mmol) was added in portions to a solution of 1,8-dibromooctane (13) (20g, 73.5 mmol) in 50 mL of DMSO at 60°C. After 30 min, the reaction was stopped and allowed to cool to room temperature. The reaction was diluted with 200 mL of diethyl ether and 200 mL of hexane and then washed with water (2 × 50 mL). The organic layer was separated, dried with sodium sulfate and concentrated. The resulting slurry was purified on silica using MeOH/CH2Cl2 (1:9) to give 15e (7.16g, 44.7%) as a colorless oil. 1H NMR (CDCl3) δ 1.35 (t, J = 7.5, 3H), 1.82 (s, 3H), 4.32 (q, J = 7.5, 2H), 7.24 (d, J = 9.0, 1H), 7.37 (d, J = 7.5, 1H), 7.48 (s, 1H).
11-Bromoundecanenitrile (15f)
15f (6.95g, 42.4%) was obtained from 1,10-dibromodecane (14) (20g, 66.6 mmol) as an oil. 1H NMR (CDCl3) δ 1.15 – 1.50 (m, 12H), 1.65 (m, 2H), 1.84 (m, 2H), 3.34 (t, J = 7.5, 2H), 3.41 (t, J = 6.0, 2H).
5,5′-(Benzylimino)dipentanenitrile (16c)
A mixture of benzylamine (0.5g, 4.67 mmol), potassium carbonate (1.94g, 14.0 mmol) and potassium iodide (0.27g, 1.63 mmol) was heated to 115°C. A solution of 5-bromopentanenitrile in 1-butanol was added drop wise. The resulting mixture was kept at 115°C for 20h. The reaction was allowed to cool to room temperature and then filtered. The solid was washed with diethyl ether (2 × 30 mL). The combined organic layers were extracted with 3N HCl (2 × 20 mL). The aqueous layer was washed with ether and basified with sodium carbonate. The resulting solution was then extracted with ether (3 × 40 mL). The combined organic layers were dried with Na2SO4 and concentrated to give 16c (1.02g, 81.1%) as an oil. 1H NMR (CDCl3) δ 1.50 – 1.70 (m, 8H), 2.26 (t, J = 6.6, 4H), 2.42 (t, J = 6.3, 4H), 3.51 (s, 2H), 7.15 – 7.32 (m, 5H).
The product was off sufficient purity and used in the next step without further purification.
7,7′-(Benzylimino)diheptanenitrile (16d)
16d (1.43g, 94.1%) was obtained from benzylamine (0.5g, 4.67 mmol) and 7-bromoheptanenitrile (1.86g, 9.80 mmol) as a colorless oil. 1H NMR (CDCl3) δ 1.20 – 1.65 (m, 16H), 2.29 (t, J = 6.9, 4H), 2.39 (t, J = 6.9, 4H), 3.52 (s, 2H), 7.29 (m, 5H).
9,9′-(Benzylimino)dinonanenitrile (16e)
16e (1.79g, 100%) was obtained from benzylamine (0.5g, 4.67 mmol) and 15e (2.04g, 9.33 mmol) as an oil. 1H NMR (CDCl3) δ 1.20 – 1.70 (m, 24H), 2.20 – 2.45 (m, 8H), 3.51 (s, 2H), 2.24 (m, 5H).
11,11′-(Benzylimino)diundecanenitrile (16f)
16f (2.0g, 84.8%) was obtained from benzylamine (0.7g, 6.53 mmol) and 15f (3.38g, 13.72 mmol). 1H NMR (CDCl3) δ 1.20 – 1.50 (m, 24H), 1.65 (m, 4H), 1.83 (m, 4H), 2.35 (t, J = 7.2, 4H), 2.90 (m, 4H), 4.16 (s, 2H), 7.43 (m, 3H), 7.64 (m, 2H).
N-(5-aminopentyl)pentane-1,5-diamine (18c)
A suspension of 16c (0.5g, 1.86 mmol) and Raney Nickel (0.5g) in ethanol (40 mL), THF (10 mL) and 2N sodium hydroxide (8 mL) was stirred under hydrogen (50 psi) for 20h. The suspension was filtered through Celite and concentrated. The resulting slurry was diluted with water (40 mL) and then extracted with CH2Cl2 (2 × 50 mL). The combined organic layers were washed with brine and dried over Na2SO4 to give 17c as an off-white oil. 17c was used in the next step without purification.
A suspension of 17c (0.35g, 1.25mmol), 10% palladium on carbon (40 mg) in ethanol (15 mL) and acetic acid (5 mL) was stirred under 50 psi hydrogen for 3h. The suspension was filtered through Celite and the filtrate was concentrated. To the resulting slurry was added 2N NaOH (20 mL) and extracted with CH2Cl2 (2 × 30 mL). The combined organic layers were washed with water and dried with Na2SO4. The solvent was evaporated in vacuo to give 18c (228 mg, 91.1% over both steps) as a clear oil. 1H NMR (CDCl3) δ 1.15 – 1.55 (m, 12H), 2.53 (t, J = 6.9, 4H), 2.62 (t, J = 6.6, 4H).
