

Abstract
Using an accelerated and consistent simian immunodeficiency virus (SIV) pigtailed macaque model of HIV associated neurological disorders, we have demonstrated that virus enters the brain during acute infection. However, neurological symptoms do not manifest until late stages of infection, suggesting that immunological mechanisms exist within the central nervous system (CNS) that control viral replication and associated inflammation. We have shown that interferon beta, a type I interferon central to viral innate immunity, is a major cytokine present in the brain during acute infection and is responsible for limiting virus infection and inflammatory cytokine expression. However, the induction and role of interferon alpha in the CNS during acute SIV infection has never been examined in this model. In the classical model of interferon signaling, interferon beta signals through the interferon α/β receptor, leading to expression of interferon alpha. Surprisingly, although interferon beta is up regulated during acute SIV infection, we found that interferon alpha is down regulated. We demonstrate that this down regulation is coupled with a suppression of signaling molecules downstream of the interferon receptor, namely tyk2, STAT1 and IRF7, as indicated by either lack of protein phosphorylation, lack of nuclear accumulation, or transcriptional and/or translational repression. In contrast to brain, interferon alpha is up regulated in lung and accompanied by activation of tyk2 and STAT1. These data provide a novel observation that during acute SIV infection in the brain there is differential signaling through the interferon α/β receptor that fails to activate expression of interferon alpha in the brain.
Introduction
HIV associated neurological diseases (HAND) are a mounting problem in HIV treatment despite the introduction of highly active antiretroviral therapy (HAART). HAART has greatly decreased the prevalence of people with HAND, however, the incidence has increased as more HIV infected individuals are living longer (1–3). HIV is thought to enter the central nervous system through a ‘trojan horse’ mechanism, where infected monocytes in the peripheral blood traffic to the brain and mature into macrophages where they produce virus and infect neighboring macrophages, resident microglia and astrocytes (4). Because of the blood brain barrier, antiretrovirals that are administered to HIV infected individuals have variable CNS penetration and do not consistently control replication (5–7). Thus the brain represents a significant viral reservoir that can be reactivated during infection and lead to neurological damage. Therefore, it is critical to understand how the innate immune response during acute infection initially controls virus replication and inflammation and how this control fails, leading to increased virus expression, inflammation and neurological disease. If therapeutics could be identified that maintain immunological control mechanisms that limit inflammation in brain, they would be candidates for adjunctive therapy with HAART.
We have developed and characterized an accelerated, consistent simian immunodeficiency virus (SIV) model of HIV AIDS and neurological disease (8). Like HIV infection in humans, SIV infection is characterized by stages of disease. In our accelerated SIV model acute infection occurs between 4 and 21 days post inoculation (p.i.). Viral load in plasma peaks at 7 days p.i. paralleled by a decrease in CD4+ T cell counts. CD4+ T cell counts rebound and the asymptomatic phase occurs from 21–42 days p.i. (9–11). SIV-infected macaques then start to develop signs of disease and develop AIDS by 84 days p.i. (9, 10). In addition, 90% of animals develop SIV associated neurological disease, as indicated by neuropathological lesions and inflammation. Analyses of the brain from these SIV-infected macaques at various stages of infection have made it possible to examine both the viral and host factors throughout the course of disease.
Using this model, we have shown that although virus enters the brain and actively replicates in macrophages as early as 4 days p.i., inflammatory changes that accompany acute infection are transient and clinical signs of neurological disease do not manifest themselves until late stages of infection (9, 10). SIV replication in the brain is down regulated during the acute infection but this does not occur in the peripheral blood (11). This suggests that virus replication is regulated differently in brain compared to the periphery.
Two of the major cytokines involved in the innate immune response to viral infections are the type I interferons, interferon alpha and beta (IFNA and IFNB). While there is only a single gene encoding the interferon beta protein in both humans and macaques, there are 13 different interferon alpha genes encoding 12 different proteins located on chromosome 9 (in humans) and 15 (in macaques) (12). In the classical interferon signaling pathway, pattern recognition receptors such as toll like receptors, nod like receptors, and cytosolic receptors such as RIG-I and MDA5, are stimulated, and these receptors trigger activation of numerous kinases such as TANK binding kinase I (TBK1), and the inhibitor of NFkB kinases (IKKi). These kinases are thought to phosphorylate the transcription factor interferon regulatory factors 3 (IRF3), which leads to its dimerization, nuclear translocation and the transcription of interferon beta (13, 14). Interferon beta is then secreted and engages one of the two subunits of the heterdimeric interferon receptor (IFNAR1 and IFNAR2) in an autocrine and paracrine manner, which leads to receptor dimerization. IFNAR1 is associated with the Janus kinase tyrosine kinase 2 (tyk2), while IFNAR2 is associated with the janus kinase 1 (JAK1). JAK1 and tyk2 phosphorylate themselves, each other and the IFN receptor subunits. Phosphorylation of the receptor creates a docking site for signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2) to bind through SH2 domains. Phosphorylated STAT1, STAT2 and interferon regulatory factor 9 (IRF9) form the ISGF3 complex and translocate into the nucleus, where it up regulates transcription of interferon regulatory factor 7 (IRF7). IRF7 then undergoes phosphorylation, dimerization, and translocation into the nucleus where it stimulates interferon alpha transcription. Interferon alpha is then secreted and continues to signal in an autocrine and paracrine manner through the IFN receptor, thus amplifying interferon production and in turn the antiviral response through a positive feedback loop (15, 16).
We have demonstrated in our SIV macaque model that interferon beta is critical in controlling virus replication and establishing virus latency during acute infection in the brain (17, 18). Interferon beta production in the brain follows the same pattern as virus replication in the brain, with peak mRNA levels at 7 days pi (18). IFN beta in turn leads to the translation of a truncated dominant negative isoform of CEBPbeta, which binds to the SIV LTR through its DNA binding domain and represses transcription (18, 19). It is thought that this mechanism is responsible for the down regulation of both viral and cytokine gene expression (9). Although the production of interferon beta usually leads to the production of interferon alpha, it has been reported that HIV infected monocytes have a reduced ability to produce interferon alpha while interferon beta levels remain the same (20). This implies that differential interferon signaling patterns may exist, which may not be surprising since interferon alpha and beta have been shown to have different activities, especially in the CNS (21). While interferon beta has been reported to be protective against autoimmune encephalitis in mice, studies have shown interferon alpha to be associated with CNS disease (22, 23). Therefore, we examined the regulation of interferon alpha in the brain in our SIV model.
Surprisingly, we found that in the brain, interferon alpha mRNA is down regulated during acute infection when interferon beta mRNA is up regulated. Furthermore, we determined that the signaling molecules tyk2, STAT1 and IRF7 are all suppressed transcriptionally, translationally and/or post translationally. This is consistent with our previous studies that demonstrated that IL-12 mRNA is downregulated during acute infection in the brain, since tyk2 is a component of the IL-12 receptor as well (9). In contrast to the brain, examination of lung, a peripheral tissue that is infected during SIV and HIV infection (24–26), demonstrated the expected type 1 interferon signaling where both interferon beta and alpha were induced. Therefore, we conclude that acute SIV infection in the brain leads to a non-classical interferon signaling pathway which results in the differential regulation of type I interferons.
Materials and Methods
Animal studies
Pigtailed macaques (Macaca nemestrina) were dual inoculated with an immunosuppressive swarm of SIV (SIV/DeltaB670) and a neurovirulent clone (SIV/17EFr) as previously described (8, 10). Macaques were euthanized without infection (6 animals) or at days 4 (6 animals), 7 (6 animals), 10 (6 animals), 14 (6 animals), 21 (6 animals), 42 (8 animals), and 56 (8 animals) p.i. in accordance with federal guidelines and approved by the institutional review board. Tissues were snap frozen at necropsy and were later used for RNA and protein isolation.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
RNA Isolation
RNA was isolated from 50 mg of snap frozen tissue from the parietal cortex, basal ganglia and lung using STAT60 reagent (IsoTex Diagnostics, Freindswood TX). RNA was treated with turbo DNase (Ambion, Austin TX) for 30 minutes at 37°C and then purified with the RNeasy kit (Qiagen, Valencia CA).