N-(7-aminoheptyl)heptane-1,7-diamine (18d)
Following the procedure for the synthesis of 18c, 18d (0.85g, 94.6%) was obtained from 16d (1.2g, 3.69 mmol). 1H NMR (CDCl3) δ 1.15 – 1.50 (m, 20H), 2.52 (t, J = 7.2, 4H), 2.61 (t, J = 6.9, 4H).
N-(9-aminononyl)nonane-1,9-diamine (18e)
Following the procedure for the synthesis of 18c, 18e (0.425g, 77.5%) was obtained from 16e (0.7g, 1.83 mmol). 1H NMR (CDCl3) δ 1.20 – 1.55 (m, 28H), 2.57 (t, J = 7.2, 4H), 2.66 (t, J= 6.9, 4H).
N-(11-aminoundecyl)undecane-1,11-diamine (18f)
Following the procedure for the synthesis of 18c, 18f (0.34g, 84.1%) was obtained from 16f (0.5g, 1.14 mmol). 1H NMR (CDCl3) δ 1.10 – 1.50 (m, 36H), 2.56 (t, J= 7.5, 4H), 2.62 (t, J = 7.2, 4H).
N,N′-(iminodiethane-2,1-diyl)bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (5a)
A solution of acid 9 (0.2g, 0.52 mmol), carbonyldiimidazole (85mg, 0.52 mmol) in 5 mL of CH2Cl2 was stirred at room temperature for 1h. TLC showed the complete consumption of the starting material. Triamine 18a (28 μL, 0.26 mmol) was added and the reaction was allowed to stir at room temperature for 3h. The reaction was diluted with CH2Cl2 and washed sequentially with NaHCO3, water and brine. The solution was dried with Na2SO4 and concentrated. The resulting slurry was purified on silica using CHCl3-MeOH-NH4OH (80:18:2) and EtOAc to give 5a (0.13g, 61.1%) as a white solid. 1H NMR (CDCl3) δ 2.35 (s, 6H), 2.90 (t, J = 6.0, 4H), 3.52 (dt, J1 = J2 = 6.0, 4H), 7.05 (d, J = 6.0, 4H), 7.22 – 7.30 (m, 10H), 7.40 (s, 2H). HRMS: C38H31Cl6N7O2, [M+H]+ calcd 828.0749, found 828.0768.
N,N′-(iminodipropane-3,1-diyl)bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (5b)
Following the procedure for the preparation of 5a, 5b was obtained from 18b in 49.5% yield. 1H NMR (CDCl3) δ 1.75 (m, 4H), 2.35 (s, 6H), 2.72 (t, J = 6.0, 4H), 3.45 (dt, J1 = 9.0, J2 = 6.0, 4H), 7.05 (d, J = 6.0, 4H), 7.28 (m, 8H), 7.40 (s, 2H), 7.54 (t, J = 3.0, 2H). HRMS: C40H35Cl6N7O2, [M+H]+ calcd 856.1062, found 856.1076.
N,N′-(iminodipentane-5,1-diyl)bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (5c)
Following the procedure for the preparation of 5a, 5c was obtained from 18c in 42.5% yield. 1H NMR (CDCl3) δ 1.37 – 1.64 (m, 12H), 2.37 (s, 6H), 2.59 (t, J = 6.0, 4H), 3.41 (dt, J1 = 9.0, J2 = 6.0, 4H), 6.96 (t, J = 3.0, 2H), 7.06 (d, J = 6.0, 4H), 7.26 (m, 8H), 7.43 (s, 2H). HRMS: C44H43Cl6N7O2, [M+H]+ calcd 912.1688, found 912.1679.
N,N′-(iminodiheptane-7,1-diyl)bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (5d)
Following the procedure for the preparation of 5a, 5d was obtained from 18d in 55.6% yield. 1H NMR (CDCl3) δ 1.34 – 1.70 (m, 20H) 2.37 (s, 6H), 2.56 (t, J = 6.0, 4H), 3.41 (dt, J1 = 6.9, J2 = 6.6, 4H), 6.95 (t, J = 3.0, 2H), 7.05 (d, J = 6.0, 4H), 7.29 (m, 8H), 7.43 (s, 2H). HRMS: C48H51Cl6N7O2, [M+H]+ calcd 968.2314, found 968.2321.
N,N′-(iminodinonane-9,1-diyl)bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (5e)
Following the procedure for the preparation of 5a, 5e was obtained from 18e in 42.8% yield. 1H NMR (CDCl3) δ 1.26 – 1.59 (m, 28H), 2.37 (s, 6H), 2.60 (t, J = 6.0, 4H), 3.40 (dt, J1 = 6.6, J2 = 6.0, 4H), 6.95 (t, J = 3.0, 2H), 7.28 (m, 8H), 7.43 (s, 2H). HRMS: C52H59Cl6N7O2, [M+H]+ calcd 1024.2940, found 1024.2972.
N,N′-(iminodiundecane-11,1-diyl)bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (5f)
Following the procedure for the preparation of 5a, 5f was obtained from 18f in 38.9% yield. 1H NMR (CDCl3) δ 1.25 – 1.61 (m, 36H), 2.37 (s, 6H), 2.61 (t, J = 7.5, 4H), 3.40 (dt, J1 = 6.9, J2 = 6.6, 4H), 6.96 (t, J = 6.0, 2H), 7.06 (d, J = 8.4, 4H), 7.29 (m, 8H), 7.42 (s, 2H). HRMS: C56H67Cl6N7O2, [M+H]+ calcd 1080.3566, found 1080.3590.
N-{2-[(2-aminoethyl)amino]ethyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (7a)
A solution of acid 9 (50 mg, 0.13 mmol) and carbonyldiimidazole (21mg, 0.13 mmol) in 5 mL of CH2Cl2 was stirred at room temperature for 30 min. TLC showed the complete consumption of starting acid. This suspension was then added to a solution of triamine 18a (41 mg, 0.39 mmol) drop wise. The resulting clear solution was allowed to stir at room temperature for 3h. The reaction was diluted with CH2Cl2 and washed sequentially with NaHCO3, water and brine. The solution was dried with Na2SO4 and concentrated. The resulting slurry was purified on silica using MeOH-CHCl3-NH4OH (80:18:2) and EtOAc to give 5a (47 mg, 89.0%) as a solid. 1H NMR (CDCl3) δ 2.36 (s, 3H), 2.72 (t, J = 5.7, 2H), 2.80 (t, J = 4.8, 2H), 2.87 (t, J = 6.0, 2H), 3.53 (dt, J1 = J2 = 6.0, 2H), 7.05 (d, J = 6.0, 2H), 7.28 (m, 5H), 7.42 (s, 1H). HRMS: C21H22Cl3N5O, [M+H]+ calcd 466.0968, found 466.0971.
N-{3-[(3-aminopropyl)amino]propyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (7b)
Following the procedure for the preparation of 7a, 7b was obtained from 18b in 67.9% yield. 1H NMR (CDCl3) δ 1.64 (m, 2H), 1.79 (m, 2H), 2.37 (s, 3H), 2.67 – 2.77 (m, 6H), 3.53 (dt, J1 = 5.7, J2 = 5.4, 2H), 7.05 (d, J = 8.4, 2H), 7.30 (m, 4H), 7.42 (s, 1H), 7.56 (m, 1H). HRMS: C23H26Cl3N5O, [M+H]+ calcd 494.1281, found 494.1284.
N-{5-[(5-aminopentyl)amino]pentyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (7c)
Following the procedure for the preparation of 7a, 7c was obtained from 18c in 44.9% yield. 1H NMR (CDCl3) δ 1.36 – 1.60 (m, 12H), 2.37 (s, 3H), 2.59 – 2.72 (m, 6H), 3.43 (dt, J1 = 9.3, J2 = 6.6, 2H), 6.95 (t, J = 3.0, 1H), 7.06 (d, J = 8.4, 2H), 7.29 (m, 4H), 7.43 (s, 1H). HRMS: C27H34Cl3N5O, [M+H]+ calcd 550.1907, found 550.1909.
N-{7-[(7-aminoheptyl)amino]heptyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (7d)
Following the procedure for the preparation of 7a, 7d was obtained from 18d in 35.2% yield. 1H NMR (CDCl3) δ 1.26 – 1.62 (m, 20H), 2.38 (s, 3H), 2.56 (m, 4H), 2.67 (t, J = 6.6, 2H), 3.41 (dt, J1 = 6.9, J2 = 6.6, 2H), 6.96 (t, J = 3.0, 1H), 7.06 (d, J = 6.9, 2H), 7.29 (m, 4H), 7.43 (s, 1H). HRMS: C31H42Cl3N5O, [M+H]+ calcd 606.2533, found 606.2536.
N-{9-[(9-aminononyl)amino]nonyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (7e)
Following the procedure for the preparation of 7a, 7e was obtained from 18e in 47.2% yield. 1H NMR (CDCl3) δ 1.18 – 1.62 (m, 28H), 2.38 (s, 3H), 2.58 (t, J = 7.2, 4H), 2.67 (t, J = 6.9, 2H), 3.40 (dt, J1 = 6.9, J2 = 6.6, 2H), 6.96 (t, J = 3.0, 1H), 7.28 (m, 4H), 7.43 (s, 1H). HRMS: C35H50Cl3N5O, [M+H]+ calcd 662.3159, found 662.3150.
N-{11-[(11-aminoundecyl)amino]undecyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (7f)
Following the procedure for the preparation of 7a, 7f was obtained from 18f in 46.7% yield. 1H NMR (CDCl3) δ 1.20 – 1.65 (m, 36H), 2.38 (s, 3H), 2.58 (t, J = 7.2, 4H), 2.67 (t, J = 6.9, 2H), 3.40 (dt, J1 = 6.9, J2 = 6.6, 2H), 6.95 (t, J = 3.0, 1H), 7.05 (d, J = 8.4, 7.28 (m, 4H), 7.43 (s, 1H). HRMS: C39H58Cl3N5O, [M+H]+ calcd 718.3785, found 718.3784.
5,5′-(Methylimino)dipentanenitrile (19a)
A pressure tube equipped with a mixture of methyl amine hydrochloride (1g, 16 mmol), 15c (3.7 mL, 32 mmol), potassium carbonate (4.4g, 32 mmol) and potassium iodide (0.53g, 3.2 mmol) in 20 mL of ethanol was heated to 110°C for 16 hours. The reaction was cooled to room temperature and the solvent was removed. The resulting slurry was partitioned between ethyl acetate and water. The organic layer was washed with brine and dried. The resulting residue was purified by chromatography on silica gel to give 19a (1.24g, 40.1%) as an off-white oil. 1H NMR (CDCl3) δ 1.55 – 1.85 (m, 8H), 2.19 (s, 3H), 2.38 (m, 8H).