IFN Alpha qRT-PCR primer development
Forward and reverse primers for interferon alpha were made based off 2 conserved regions between an alignment of all subtypes. However, for the forward primer, 3 alpha subtypes (alphas 6, 8 and 13) had 1–2 nucleotide differences within the middle of the conserved region, so a mixture of 4 primers were made that contained exact sequences for alphas 6, 8, 13 and the 10 subtypes which had the same sequence. Similarly, for the reverse primer, alphas 13, 4 and 2 had 1–2 nucleotides differences within the middle of the conserved region, so a mixture of primers were made using the same approach. Although a third conserved region exists between the two primers, the sequence would not yield a functional probe, so SYBR green chemistry was used for all qRT-PCR reactions. In order to verify that these primers would amplify all subtypes, RNA was isolated from 6 animals (3 uninfected and 3 SIV infected) and then reverse transcribed to cDNA using SSIII reverse transcriptase (Invitrogen, Carlsbad CA). PCR was done on the cDNA from each animal using the IFNA primers using High Fidelity PCR Supermix (Invitrogen, Carlsbad CA) and then the amplification product was cloned into the vector pcr 2.1 using TOPO TA cloning kit (Invitrogen). Between 26 and 96 colonies were sequenced from each animal, making a total of 467 colonies for sequence analysis. 12/13 IFNA subtypes were identified using these primers, with the exception being IFNA 21 (data not shown). Therefore, IFNA real time pcr results indicate levels of expression for all subtypes except IFNA 21.
IFNA qRT-PCR
1 ug of total RNA was reverse transcribed into cDNA using Superscrpt III reverse transcriptase (Invitrogen, Carlsbad CA). cDNA was then incubated with RNaseH (Invitrogen, Carlsbad CA) for 20 minutes at 37°C. 2–4 ul of the cDNA stock was used for real time pcr using the SYBR green mix (Qiagen, Valencia CA) in a Chromo4 thermocycler (Biorad, Hercules CA). Reactions were run in triplicate with appropriate controls, including reactions that lacked DNA template or reverse transcriptase enzyme, and then normalized to GAPDH run in parallel wells. Several samples were also normalized to 18S ribosomal RNA to ensure the same results using two different housekeeping genes. All qRT-PCR samples were analyzed using the ΔΔCt method (27) and expressed as a fold induction over the average of uninfected controls.
qRT-PCR for all other genes
200 ng of RNA was reverse transcribed into cDNA using Superscript III reverse transcriptase (Invitrogen, Carlsbad CA) and then analyzed by real time PCR using Multiplex NoRox mix (Qiagen, Valencia CA) in a Chromo4 Thermocycler (Biorad, Hercules CA). Cellular RNA levels were normalized using 18S ribosomal RNA. Reactions were run in triplicate with controls that lacked either DNA template or reverse transcriptase enzyme. All qRT-PCR samples were analyzed using the ΔΔCt method (27) and expressed as a fold induction over the average of uninfected controls.
Primer and Probe Sequences
Sequences for all primers were based on the rhesus genome (Macaca mulatta) obtained from GenBank. All forward and reverse primer and probe sequences are expressed in the 5′ - 3′ direction. IFNA Forward: TCCTCCATGAGGTGATCCAGCAGACCT (Alpha 6), TCCTCCATGAGATAATCCAGCAGACCT (Alpha 8), TCCTCCATGAGCTGATCCAGCAGACCT (Alpha 13), TCCTCCATGAGATGATCCAGCAGACCT (all other alphas); Reverse: GATCTCATAATTTCTGCTCTGACAACC Alpha 2, GATCTCATGATTTCTGTTCTGACAACC Alpha 4, GATCTCATGATTTCTGCTCTGATAACC Alpha 13, GATCTCATGATTTCTGCTCTGACAACC all other alphas. GAPDH primers and probe sequences: F: AGCCTCAAGATCATCAGCAATG, probe: CCAACTGCTTAGCACCCCTGGCC, R: ATGGACTGTGGTCATGAGTCCTT. IL-12 and 18S primers and probes were described elsewhere (7). Tyk2 primers were as follows: F: TTCAATCTCTTCGCCCTCTTC, probe: CCCAAACCACATCCTAGAGATCCCCA, R: CCGGAAATAAAACCTCATGCG. STAT1 primer and probe sequences: F: TGAACTTACCCAGAATGCCC, probe: CTCTGCTGTCTCCGCTTCCACTC, R: CAGACTCTCTGCAACTATGGTG. TRAIL primer and probe sequences: F: GAGAGTATGAACAGCCCCTG, probe: TGGCAACTCCGTCAGCTCGTTAG, R: AGGACCTCTTTCTCTCACTAGG. IRF7 primers and probe sequences: F: GTGGCTCCCCCTGCTATAC, probe: TGGGCTTCGGGCAGGACCTGTCAG, R: GCTCCAGCTTCACCAGGAC.
Western Blots and Antibodies
Protein lysates were made by using 100 mg punches of parietal cortex and lung snap frozen tissues and homogenized in RIPA buffer (0.25% NaDeoxyCholate, 0.1% SDS, 25 mM Tris pH 7.4, 150 mM NaCl, I mM EDTA, and 1% NP40 in water) with protease and phosphatase inhibitors (Calbiochem, Darmstadt, Germany), 1 mM DTT and 0.1 nM PMSF. Protein lysates were sonicated for 10 seconds every 10 minutes for 30 minutes on ice and then spun down to pellet cellular debris. Protein was quantitated using Biorad Protein Assay kit. Protein lysates were run on 10% Tris-HCl criterion gels (BioRad, Hercules CA) and then transferred onto polyvinylidene difluoride (PVDF) membranes using the iBlot apparatus (Invitrogen, Carlsbad CA). For all western blots except pTyk2, membranes were blocked in 5% milk for 1 hour and then probed with primary antibody overnight in 5% milk. Blots were washed 3X for 15 minutes with TBST and then incubated with horseradish peroxidase (HRP) conjugated secondary antibodies of the correct isotype that were diluted in 5% milk. Blots were then washed 3X with TBST for 20 minutes and were developed using either ECL Advance (Amersham, Piscataway NJ) substrate (tyk2, pTyk2, pSTAT3, pSTAT1), Supersignal West Pico (Pierce, Rockford IL) substrate (GAPDH, STAT1, IRF7), or Supersignal West Dura (Pierce, Rockford IL) substrate (TFIID). For pTyk2, blots were washed 3X for 15 minutes with TBST after blocking in 5% milk, and primary antibody was diluted in 5% BSA. All further steps were carried out in the same way as other blots, with the exception that all washes from that point on were done for 5 minutes. Antibodies and concentrations were as follows: (primary and secondary respectively): Tyk2 (Abcam, Cambridge MA) 1:200, 1:5000; GAPDH (Santa Cruz, Santa Cruz CA) 1:5000, 1:5000, pSTAT3 S727 (Santa Cruz, Santa Cruz CA) 1:500, 1:5000, pTyk2 Y1054/1055 (Cell Signaling, Danvers MA) 1:500, 1:2000, STAT1 (Santa Cruz, Santa Cruz CA): 1:1000, 1:5000, pSTAT1 Y701 (Cell Signalling, Danvers MA): 1:1000, 1:5000, TFIID (TBP, SI-1) (Santa Cruz, Santa Cruz CA) 1:1000, 1:5000, IRF7 (Abcam, Cambridge MA) 1:4000, 1:10,000. Western blot quantitations were carried out by scanning the developed film using a CanoScan 8400F scanner and then the images were quantitated using ImageQuant TL 7.0 software.
Cells and Reagents
Primary macaque macrophages were isolated from whole blood as previously reported (19). Cells were allowed to differentiate for 7 days in culture. On day 7, cells were stimulated with 100 units/mL of IFNB (PBL) for 5, 10 and 15 minutes. Nuclear and cytoplasmic cellular lysates were prepared using the Nuclear Isolation kit (Marligen, Ijamsville MD).
Nuclear and cytoplasmic lysate preparation from tissue
Nuclear and cytoplasmic lysates were prepared from the parietal cortex of 2 uninfected and 8 SIV infected macaques (4 from 7 days p.i and 4 from 10 days p.i). 100 mg of snap frozen parietal cortex was homogenized in 800 ul total of Buffer A (400 ul) and Buffer B (400 ul) from the Nuclei Enrichment kit for Tissue (Pierce, Rockford IL) with protease and phosphatase inhibitors (Calbiochem, Darmstadt, Germany), 1 mM DTT and 0.1 nM PMSF. Samples sat on ice for 10 minutes and non homogenized debris was pelleted. 25 ul of detergent solution (Marligen Nuclear Isolation kit, Ijamsville MD) was added to the supernatant and nuclear lysates were prepared via the manufacturers protocol (Marligen, Ijamsville MD). Protein was quantitated using the Biorad Protein Assay kit (Biorad, Hercules CA).
Statistical Analysis
The data from the qRT-PCR and western blot analysis were analyzed using the non parametric Mann Whitney test. Correlations were determined using the Spearman non parametric test.
Results
Interferon alpha is down regulated in the brain but not lung during acute SIV infection
We have previously reported that during acute SIV infection in the brain, interferon beta RNA and protein are significantly up regulated (Figure 1A, data from (9)). However, the expression of interferon alpha had not been examined. To examine the pattern of expression of interferon alpha in the brain during SIV infection, RNA was isolated from the parietal cortex of pigtailed macaques that were sacrificed at different times after infection. The parietal cortex (hereon referred to as brain) was chosen because previous reports from the lab have looked at this region in the context of SIV infection (11). Expression of interferon alpha mRNA was quantiated by quantitative reverse transcriptase PCR (qRT-PCR) using primers that spanned regions that were conserved amongst all interferon alpha subtypes. We validated that these primers detected all subtypes as described in the Materials and Methods. RNA levels were expressed as a ratio of the infected brain RNA over the average of uninfected controls using the ΔΔCt method (27). Interestingly, during acute infection, there is a significant down regulation (p=0.0101) of interferon alpha mRNA expression, with its nadir level at 7 days p.i. (Figure 1B). There is on average a 65% fold decrease in interferon alpha mRNA expression as compared to uninfected controls. Levels of interferon alpha mRNA expression do not peak above uninfected controls until 56 days p.i.. This is in contrast to interferon beta mRNA levels, which shows a reciprocal expression pattern (Figure 1C). Viral RNA in the brain during these same time points has been previously reported (9). In addition, we previously demonstrated that interleukin-12 (IL-12) mRNA is also down regulated in the brain (Figure 1D, data from (9)), which is in contrast to many other cytokines which are up regulated during acute infection.
Figure 1.

Interferon alpha and IL-12 mRNA are down regulated in the brain, but up regulated in the lung during acute SIV infection. RNA was isolated from the brain and lung from SIV infected pigtailed macaques that were sacrificed at days 4, 7, 10, 14, 21, 42, and 56 as well as 6 uninfected controls and analyzed by qRT-PCR. Each animal is represented by a black dot, and dashes connect medians. Values are normalized to GAPDH (for alpha) and 18S ribosomal RNA (for IL-12) and are expressed as a ratio over the average of the uninfected controls (fold induction) using the ΔΔCt method (14). A. interferon beta mRNA expression in the brain. B and E, interferon alpha mRNA expression in brain and lung (respectively) * p=0.0101, # p=0.0519. Primers were made against conserved regions between all subtypes. C, overlay of the medians for mRNA expression of interferon beta and interferon alpha during SIV infection in the brain. D and F, IL-12 RNA expression in brain and lung, respectively.
To determine whether the pattern of interferon signaling was specific to the brain, we examined interferon alpha expression in the periphery. We first examined interferon alpha expression in peripheral blood mononuclear cells (PBMCs) obtained at necropsy from the same animals used in the brain studies, however no induction was detected (data not shown). This is not surprising since plasmacytoid dendritic cells, the major source of interferon alpha in PBMCs, rapidly migrate to lymph nodes during acute infection (28, 29). We next examined interferon alpha expression in the lung as a peripheral comparison, since, like the brain, macrophages are the major cell infected by SIV and HIV (24–26). Furthermore, it has been reported in our SIV model that viral infection in the lung is followed by induction of interferon beta during acute infection (30). RNA was isolated from lung of the same animals used in the brain studies and qRT-PCR was performed. We measured IFN alpha mRNA levels in the lung and found that unlike the brain, levels are increased during acute infection (p=0.0519) (Figure 1E). In addition, we examined the level of IL-12 mRNA because it is the only other cytokine that we have measured that is down regulated in the brain during acute infection. Similar to interferon alpha, IL-12 mRNA expression increased in the lung during acute infection. (Figure 1F). These results indicate that SIV infection leads to a differential IFN signaling pattern during acute infection in the brain and lung.
Transcriptional and translational expression of Tyk2 is impaired in the brain
Because IFN alpha and IL-12 are the only cytokines in the brain that we have examined that are down regulated during acute infection, the signaling pathways of the two cytokines were compared to identify signaling molecules that are involved in the regulation of both cytokines. Like type I IFNs, it has been reported that IL-12 can signal in a positive feedback loop with interferon gamma as the main mediator (31, 32). Both IFN alpha and IL-12 signal through receptors that are associated with specific tyrosine kinases that are then activated by phosphorylation and signal through the JAK-STAT pathway. Tyk2 is associated with both the IL-12 and IFNα/β receptors. Because interferon alpha production is maintained and amplified in a positive feedback loop through the interferon receptor, we examined tyk2 mRNA expression levels by qRT-PCR in the brains of the same SIV-infected macaques in Figure 1. At 4 days p.i., tyk2 mRNA was significantly (p=0.0022) down regulated by roughly 70% (Figure 2A) and by 10 days post infection, tyk2 mRNA levels returned to uninfected control levels. To determine if this transcriptional down regulation correlated with protein levels, protein homogenates were made from the brain and tyk2 protein levels were assayed by western blot. We found that levels of tyk2 protein were significantly down regulated at days 4 (p=0.0087), 7 (p= 0.015) and 10 (p=0.0022) p.i. (Figure 2B). In parallel, we examined tyk2 levels in the lung during acute infection when both interferon alpha and IL-12 are induced. We found that both tyk2 mRNA and protein are up regulated (Figures 2C and 2D respectively), thus illustrating that interferon alpha signaling is specifically suppressed in the brain.
Figure 2.

Tyk2 mRNA and protein expression is down regulated during acute SIV infection in brain. A. qRT-PCR analysis for tyk2 on RNA isolated from the brain of pigtailed macaques sacrificed between days 4 and 56 p.i. including uninfected controls. 5–6 animals used per group. Values are normalized to 18S ribosomal RNA and expressed as fold induction. Medians are connected by red dashes. * p=0.0649, ** p= 0.0022. B. Western blot analysis of tyk2 protein in lysates from brain of pigtailed macaques sacrificed between days 4 and 10 p.i. including uninfected controls. 6 animals were used per group. All are normalized to GAPDH and expressed as fold induction over uninfected. Medians are connected by red dashes. * p=0.0087, **p=0.0152, *** p=0.0022 C. qRT-PCR analysis for tyk2 on RNA isolated from the lungs of the same pigtailed macaques sacrificed between days 4 and 14 p.i including uninfected controls. Values normalized to 18S ribosomal RNA. Medians are connected by red dashes. *p=0.0317, # p=0.0635. D. Western blot analysis of tyk2 protein on lysates made from lung of pigtailed macaques sacrificed on day 4 p.i including uninfected controls. Values normalized to GAPDH and expressed as fold induction. Medians are indicated by red dashes. * p=0.0571. E. Western blot analysis of ptyk2 (Y1054/1055) and P-STAT3 (S727) on the same protein lysates, as well as a lysate made from pigtailed macrophages stimulated with interferon beta (100 units/mL) for 10 minutes (designated “+”).