7,7′-(Methylimino)diheptanenitrile (19b)
19b was synthesized from 15d in 48.8% yield following procedure for 19a. 1H NMR (CDCl3) 1.25 -1.70 (m, 16H), 2.18 (s, 3H), 2.33 (m, 8H).
9,9′-(Methylimino)dinonanenitrile (19c)
19c was synthesized from 15e in 55.0% yield following procedure for 19a. 1H NMR (CDCl3) 1.25 – 1.50 (m, 20H), 1.65 (m, 4H), 2.19 (s, 3H), 2.31 (m, 8H).
11,11′-(Methylimino)diundecanenitrile (19d)
19d was synthesized from 15f in 42.5% yield following procedure for 19a. 1H NMR (CDCl3) 1.20 – 1.50 (m, 24H), 1.66 (m, 4H), 1.85 (m, 4H), 2.36 (t, J = 3.9, 4H), 2.78 (s, 3H), 3.01 (t, J = 8.4, 4H).
N-(5-aminopentyl)-N-methylpentane-1,5-diamine (20a)
20a was synthesized from 19a in 59.0% yield following procedure for 17c. 1H NMR (CDCl3) δ 1.15 – 1.55 (m, 12H), 2.20 (s, 3H), 2.31 (t, J = 7.5, 4H), 2.69 (t, J = 6.9, 4H).
N-(7-aminoheptyl)-N-methylheptane-1,7-diamine (20b)
20b was synthesized from 19b in 73.8% yield following procedure for 17c. 1H NMR (CDCl3) δ 1.20 – 1.50 (m, 24H), 2.18 (s, 3H), 2.29 (t, J = 7.5, 4H), 2.67 (t, J = 6.9, 4H).
N-(9-aminononyl)-N-methylnonane-1,9-diamine (20c)
20c was synthesized from 19c in 99.0% yield following procedure for 17c. 1H NMR (CDCl3) δ 1.20 – 1.55 (m, 28H), 2.19 (s, 3H), 2.29 (t, J = 7.8, 4H), 2.67 (t, J = 6.9, 4H).
N-(11-aminoundecyl)-N-methylundecane-1,11-diamine (20d)
20d was synthesized from 19d in 79.0% yield following procedure for 17c. 1H NMR (CDCl3) δ 1.20 – 1.50 (m, 36H), 2.19 (s, 3H), 2.29 (t, J = 7.8, 4H), 2.67 (t, J = 6.9, 4H).
N,N′-[(methylimino)dipentane-5,1-diyl]bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (6a)
Following the procedure for the preparation of 5a, 6a was obtained from 20a in 50.2% yield. 1H NMR (CDCl3) δ 1.37 – 1.65 (m, 12H), 2.24 (s, 3H), 2.38 (m, 10H), 3.41 (dt, J1 = 6.6, J2 = 6.0, 4H), 7.00 (t, J = 3.0, 2H), 7.06 (d, J = 6.0, 4H), 7.28 (m, 8H), 4.23 (s, 2H). HRMS: C45H45Cl6N7O2, [M+H]+ calcd 926.1844, found 926.1866.
N,N′-[(methylimino)diheptane-7,1-diyl]bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (6b)
Following the procedure for the preparation of 5a, 6b was obtained from 20b in 40.3% yield. 1H NMR (CDCl3) δ 1.26 -1.60 (m, 20H), 2.18 (s, 3H), 2.28 (t, J = 6.0, 4H), 2.37 (s, 6H), 3.41 (dt, J1 = 6.6, J2 = 6.0, 4H), 6.95 (t, J = 3.0, 2H), 7.06 (d, J = 6.0, 4H), 7.28 (m, 8H), 7.42 (s, 2H). HRMS: C49H53Cl6N7O2, [M+H]+ calcd 982.2470, found 982.2482.
N,N′-[(methylimino)dinonane-9,1-diyl]bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (6c)
Following the procedure for the preparation of 5a, 6c was obtained from 20c in 53.5% yield. 1H NMR (CDCl3) δ 1.28 – 1.50 (m, 24H), 1.50 – 1.65 (m, 4H), 2.19 (s, 3H), 2.29 (t, J = 7.8, 4H), 2.38 (s, 6H), 3.40 (dt, J1 = 6.9, J2 = 6.6, 4H), 6.96 (t, J = 3.0, 2H), 7.06 (d, J = 8.4, 4H), 7.29 (m, 8H), 7.43 (s, 2H). HRMS: C53H61Cl6N7O2, [M+H]+ calcd 1038.3096, found 1038.3094.
N,N′-[(methylimino)diundecane-11,1-diyl]bis[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (6d)
Following the procedure for the preparation of 5a, 6d was obtained from 20d in 43.8% yield. 1H NMR (CDCl3) δ 1.20 – 1.65 (m, 36H), 2.29 (s, 3H), 2.41 (m, 10H), 3.42 (dt, J1 = 9.0, J2 = 6.0, 4H), 6.94 (t, J = 3.0, 2H), 7.05 (d, J = 9.0, 4H), 7.28 (m, 8H), 7.42 (s, 2H). HRMS: C57H69Cl6N7O2, [M+H]+ calcd 1094.3722, found 1094.3715.
N-{5-[(5-aminopentyl)(methyl)amino]pentyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (8a)
Following the procedure for the preparation of 7a, 8a was obtained from 20a in 60.8% yield. 1H NMR (CDCl3) δ 1.22 – 1.70 (m, 12H), 2.20 (s, 3H), 2.32 (t, J = 7.5, 4H), 2.37 (s, 3H), 2.69 (t, J = 6.9, 2H), 2.42 (dt, J1 = 6.9, J2 = 6.6, 2H), 6.99 (t, J = 3.0, 1H), 7.06 (d, J = 6.6, 2H), 7.29 (m, 4H), 7.43 (s, 1H). HRMS: C28H36Cl3N5O, [M+H]+ calcd 564.2064, found 564.2073.
N-{7-[(7-aminoheptyl)(methyl)amino]heptyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (8b)
Following the procedure for the preparation of 7a, 8b was obtained from 20b in 29.5% yield. 1H NMR (CDCl3) δ 1.20 – 1.65 (m, 20H), 2.19 (s, 3H), 2.29 (t, J = 7.8, 4H), 2.38 (s, 3H), 2.67 (t, J = 6.6, 2H), 2.41 (dt, J1 = 6.9, J2 = 6.6, 2H), 6.95 (t, J = 3.0, 1H), 7.06 (d, J = 6.9, 2H), 7.28 (m, 4H), 7.43 (s, 1H). HRMS: C32H44Cl3N5O, [M+H]+ calcd 620.2690, found 620.2687.
N-{9-[(9-aminononyl)(methyl)amino]nonyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (8c)
Following the procedure for the preparation of 7a, 8c was obtained from 20c in 34.9% yield. 1H NMR (CDCl3) δ 1.20 – 1.65 (m, 28H), 2.19 (s, 3H), 2.29 (t, J = 9.0, 4H), 2.38 (s, 3H), 2.67 (s, 3H), 3.41 (dt, J1 = 9.0, J2 = 6.0, 2H), 6.95 (t, J = 3.0, 1H), 7.05 (d, J = 9.0, 2H), 7.28 (m, 4H), 7.43 (s, 1H). HRMS: C36H52Cl3N5O, [M+H]+ calcd 676.3316, found 676.3312.
N-{11-[(11-aminoundecyl)(methyl)amino]undecyl}-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (8d)
Following the procedure for the preparation of 7a, 8d was obtained from 20b in 40.6% yield. 1H NMR (CDCl3) δ 1.20 – 1.60 (m, 36H), 2.20 (s, 3H), 2.29 (t, J = 7.8, 4H), 2.38 (s, 3H), 3.40 (dt, J1 = 6.9, J2 = 6.6, 2H), 6.95 (t, J = 3.0, 1H), 7.07 (d, J = 6.6, 2H), 7.29 (m, 4H), 7.43 (s, 1H). HRMS: C40H60Cl3N5O, [M+H]+ calcd 732.3942, found 732.3947.
Receptor Binding Assays
CB1 and CB2 Receptor Binding Assays
The CB1 receptor binding assay involved membranes isolated from a HEK-293 expression system whereas the CB2 receptor was expressed in CHO-K1 cells (Sigma-Aldrich Chemical Co., St. Louis, Mo). The methods used for performing binding assays in transfected cells expressing human CB1 or CB2 receptors were similar to those previously described for rat brain membrane preparations.25, 35 Binding was initiated with the addition of 40 μg of cell membrane proteins to assay tubes containing [3H]CP-55,940 (ca. 130 Ci/mmol) or [3H]-1 (ca. 22.4 Ci/mmol), a test compound (for displacement studies), and a sufficient quantity of buffer (50 mM Tris•HCl, 1 mM EDTA, 3 mM MgCl2, 5 mg/mL BSA, pH 7.4) to bring the total incubation volume to 0.5 mL. All assays were performed in polypropylene test tubes. In the displacement assays, the concentrations of [3H]CP-55,940 and [3H]-1 7.2 nM and 20 nM, respectively. Nonspecific binding was determined by the inclusion of 10 μM unlabeled CP-55,940, or 1. All cannabinoid analogs were prepared by suspension in buffer A from a 1 mg/mL ethanol stock. Following incubation at 30 °C for 1 h, binding was terminated by vacuum filtration through GF/C glass fiber filter plates (Packard, Meriden, CT, pretreated in buffer B for at least 1 h) in a 96-well sampling manifold (Millipore, Bedford, MA). Reaction vessels were washed twice with 4 mL of ice cold buffer (50 mM Tris•HCl, 1 mg/mL BSA). The filter plates were air-dried and sealed on the bottom. Liquid scintillate was added to the wells and the top sealed. After incubating the plates in cocktail for at least 2 h, the radioactivity present was determined by liquid scintillation spectrometry. Assays were done in duplicate, and results represent combined data from three to six independent experiments. Saturation and displacement data were analyzed by unweighted nonlinear regression of receptor binding data. For displacement studies, curve-fitting and IC50 calculation were done with GraphPad Prism (GraphPad Software, Inc., San Diego, CA), which fits the data to one and two-site models and compares the two fits statistically.