Although these data indicate that tyk2 is suppressed at both the transcriptional and translational level in SIV-infected brain, residual protein was still present and it was possible that this residual protein could be activated. Therefore, we assessed the phosphorylation status at Y1054/1055 of tyk2 at 4, 7 and 10 days p.i. by western blot. We detected no phosphorylated tyk2 protein (Figure 2E) in brain at these times p.i. To ensure that the lack of phospo-tyk2 signal was not due to an insufficient amount of protein analyzed, western blots were performed with up to 100ug of protein lysate from brain, and we consistently found that no phospo-tyk2 could be detected (data not shown). Protein lysates made from interferon beta stimulated macrophages isolated from pigtailed macaques were used as a positive control for tyk2 phosphorylation (Figure 2E).
In addition to tyk2 protein phosphorylation, we also examined the level of phosphorylated STAT3 (S727) to ensure that there were no technical difficulties in the detection of phosphorylated proteins in the brain lysates. STAT3 is involved in IL-10 signaling and this cytokine is known to be up regulated in the brain during acute infection (9). We detected P-STAT3 in brain of all of the animals during this time, thus controlling for the detection of phospho protein (Figure 2E). These data indicate that tyk2 is suppressed at both the transcriptional and translational levels and that the lack of tyk2 protein phosphorylation further suggests that the residual tyk2 protein was not in an active state to signal.
STAT1 is transcriptionally and translationally up regulated
In the classical interferon signaling pathway, tyk2 mediated IFN receptor phosphorylation creates a docking site for STAT1. Here, STAT1 is phosphorylated and forms the ISGF3 complex with STAT2 and IRF9, where it translocates into the nucleus and leads to the transcription of genes necessary for the antiviral response, including interferon alpha. We first measured STAT1 mRNA by qRT-PCR and protein by western blot analysis to determine whether or not this transcription factor is expressed in the brain during acute infection. We found that STAT1 mRNA and protein are significantly (p=0.0079, p=0.002 respectively) up regulated by 7 days p.i. (Figures 3A and 3B), thus indicating that this protein is indeed present in the brain.
Figure 3.

STAT1 mRNA and protein expression is up regulated during acute SIV infection. A. qRT-PCR analysis for STAT1 was done on RNA isolated from brain of pigtailed macaques sacrificed on days 4–21 p.i including uninfected controls. 6 animals were used for each group. Values are expressed as a fold induction normalized to 18S ribosomal RNA. Medians are indicated by red dashes. **p=0.0079 B. Western blot analysis for STAT1 done on protein lysates made from brain of macaques sacrificed on days 4–10 p.i. including uninfected controls (6 animals per group). Values are expressed as fold induction normalized to GAPDH. Medians are indicated by red dashes. **p=0.0022
STAT1 phosphorylation and nuclear accumulation do not occur in SIV-infected brain despite the increase in mRNA and Protein
Because STAT1 RNA and protein are up regulated during acute infection in brain, we determined if the protein was in an active state, since this would indicate the activation of signaling downstream of tyk2. Therefore, we examined phosphorylation at Y701, which is required for interferon signaling, at 4, 7 and 10 days p.i by western blot. We were unable to detect tyrosine phosphorylation of STAT1, confirming a lack of STAT1 activation and upstream kinase activity (Figure 4A). Western blots were also performed using up to 75 ug of protein lysates from macaque brain to ensure that sufficient amounts of protein were analyzed (data not shown). Further, as a control, western blot analyses were done for phosphorylated STAT3, which we were readily able to detect in the brain lysates (Figure 2E). Interferon beta stimulated macrophages isolated from pigtailed macaques were used as a positive control to ensure species cross reactivity of the antibody (Figure 4A). In contrast to the brain, phosphorylated STAT1 was detected in the lung at 4 days p.i. (Figure 4B).
Figure 4.

Lack of STAT1 phosphorylation and nuclear accumulation in the brain during acute infection. A, western blot analysis of pSTAT1 (Y701) on the brain of pigtailed macaques as well as a lysate made from interferon beta stimulated macrophages harvested after 10 minutes (last sample designated “+”). B. western blot analysis of STAT1 and pSTAT1 (Y701) in lung protein lysates made from pigtailed macaques (5 animals) sacrificed at 4 days p.i and 1 uninfected control. C. nuclear (N) and cytoplasmic (C) lysates were made from the brain from animals sacrificed at 7 and 10 days p.i (4 each) as well as 2 uninfected controls. Western blot analysis for STAT1, GAPDH and TFIID were done on lysates. D. nuclear (N) and cytoplasmic (C) lysates were made from pigtailed macaque macrophages that were either untreated or treated with interferon beta for 5, 10 and 15 minutes. Western blot analysis for STAT1, GAPDH, and TFIID were done on lysates.
We then analyzed the cellular localization of STAT1 in nuclear and cytoplasmic protein extracts made from brain of SIV infected macaques. Since activation of STAT1 by phosphorylation results in nuclear translocation we would expect to see little or no STAT1 in nuclear extracts. Consistent with our results that STAT1 is not phosphorylated, western blot analysis revealed no STAT1 nuclear accumulation at days 7 and 10 p.i. (Figure 4C). GAPDH, a cytoplasmic protein, and TFIID, a nuclear protein, were used to assess the purity of the nuclear and cytoplasmic lysates of brain. As a positive control, nuclear and cytoplasmic lysates were prepared from pigtailed macaque macrophages stimulated with interferon beta for 5, 10 and 15 minutes and STAT1 nuclear accumulation was detected (Figure 4D). Therefore, although an antiviral response is initiated which results in transcriptional and translational up regulation of STAT1, activation of STAT1, as indicated by phosphorylation and nuclear accumulation, could not be detected. This suggests that signaling downstream of the interferon receptor is impaired in the brain.
TYK2 mediated gene transcription is suppressed
In order to assess the effect of tyk2 suppression on the regulation of downstream gene expression, we next examined another gene that is regulated by tyk2, TNF related apoptosis inducing ligand (TRAIL). It has previously been reported that TRAIL is one of the few interferon stimulated genes whose transcriptional induction by interferon beta is ablated in the absence of tyk2 (33). Therefore, we measured TRAIL mRNA expression levels in brain from the same SIV-infected macaques (Figure 5A). TRAIL mRNA was not induced during acute infection and did not significantly increase until 56 days p.i. (p= 0.017), the time at which tyk2 and interferon alpha mRNA is induced (Figure 5A). Furthermore, using Spearman correlation analysis, there is a significant correlation (R=0.442, p=0.0043) between tyk2 and TRAIL mRNA expression, providing further support for the relationship between tyk2 and TRAIL regulation. This lack of induction of TRAIL in brain is in contrast to the lung. Quantitation of TRAIL mRNA in lung demonstrated that it is up regulated at 4 days p.i. (Figure 5B). These data provide strong evidence that the tyk2-mediated interferon signaling pathway is suppressed during acute SIV-infection in brain compared to a peripheral tissue such as lung, suggesting that there is differential regulation of this pathway in vivo in a tissue-specific manner.
Figure 5.