GTP-γ-[35S] assay
GTP-γ-[35S] assays were performed to determine the ability of target compounds to shift the binding curves of the agonist CP55,940 or 1. Reaction mixtures consisted of either CP55,940 (2.5 pM to 25 μM) or 1 (10 pM to 100 μM), 20 μM GDP, and 100 pM GTP-γ-[35S] in 50 mM Tris•HCl, pH 7.4, 1 mM EDTA, 5 mM MgCl2, 100 mM NaCl, and 1 mg/mL BSA. The effects of these compounds on agonist binding were compared at concentrations of 1, 10, and 100 nM vs. reactions with no antagonist in a final reaction mixture volume of 0.5 mL. Binding was determined using membrane preparations as previously described. Data analysis was performed using global nonlinear regression analysis of the dose–response curves (Prism, GraphPad), and pA2 values were calculated. The calculations were performed with the slope of the Schild line constrained to 1, as well as unconstrained, and an F-test (P < 0.05) was used to determine the best model.
Calcium mobilization assay
Calcium mobilization was performed in CHO cells co-expressing Gα16 protein and the human CB1 receptor cDNAs. Activation of CB1 receptor leads to coupling of this receptor to the promiscuous Gα16 protein and consequent mobilization of intracellular calcium. In the assay, the apparent agonist dissociation equilibrium constants (Ke) value of each compound was determined by running a 6-point half-log CP55,940 concentration response curve in the presence and absence of a single concentration of antagonist.44 The concentration of antagonist was chosen such that it caused at least a 2-fold increase (shift to the right) in the CP55,940 curve but did not exceed 10 μM to retain pharmacological relevance. A three-parameter logistic equation was fit to the concentration response data with Prism (GraphPad Software; San Diego, CA) to calculate Ke. These values were reported as the mean ± SEM from at least three independent experiments. 1 was employed as the positive control (antagonist) for inhibition of CB1 activity.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health/National Institute on Drug Abuse (NIDA) Grants DA019217 (B.F.T.), DA026582 (Y.Z.) and NIAAA grant AA017514 (R.M.).
Footnotes
Supporting Information Available: HPLC data of target compounds (5a-f, 6a-d, 7a-f and 8a-d). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.De Petrocellis L, Cascio MG, Di Marzo V. The endocannabinoid system: a general view and latest additions. Br J Pharmacol. 2004;141(5):765–74. doi: 10.1038/sj.bjp.0705666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58(3):389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackie K. Cannabinoid receptor homo- and heterodimerization. Life Sci. 2005;77(14):1667–73. doi: 10.1016/j.lfs.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 4.Wager-Miller J, Westenbroek R, Mackie K. Dimerization of G protein-coupled receptors: CB1 cannabinoid receptors as an example. Chem Phys Lipids. 2002;121(1-2):83–9. doi: 10.1016/s0009-3084(02)00151-2. [DOI] [PubMed] [Google Scholar]
- 5.George SR, O'Dowd BF, Lee SP. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov. 2002;1(10):808–20. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- 6.Lee SP, O'Dowd BF, Ng GY, Varghese G, Akil H, Mansour A, Nguyen T, George SR. Inhibition of cell surface expression by mutant receptors demonstrates that D2 dopamine receptors exist as oligomers in the cell. Mol Pharmacol. 2000;58(1):120–8. doi: 10.1124/mol.58.1.120. [DOI] [PubMed] [Google Scholar]
- 7.Franco R, Casado V, Cortes A, Mallol J, Ciruela F, Ferre S, Lluis C, Canela EI. G-protein-coupled receptor heteromers: function and ligand pharmacology. Br J Pharmacol. 2008;153 1:S90–8. doi: 10.1038/sj.bjp.0707571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rios CD, Jordan BA, Gomes I, Devi LA. G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol Ther. 2001;92(2-3):71–87. doi: 10.1016/s0163-7258(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 9.Minneman KP. Heterodimerization and surface localization of G protein coupled receptors. Biochem Pharmacol. 2007;73(8):1043–50. doi: 10.1016/j.bcp.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milligan G. G protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol. 2004;66(1):1–7. doi: 10.1124/mol.104.000497.. [DOI] [PubMed] [Google Scholar]