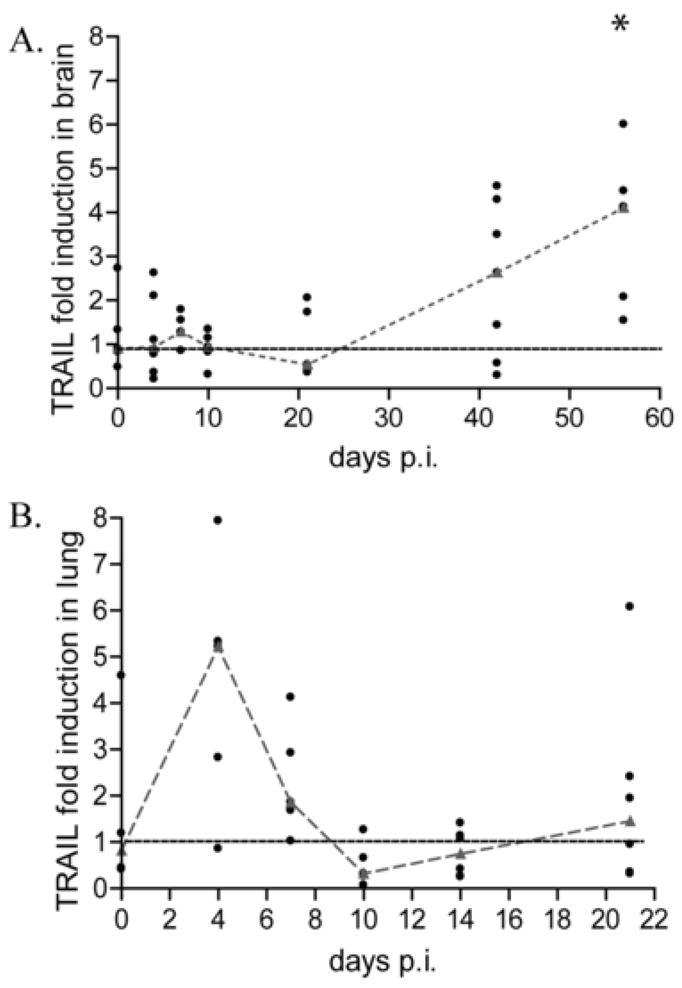
TRAIL mRNA expression is not induced in brain of pigtailed macaques during acute SIV infection. A, qRT-PCR analysis for TRAIL done on RNA isolated from brain from pigtail macaques sacrificed between days 4 and 56 p.i including uninfected controls (6 animals per group). * p=0.0157. B, qRT-PCR analysis for TRAIL done on RNA from lung isolated macaques sacrificed between days 4 and 21 days p.i. Values are normalized to 18S ribosomal RNA and represented as fold inductions over uninfected controls. Medians are connected by red dashes.
IRF7 expression is blocked at the level of translation
In addition to tyk2 and STAT1, IRF7 is another major protein in the pathway that regulates IFN alpha expression. IRF7 mRNA transcription is most notably induced by the ISGF3 complex, composed of STAT1, STAT2 and IRF9. Additionally, it has also been reported to be induced in a STAT1 independent manner through NFkB (34, 35). IRF7 serves as the transcription factor mediating interferon alpha expression. Therefore, we measured expression of IRF7 mRNA in the brain during acute infection of the same animals. Surprisingly, IRF7 mRNA is significantly up regulated (p=0.01) 20 fold by days 4, 7 and 10 p.i. and over 200 fold at days 14 and 21 p.i (Figure 6A). However, the regulation of mRNA during acute infection does not correlate with IRF7 protein expression. Western blot analysis revealed that IRF7 protein levels do not increase relative to uninfected controls between 4 and 21 days p.i. (Figure 6B). These results suggest that there is an additional block in the interferon signaling pathway that occurs post IRF7 mRNA transcription preventing translation of the protein.
Figure 6.

There is a post transcriptional block in IRF7 expression during acute SIV infection. A, qRT-PCR analysis for IRF7 on RNA isolated from brain from pigtailed macaques sacrificed between days 4 and 21 p.i. including uninfected controls (6 animals per group). Values are normalized to 18S ribosomal RNA and expressed as fold induction. Medians are connected by red dashes. # p=0.01. B, Protein lysates were made from brain of the same animals and western blot analysis was done for IRF7. Values are normalized to 18S ribosomal RNA and expressed as a fold induction over uninfected animals run on the same blot as the sample. Medians are indicated by red dashes.
Discussion
In this study, we report a novel observation that SIV infection in the brain leads to repression of a branch in the interferon signaling pathway, where interferon alpha mRNA expression is significantly down regulated during acute infection. Interferon alpha mRNA levels rebound during late stage of infection, when inflammatory cytokines increase and there is infiltration of lymphocytes (8). This expression pattern rivals that of interferon beta, which is up regulated during acute infection at the same time as the nadir of IFN alpha levels (9). We show that this transcriptional down regulation of interferon alpha is associated with suppression of signaling elements downstream of the IFN α/β receptor. Tyk2, a janus tyrosine kinase that is associated with the interferon receptor, is suppressed at both the transcriptional and translational levels by 4 days p.i. Despite residual tyk2 protein present in the brain, it is not phosphorylated and thus, in an inactive state. We also demonstrate that the lack of tyk2 activation affects the regulation of downstream genes such as TRAIL. Moreover, STAT1, which is a protein phosphorylated by the IFN receptor associated tyrosine kinases, while up regulated at the mRNA and protein levels during SIV acute infection, is neither phosphorylated, nor does it accumulate in the nucleus, signifying a lack of activation. Another important finding is that IRF7, a transcription factor mediating interferon alpha production, is up regulated at the transcriptional level, but protein levels remain comparable to uninfected controls throughout acute infection. This indicates that in addition to lack of activation of tyk2, there is yet another block in the interferon signaling pathway that occurs post IRF7 transcription. We demonstrate that suppression of IFN signaling was characteristic of SIV infection in the brain, since the same signaling mediators (tyk2 and STAT1) in the lung were activated, and IFN alpha as well as TRAIL were produced. This indicates that the type I interferon response to acute SIV infection occurs in a tissue specific manner. Taken together, these data demonstrate that innate antiviral pathways are differentially regulated during acute SIV infection specifically in the brain, resulting in a partial impairment of interferon signaling.
One of the important reasons for looking at the lung was to discover what the in vivo responses for interferon alpha and tyk2 were when the innate immune response is induced. We found that in the lung, the magnitude of the fold increase for interferon alpha is the same as the magnitude of the fold decrease in the brain. The same pattern exists for tyk2 mRNA and protein when comparing the brain and lung. Comparable fold changes are also exhibited when looking at other cytokines in the brain and lung, such as IL-12 and IFN beta. While a 2 fold change in either direction may seem like a small magnitude, we have found that this is enough to activate robust downstream activity. For example, we reported that just a 2 fold up regulation of interferon beta was sufficient to induce a 60-fold induction of Mx RNA, which is an interferon stimulated gene (9). This concept of low levels of interferons stimulating physiological events has been previously reported in the literature both in vivo and in vitro (36, 37). This is likely because interferons activate hundreds of downstream genes that are involved in immune processes, including inflammation, cellular differentiation, proliferation, and antiviral protein production. Excessive production of interferons, therefore, can lead to extensive cellular damage and immune dysfunction. In vivo, it is extremely important to maintain a balance between immune activation and attenuation, which most likely explains the magnitudes of change we see for the different cytokines.
The interferon signaling pathway can be divided into an “early” phase where interferon beta is induced via the transcription factor IRF3, and a “late” phase where interferon alpha is produced after activation of signaling factors downstream of the interferon receptor (38). Our data demonstrates that the late phase of this pathway is altered in the SIV infected brain during the acute infection. This pattern has been previously observed in HIV infected monocytes. While they have an impaired ability to produce interferon alpha, production of interferon beta remains normal (20).
We focused on tyk2 as a candidate for mediating the block in interferon alpha transcription because it is a common component of the IL-12 receptor, and IL-12 is the only other cytokine examined that is down regulated during acute SIV infection in brain. Tyk2 is a 135 kD Janus kinase associated with IFNAR1 and is activated upon receptor engagement by type I interferons, where it not only phosphorylates itself (Y1054/1055) but also the IFN receptor, jump starting the IFN mediated JAK/STAT pathway (39). Interestingly, there are published reports that implicate tyk2 specifically in interferon alpha, as opposed to interferon beta, signaling (40, 41). Several studies have reported that tyk2 deficient cells are completely unresponsive to interferon alpha, while binding of interferon beta is not only intact, but still results in signaling (33, 40–43). It has also been suggested that tyk2 plays a structural role particularly in receptor engagement of interferon alpha (44). This was further supported by the observation that while tyk2 deficiency abrogated interferon alpha receptor engagement, cells that contained the inactive kinase restored partial interferon alpha engagement and signaling (41). Furthermore, although both type I IFNs bind to the same receptor, interferon alpha binds with a lower affinity to IFNAR1 than interferon beta, resulting in the need for a higher concentration of IFNAR1 to retain adequate interferon alpha binding (45). Studies have demonstrated that tyk2 stabilizes the expression and increases the half-life of IFNAR1, and that this in turn has lead to increased interferon alpha receptor engagement (42). Most studies report that tyk2is regulated at the level of either phosphorylation by phosphatases and kinases, or protein degradation by SOCS proteins as a way to modulate the interferon response once initiated (46–48). However, in our studies, we found tyk2 is down regulated at the levels of transcription, translation, and phosphorylation as early as 4 days p.i. in brain, which is when virus replication is first detected in the CNS. This suggests that either viral infection or pathways activated by the virus are triggering an inhibitory mechanism that prevents interferon signaling through the type I IFN receptor specifically in the brain. This is further supported by the lack of TRAIL induction, which is a gene dependent upon tyk2 activation. Since interferon alpha production is regulated in a positive feedback loop, the direct implication that tyk2 has on interferon alpha signaling suggests that the downregulation and inactivaton of tyk2 during acute SIV infection is likely to be responsible for the selective down regulation of interferon alpha in brain.
One question that arises from our studies is if the JAK/STAT signaling pathway downstream of the interferon receptor is indeed suppressed, how is it possible that interferon stimulated genes (ISG) such as IRF7 and MxA (9) are up regulated? Previously published work in the field has described similar results when ISG expression is activated despite disruption in JAK/STAT signaling. STAT1 deficient mice show an increase in IRF7 RNA expression in response to LCMV infection (35) and this was attributed to signaling through TNF-α which activates NFkB. This in turn binds NFkB binding site sequences in the IRF7 promoter region, activating transcription (34). In our macaque model, TNF-α mRNA expression is up regulated during acute infection (9), so it is possible that IRF7 transcription is mediated through an NF-kB dependant pathway. Furthermore, in cells either deficient in tyk2 or cells expressing an inactive form of tyk2, stimulation with interferon beta resulted in up regulation of almost all of the same interferon stimulated genes as control cells with one of the exceptions being TRAIL (49). In addition, fibroblasts and splenocytes from tyk2 deficient mice were still able to respond to high concentrations of interferon alpha as indicated by up regulation of the interferon stimulated genes IRF1 and MHC class I, as well as protection against VSV (50). These data can be explained by the fact that alternative pathways stimulated by type I inteferon that do not signal through the JAK/STAT pathway exist and can mount an antiviral response. These pathways include p38 MAP kinase and PI3K (49). It has been reported that type I interferons activate both p38 MAP kinase and PI3K kinase, and pharmacological kinase inhibition results in the down regulation of type I IFN mediated transcription (51–53). Furthermore, embryonic fibroblasts from p38 knockout mice had a reduced ability to activate transcription of genes with interferon response elements after stimulation with type I IFN (53). The activation of the MAPK pathway in response to type I IFN was reported to be completely independent of STAT1 and STAT3 phophorylation, indicating a signaling cascade separate from the classical JAK/STAT (53). In addition, interferon alpha mediated antiviral effects against EMCV were reduced in cells treated with a p38 pharmacological inhibitor (54). It has been shown in our macaque model that both phosphorylated ERK and phosphorylated p38 expression are up regulated during acute infection in the brain (55). Taken together, these reports strongly suggest that in the brain of SIV-infected macaques, the up regulation of interferon beta during acute infection may be activating alternative pathways that do not involve JAK or STAT activation leading to the production of ISGs such as MxA.
The regulation of STAT1 and IRF7 in our SIV model is very striking. What is surprising is that although we did not detect phosphorylation of STAT1 (Y701) during acute infection in the brain, there was a significant increase in transcription and translation of this protein. However, it has been reported that unphosphorylated STAT1 does have several biological functions (reviewed in (56)). It associates with IRF1 to activate transcription of LMP2, as well as being involved in TNF-Alpha mediated apoptosis (57–59). Furthermore, microarray analysis showed similar expression patterns in cells expressing STAT1 and a mutant STAT1 that is unable to become tyrosine phosphorylated (58). Moreover, serine phosphorylation of STAT1 (S727) also regulates gene transcription that is independent of Y701 signaling (59–61). Therefore, it is possible that the transcriptional and translation up regulation of STAT1 serves an alternative function from the interferon response. Furthermore, although IRF7 transcription is induced 20 fold by day 7 and 200 fold by day 14, protein level remains constant. This suggests that there is not only a post transcriptional block in IRF7 production, but also a compensatory mechanism that drives transcription of IRF7 mRNA when no protein is produced.
The specificity in determining which parts of the interferon response are activated (such as interferon beta and MxA) and which are not (such as interferon alpha, tyk2, and TRAIL) most likely results from the particular microenvironment where infection occurs. For example, our results comparing IFN signaling in the brain and the lung suggest that down modulation of the pathway is specific to the brain and not more generally to SIV infection. The brain, unlike many other tissues, contains neurons that are not renewed and have been shown to be vulnerable to inflammation and inflammatory cytokines (reviewed in (62)). However, the CNS must mount innate immune responses to pathogenic insult to reduce both infections and the inflammatory processes that accompany them which damage or kill neurons (63, 64), thus illustrating that there must be an immunological balance. In our studies, interferon beta is up regulated at the peak of viral infection, and this is accompanied by a striking down regulation of both interferon alpha and tyk2 mRNA as soon as virus enters the brain at 4 days p. i. It is possible that this differential type I interferon signaling pattern is an inherent characteristic of the primate CNS, since it has been reported that while interferon beta is a beneficial cytokine in the CNS in terms of its antiviral and antiproliferative effects, interferon alpha and tyk2 are both harmful. For example, transgenic mice that chronically express interferon alpha in the brain developed a marked inflammatory response resulting in neurodegeneration and encephalitis (22). Two human genetic neurological disorders, Aicardi-Goutierres syndrome and Cree encephalitis, which are characterized by neurodegeneration, brain lesions and ongoing inflammatory processes, are associated with elevated and persistent levels of interferon alpha in the CNS (65, 66). Furthermore, hepatits C virus infected patients receiving interferon alpha treatment have been reported to suffer from neuropsychiatric side effects including depression, seizures, and EEG changes (67–70). Tyk2 has been found by genome wide association studies to be associated with the CNS inflammatory disease multiple sclerosis (71, 72). Mutations in the tyk2 gene that result in reduced kinase activity are reported to be protective against multiple sclerosis (71). Additionally, studies in mice report that attenuation of the tyk2 protein results in increased CNS repair in response to Theiler’s murine encephalomyelitis virus (73).