- 11.Milligan G. G-protein-coupled receptor heterodimers: pharmacology, function and relevance to drug discovery. Drug Discov Today. 2006;11(11-12):541–9. doi: 10.1016/j.drudis.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 12.Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A. 2005;102(52):19208–13. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Portoghese PS. From models to molecules: opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. J Med Chem. 2001;44(14):2259–69. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- 14.Messer WS., Jr Bivalent ligands for G protein-coupled receptors. Curr Pharm Des. 2004;10(17):2015–20. doi: 10.2174/1381612043384213. [DOI] [PubMed] [Google Scholar]
- 15.Peng X, Neumeyer JL. Kappa receptor bivalent ligands. Curr Top Med Chem. 2007;7(4):363–73. doi: 10.2174/156802607779941251. [DOI] [PubMed] [Google Scholar]
- 16.Zheng W, Lei L, Lalchandani S, Sun G, Feller DR, Miller DD. Yohimbine dimers exhibiting binding selectivities for human alpha2a- versus alpha2b-adrenergic receptors. Bioorg Med Chem Lett. 2000;10(7):627–30. doi: 10.1016/s0960-894x(00)00068-8. [DOI] [PubMed] [Google Scholar]
- 17.Lalchandani SG, Lei L, Zheng W, Suni MM, Moore BM, Liggett SB, Miller DD, Feller DR. Yohimbine dimers exhibiting selectivity for the human alpha 2C-adrenoceptor subtype. J Pharmacol Exp Ther. 2002;303(3):979–84. doi: 10.1124/jpet.102.039057. [DOI] [PubMed] [Google Scholar]
- 18.Tamiz AP, Bandyopadhyay BC, Zhang J, Flippen-Anderson JL, Zhang M, Wang CZ, Johnson KM, Tella S, Kozikowski AP. Pharmacological and behavioral analysis of the effects of some bivalent ligand-based monoamine reuptake inhibitors. J Med Chem. 2001;44(10):1615–22. doi: 10.1021/jm000552s. [DOI] [PubMed] [Google Scholar]
- 19.Russo O, Berthouze M, Giner M, Soulier JL, Rivail L, Sicsic S, Lezoualc'h F, Jockers R, Berque-Bestel I. Synthesis of specific bivalent probes that functionally interact with 5-HT(4) receptor dimers. J Med Chem. 2007;50(18):4482–92. doi: 10.1021/jm070552t. [DOI] [PubMed] [Google Scholar]
- 20.Meltzer PC, Kryatova O, Pham-Huu DP, Donovan P, Janowsky A. The synthesis of bivalent 2beta-carbomethoxy-3beta-(3,4-dichlorophenyl)-8-heterobicyclo[3.2.1]octane s as probes for proximal binding sites on the dopamine and serotonin transporters. Bioorg Med Chem. 2008;16(4):1832–41. doi: 10.1016/j.bmc.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christopoulos A, Grant MK, Ayoubzadeh N, Kim ON, Sauerberg P, Jeppesen L, El-Fakahany EE. Synthesis and pharmacological evaluation of dimeric muscarinic acetylcholine receptor agonists. J Pharmacol Exp Ther. 2001;298(3):1260–8. [PubMed] [Google Scholar]
- 22.Rajeswaran WG, Cao Y, Huang XP, Wroblewski ME, Colclough T, Lee S, Liu F, Nagy PI, Ellis J, Levine BA, Nocka KH, Messer WS., Jr Design, synthesis, and biological characterization of bivalent 1-methyl-1,2,5,6-tetrahydropyridyl-1,2,5-thiadiazole derivatives as selective muscarinic agonists. J Med Chem. 2001;44(26):4563–76. doi: 10.1021/jm0102405. [DOI] [PubMed] [Google Scholar]
- 23.Beardsley PM, Thomas BF. Current evidence supporting a role of cannabinoid CB1 receptor (CB1R) antagonists as potential pharmacotherapies for drug abuse disorders. Behav Pharmacol. 2005;16(5-6):275–96. doi: 10.1097/00008877-200509000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Bifulco M, Grimaldi C, Gazzerro P, Pisanti S, Santoro A. Rimonabant: just an antiobesity drug? Current evidence on its pleiotropic effects. Mol Pharmacol. 2007;71(6):1445–56. doi: 10.1124/mol.106.033118. [DOI] [PubMed] [Google Scholar]
- 25.Francisco ME, Seltzman HH, Gilliam AF, Mitchell RA, Rider SL, Pertwee RG, Stevenson LA, Thomas BF. Synthesis and structure-activity relationships of amide and hydrazide analogues of the cannabinoid CB(1) receptor antagonist N-(piperidinyl)- 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxami de (SR141716) J Med Chem. 2002;45(13):2708–19. doi: 10.1021/jm010498v. [DOI] [PubMed] [Google Scholar]
- 26.Thomas BF, Francisco ME, Seltzman HH, Thomas JB, Fix SE, Schulz AK, Gilliam AF, Pertwee RG, Stevenson LA. Synthesis of long-chain amide analogs of the cannabinoid CB1 receptor antagonist N-(piperidinyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyra zole-3-carboxamide (SR141716) with unique binding selectivities and pharmacological activities. Bioorg Med Chem. 2005;13(18):5463–74. doi: 10.1016/j.bmc.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Dyck B, Goodfellow VS, Phillips T, Grey J, Haddach M, Rowbottom M, Naeve GS, Brown B, Saunders J. Potent imidazole and triazole CB1 receptor antagonists related to SR141716. Bioorg Med Chem Lett. 2004;14(5):1151–4. doi: 10.1016/j.bmcl.2003.12.068. [DOI] [PubMed] [Google Scholar]