In contrast, interferon beta has been reported to be neuroprotective through the secretion of nerve growth factors, as well as anti-inflammatory by limiting T cell differentiation and increasing production of anti-inflammatory cytokines (23, 74–76). This is further supported by reports showing that interferon beta knockout mice have an increased chance of developing experimental autoimmune encephalitis (23). Therefore, it is possible that the primate brain has evolved a mechanism to illicit an antiviral response through the production of interferon beta, but prevents inflammatory damage by down regulating interferon alpha and tyk2. This is most likely characteristic of the early stages of CNS infection, before infiltration of peripheral cells that may serve to overcome the block of interferon alpha and tyk2 activation.
Our studies indicate that the interferon pathway consists of a complex network of branches that are each regulated in a tissue specific way. We found that viral infection does not result in a collective up regulation of all known interferons and interferon stimulated genes. Rather, a fine tuned specificity exists that results in the differential regulation of antiviral genes that appear to be dependent upon cell and tissue type. Thus, it may not be surprising that TRAIL, a gene whose primary function involves apoptosis, is not activated in the brain, a tissue consisting of non renewable cells, but is activated in the lung where there is ongoing cellular turnover. Because astrocytes are the immunomodulatory cells of the CNS and are only present in the brain, it is possible that these cells are mediating the specific IFN response to acute SIV infection. The multiple signaling pathways that can be activated in response to interferon beta and viral infections, such as JAK/STAT, MAPK and PI3K, provide subtle levels of regulation that can tailor the antiviral response in a tissue and cell type specific way.
Acknowledgments
The authors would like to acknowledge all of the members of the retrovirus lab for their thought provoking knowledge and valuable insights.
Footnotes
This work was supported by grants from the NIH to JEC (NS047984, NS055648 and MH070306).
References
- 1.Maschke M, Kastrup O, Esser S, Ross B, Hengge U, Hufnagel A. Incidence and prevalence of neurological disorders associated with HIV since the introduction of highly active antiretroviral therapy (HAART) J Neurol Neurosurg Psychiatry. 2000;69:376–380. doi: 10.1136/jnnp.69.3.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, Sacktor N. Human immunodeficiency virus-associated dementia: an evolving disease. J Neurovirol. 2003;9:205–221. doi: 10.1080/13550280390194109. [DOI] [PubMed] [Google Scholar]
- 3.Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002;8:136–142. doi: 10.1080/13550280290049615. [DOI] [PubMed] [Google Scholar]
- 4.Fischer-Smith T, Rappaport J. Evolving paradigms in the pathogenesis of HIV-1-associated dementia. Expert Rev Mol Med. 2005;7:1–26. doi: 10.1017/S1462399405010239. [DOI] [PubMed] [Google Scholar]
- 5.Marra CM, Zhao Y, Clifford DB, Letendre S, Evans S, Henry K, Ellis RJ, Rodriguez B, Coombs RW, Schifitto G, McArthur JC, Robertson K. Impact of combination antiretroviral therapy on cerebrospinal fluid HIV RNA and neurocognitive performance. AIDS. 2009;23:1359–1366. doi: 10.1097/QAD.0b013e32832c4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis RJ, Rosario D, Clifford DB, McArthur JC, Simpson D, Alexander T, Gelman BB, Vaida F, Collier A, Marra CM, Ances B, Atkinson JH, Dworkin RH, Morgello S, Grant I. Continued high prevalence and adverse clinical impact of human immunodeficiency virus-associated sensory neuropathy in the era of combination antiretroviral therapy: the CHARTER Study. Arch Neurol. 67:552–558. doi: 10.1001/archneurol.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Letendre SL, Ellis RJ, Ances BM, McCutchan JA. Neurologic complications of HIV disease and their treatment. Top HIV Med. 18:45–55. [PMC free article] [PubMed] [Google Scholar]
- 8.Mankowski JL, Clements JE, Zink MC. Searching for clues: tracking the pathogenesis of human immunodeficiency virus central nervous system disease by use of an accelerated, consistent simian immunodeficiency virus macaque model. J Infect Dis. 2002;186(Suppl 2):S199–208. doi: 10.1086/344938. [DOI] [PubMed] [Google Scholar]
- 9.Witwer KW, Gama L, Li M, Bartizal CM, Queen SE, Varrone JJ, Brice AK, Graham DR, Tarwater PM, Mankowski JL, Zink MC, Clements JE. Coordinated regulation of SIV replication and immune responses in the CNS. PLoS One. 2009;4:e8129. doi: 10.1371/journal.pone.0008129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zink MC, Suryanarayana K, Mankowski JL, Shen A, Piatak M, Jr, Spelman JP, Carter DL, Adams RJ, Lifson JD, Clements JE. High viral load in the cerebrospinal fluid and brain correlates with severity of simian immunodeficiency virus encephalitis. J Virol. 1999;73:10480–10488. doi: 10.1128/jvi.73.12.10480-10488.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clements JE, Babas T, Mankowski JL, Suryanarayana K, Piatak M, Jr, Tarwater PM, Lifson JD, Zink MC. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J Infect Dis. 2002;186:905–913. doi: 10.1086/343768. [DOI] [PubMed] [Google Scholar]
- 12.Szubin R, Chang WL, Greasby T, Beckett L, Baumgarth N. Rigid interferon-alpha subtype responses of human plasmacytoid dendritic cells. J Interferon Cytokine Res. 2008;28:749–763. doi: 10.1089/jir.2008.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 1998;425:112–116. doi: 10.1016/s0014-5793(98)00210-5. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem. 2007;141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 16.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barber SA, Herbst DS, Bullock BT, Gama L, Clements JE. Innate immune responses and control of acute simian immunodeficiency virus replication in the central nervous system. J Neurovirol. 2004;10(Suppl 1):15–20. doi: 10.1080/753312747. [DOI] [PubMed] [Google Scholar]
- 18.Barber SA, Gama L, Dudaronek JM, Voelker T, Tarwater PM, Clements JE. Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus-macaque model. J Infect Dis. 2006;193:963–970. doi: 10.1086/500983. [DOI] [PubMed] [Google Scholar]
- 19.Dudaronek JM, Barber SA, Clements JE. CUGBP1 is required for IFNbeta-mediated induction of dominant-negative CEBPbeta and suppression of SIV replication in macrophages. J Immunol. 2007;179:7262–7269. doi: 10.4049/jimmunol.179.11.7262. [DOI] [PubMed] [Google Scholar]
- 20.Meltzer MS, Baca L, Turpin JA, Kalter DC, Dieffenbach C, Friedman RM, Gendelman HE. Regulation of cytokine and viral gene expression in monocytes infected with the human immunodeficiency virus. Pathobiology. 1991;59:209–213. doi: 10.1159/000163647. [DOI] [PubMed] [Google Scholar]
- 21.Griffin DE. Immune responses to RNA-virus infections of the CNS. Nat Rev Immunol. 2003;3:493–502. doi: 10.1038/nri1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL. Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol. 1998;161:5016–5026. [PubMed] [Google Scholar]
- 23.Teige I, Treschow A, Teige A, Mattsson R, Navikas V, Leanderson T, Holmdahl R, Issazadeh-Navikas S. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol. 2003;170:4776–4784. doi: 10.4049/jimmunol.170.9.4776. [DOI] [PubMed] [Google Scholar]