- 28.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 29.Portoghese PS, Larson DL, Sayre LM, Yim CB, Ronsisvalle G, Tam SW, Takemori AE. Opioid agonist and antagonist bivalent ligands. The relationship between spacer length and selectivity at multiple opioid receptors. J Med Chem. 1986;29(10):1855–61. doi: 10.1021/jm00160a010. [DOI] [PubMed] [Google Scholar]
- 30.Neumeyer JL, Zhang A, Xiong W, Gu XH, Hilbert JE, Knapp BI, Negus SS, Mello NK, Bidlack JM. Design and synthesis of novel dimeric morphinan ligands for kappa and micro opioid receptors. J Med Chem. 2003;46(24):5162–70. doi: 10.1021/jm030139v. [DOI] [PubMed] [Google Scholar]
- 31.Peng X, Knapp BI, Bidlack JM, Neumeyer JL. Synthesis and preliminary in vitro investigation of bivalent ligands containing homo- and heterodimeric pharmacophores at mu, delta, and kappa opioid receptors. J Med Chem. 2006;49(1):256–62. doi: 10.1021/jm050577x. [DOI] [PubMed] [Google Scholar]
- 32.Soriano A, Ventura R, Molero A, Hoen R, Casado V, Cortes A, Fanelli F, Albericio F, Lluis C, Franco R, Royo M. Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. J Med Chem. 2009;52(18):5590–602. doi: 10.1021/jm900298c. [DOI] [PubMed] [Google Scholar]
- 33.Bonger KM, van den Berg RJ, Heitman LH, IJ AP, Oosterom J, Timmers CM, Overkleeft HS, van der Marel GA. Synthesis and evaluation of homo-bivalent GnRHR ligands. Bioorg Med Chem. 2007;15(14):4841–56. doi: 10.1016/j.bmc.2007.04.065. [DOI] [PubMed] [Google Scholar]
- 34.Wiley JL, Jefferson RG, Grier MC, Mahadevan A, Razdan RK, Martin BR. Novel pyrazole cannabinoids: insights into CB(1) receptor recognition and activation. J Pharmacol Exp Ther. 2001;296(3):1013–22. [PubMed] [Google Scholar]
- 35.Zhang Y, Burgess JP, Brackeen M, Gilliam A, Mascarella SW, Page K, Seltzman HH, Thomas BF. Conformationally constrained analogues of N-(piperidinyl)-5-(4-chlorophenyl)-1-(2,4- dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716): design, synthesis, computational analysis, and biological evaluations. J Med Chem. 2008;51(12):3526–39. doi: 10.1021/jm8000778. [DOI] [PubMed] [Google Scholar]
- 36.Seltzman HH, Carroll FI, Burgess JP, Wyrick CD, Burch DF. Tritiation of SR141716 by metallation-iodination-reduction: tritium-proton nOe study. Journal of Labelled Compounds & Radiopharmaceuticals. 2002;45(1):59–70. [Google Scholar]
- 37.Wiley JL, Wang P, Mahadevan A, Razdan RK, Martin BR. Pyrazole Cannabinoids With Non-CB1, Non-CB2 Activity. International Cannabinoid Research Society 19th Symposium; St Charles, Illinois, USA. 2009. [Google Scholar]
- 38.Portoghese PS, Edward E. Smissman-Bristol-Myers Squibb Award Address. The role of concepts in structure-activity relationship studies of opioid ligands. J Med Chem. 1992;35(11):1927–37. doi: 10.1021/jm00089a001. [DOI] [PubMed] [Google Scholar]
- 39.Neumeyer JL, Peng X, Knapp BI, Bidlack JM, Lazarus LH, Salvadori S, Trapella C, Balboni G. New opioid designed multiple ligand from Dmt-Tic and morphinan pharmacophores. J Med Chem. 2006;49(18):5640–3. doi: 10.1021/jm0605785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonger KM, van den Berg RJ, Knijnenburg AD, Heitman LH, Ijzerman AP, Oosterom J, Timmers CM, Overkleeft HS, van der Marel GA. Synthesis and evaluation of homodimeric GnRHR antagonists having a rigid bis-propargylated benzene core. Bioorg Med Chem. 2008;16(7):3744–58. doi: 10.1016/j.bmc.2008.01.054. [DOI] [PubMed] [Google Scholar]
- 41.Bhushan RG, Sharma SK, Xie Z, Daniels DJ, Portoghese PS. A bivalent ligand (KDN-21) reveals spinal delta and kappa opioid receptors are organized as heterodimers that give rise to delta(1) and kappa(2) phenotypesSelective targeting of delta-kappa heterodimers. J Med Chem. 2004;47(12):2969–72. doi: 10.1021/jm0342358. [DOI] [PubMed] [Google Scholar]
- 42.Daniels DJ, Kulkarni A, Xie Z, Bhushan RG, Portoghese PS. A bivalent ligand (KDAN-18) containing delta-antagonist and kappa-agonist pharmacophores bridges delta2 and kappa1 opioid receptor phenotypes. J Med Chem. 2005;48(6):1713–6. doi: 10.1021/jm034234f. [DOI] [PubMed] [Google Scholar]
- 43.Xie Z, Bhushan RG, Daniels DJ, Portoghese PS. Interaction of bivalent ligand KDN21 with heterodimeric delta-kappa opioid receptors in human embryonic kidney 293 cells. Mol Pharmacol. 2005;68(4):1079–86. doi: 10.1124/mol.105.012070. [DOI] [PubMed] [Google Scholar]
- 44.Kosterlitz HW, Lees GM, Wallis DI, Watt AJ. Non-specific inhibitory effects of morphine-like drugs on transmission in the superior cervical ganglion and guinea-pig isolated ileum. Br J Pharmacol. 1968;34(3):691P–692P. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.