- 24.Mankowski JL, Carter DL, Spelman JP, Nealen ML, Maughan KR, Kirstein LM, Didier PJ, Adams RJ, Murphey-Corb M, Zink MC. Pathogenesis of simian immunodeficiency virus pneumonia: an immunopathological response to virus. Am J Pathol. 1998;153:1123–1130. doi: 10.1016/S0002-9440(10)65656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baskin GB, Murphey-Corb M, Martin LN, Soike KF, Hu FS, Kuebler D. Lentivirus-induced pulmonary lesions in rhesus monkeys (Macaca mulatta) infected with simian immunodeficiency virus. Vet Pathol. 1991;28:506–513. doi: 10.1177/030098589102800607. [DOI] [PubMed] [Google Scholar]
- 26.van’t Wout AB, Ran LJ, Kuiken CL, Kootstra NA, Pals ST, Schuitemaker H. Analysis of the temporal relationship between human immunodeficiency virus type 1 quasispecies in sequential blood samples and various organs obtained at autopsy. J Virol. 1998;72:488–496. doi: 10.1128/jvi.72.1.488-496.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Barratt-Boyes SM, Wijewardana V, Brown KN. In acute pathogenic SIV infection plasmacytoid dendritic cells are depleted from blood and lymph nodes despite mobilization. J Med Primatol. 2010;39:235–242. doi: 10.1111/j.1600-0684.2010.00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown KN, Wijewardana V, Liu X, Barratt-Boyes SM. Rapid influx and death of plasmacytoid dendritic cells in lymph nodes mediate depletion in acute simian immunodeficiency virus infection. PLoS Pathog. 2009;5:e1000413. doi: 10.1371/journal.ppat.1000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barber SA, Gama L, Li M, Voelker T, Anderson JE, Zink MC, Tarwater PM, Carruth LM, Clements JE. Longitudinal analysis of simian immunodeficiency virus (SIV) replication in the lungs: compartmentalized regulation of SIV. J Infect Dis. 2006;194:931–938. doi: 10.1086/507429. [DOI] [PubMed] [Google Scholar]
- 31.Ma X, Chow JM, Gri G, Carra G, Gerosa F, Wolf SF, Dzialo R, Trinchieri G. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J Exp Med. 1996;183:147–157. doi: 10.1084/jem.183.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida A, Koide Y, Uchijima M, Yoshida TO. IFN-gamma induces IL-12 mRNA expression by a murine macrophage cell line, J774. Biochem Biophys Res Commun. 1994;198:857–861. doi: 10.1006/bbrc.1994.1122. [DOI] [PubMed] [Google Scholar]
- 33.Rani MR, Pandalai S, Shrock J, Almasan A, Ransohoff RM. Requirement of catalytically active Tyk2 and accessory signals for the induction of TRAIL mRNA by IFN-beta. J Interferon Cytokine Res. 2007;27:767–779. doi: 10.1089/jir.2007.0005. [DOI] [PubMed] [Google Scholar]
- 34.Lu R, Moore PA, Pitha PM. Stimulation of IRF-7 gene expression by tumor necrosis factor alpha: requirement for NFkappa B transcription factor and gene accessibility. J Biol Chem. 2002;277:16592–16598. doi: 10.1074/jbc.M111440200. [DOI] [PubMed] [Google Scholar]
- 35.Ousman SS, Wang J, Campbell IL. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J Virol. 2005;79:7514–7527. doi: 10.1128/JVI.79.12.7514-7527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abel K, Alegria-Hartman MJ, Rothaeusler K, Marthas M, Miller CJ. The relationship between simian immunodeficiency virus RNA levels and the mRNA levels of alpha/beta interferons (IFN-alpha/beta) and IFN-alpha/beta-inducible Mx in lymphoid tissues of rhesus macaques during acute and chronic infection. J Virol. 2002;76:8433–8445. doi: 10.1128/JVI.76.16.8433-8445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radvanyi LG, Banerjee A, Weir M, Messner H. Low levels of interferon-alpha induce CD86 (B7.2) expression and accelerates dendritic cell maturation from human peripheral blood mononuclear cells. Scand J Immunol. 1999;50:499–509. doi: 10.1046/j.1365-3083.1999.00625.x. [DOI] [PubMed] [Google Scholar]
- 38.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 39.Colamonici OR, Uyttendaele H, Domanski P, Yan H, Krolewski JJ. p135tyk2, an interferon-alpha-activated tyrosine kinase, is physically associated with an interferon-alpha receptor. J Biol Chem. 1994;269:3518–3522. [PubMed] [Google Scholar]
- 40.Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell. 1992;70:313–322. doi: 10.1016/0092-8674(92)90105-l. [DOI] [PubMed] [Google Scholar]
- 41.Velazquez L, Mogensen KE, Barbieri G, Fellous M, Uze G, Pellegrini S. Distinct domains of the protein tyrosine kinase tyk2 required for binding of interferon-alpha/beta and for signal transduction. J Biol Chem. 1995;270:3327–3334. doi: 10.1074/jbc.270.7.3327. [DOI] [PubMed] [Google Scholar]
- 42.Uze G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Curr Top Microbiol Immunol. 2007;316:71–95. doi: 10.1007/978-3-540-71329-6_5. [DOI] [PubMed] [Google Scholar]
- 43.Gauzzi MC, Barbieri G, Richter MF, Uze G, Ling L, Fellous M, Pellegrini S. The amino-terminal region of Tyk2 sustains the level of interferon alpha receptor 1, a component of the interferon alpha/beta receptor. Proc Natl Acad Sci U S A. 1997;94:11839–11844. doi: 10.1073/pnas.94.22.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Platis D, Foster GR. Activity of hybrid type I interferons in cells lacking Tyk2: a common region of IFN-alpha 8 induces a response, but IFN-alpha2/8 hybrids can behave like IFN-beta. J Interferon Cytokine Res. 2003;23:655–666. doi: 10.1089/107999003322558791. [DOI] [PubMed] [Google Scholar]
- 45.Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, Uze G, Schreiber G. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol. 2006;26:1888–1897. doi: 10.1128/MCB.26.5.1888-1897.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu H, An H, Hou J, Han C, Wang P, Yu Y, Cao X. Phosphatase PTP1B negatively regulates MyD88- and TRIF-dependent proinflammatory cytokine and type I interferon production in TLR-triggered macrophages. Mol Immunol. 2008;45:3545–3552. doi: 10.1016/j.molimm.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 47.Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, Salmeen A, Barford D, Tonks NK. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem. 2001;276:47771–47774. doi: 10.1074/jbc.C100583200. [DOI] [PubMed] [Google Scholar]
- 48.Gingras S, Parganas E, de Pauw A, Ihle JN, Murray PJ. Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J Biol Chem. 2004;279:54702–54707. doi: 10.1074/jbc.M411043200. [DOI] [PubMed] [Google Scholar]
- 49.Rani MR, Ransohoff RM. Alternative and accessory pathways in the regulation of IFN-beta-mediated gene expression. J Interferon Cytokine Res. 2005;25:788–798. doi: 10.1089/jir.2005.25.788. [DOI] [PubMed] [Google Scholar]
- 50.Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, Yamashita M, Numata A, Takase K, Kobayashi S, Shibata S, Asano Y, Gondo H, Sekiguchi K, Nakayama K, Nakayama T, Okamura T, Okamura S, Niho Y. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. 2000;13:561–571. doi: 10.1016/s1074-7613(00)00055-8. [DOI] [PubMed] [Google Scholar]
- 51.Uddin S, Fish EN, Sher DA, Gardziola C, White MF, Platanias LC. Activation of the phosphatidylinositol 3-kinase serine kinase by IFN-alpha. J Immunol. 1997;158:2390–2397. [PubMed] [Google Scholar]
- 52.Uddin S, Majchrzak B, Woodson J, Arunkumar P, Alsayed Y, Pine R, Young PR, Fish EN, Platanias LC. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. 1999;274:30127–30131. doi: 10.1074/jbc.274.42.30127. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Deng H, Zheng D, Li H. Role of p38 mitogen-activated protein kinase in mediating monocyte chemoattractant protein-1 in human umbilical vein endothelial cells. Chin Med Sci J. 2004;19:71. [PubMed] [Google Scholar]
- 54.Mayer IA, Verma A, Grumbach IM, Uddin S, Lekmine F, Ravandi F, Majchrzak B, Fujita S, Fish EN, Platanias LC. The p38 MAPK pathway mediates the growth inhibitory effects of interferon-alpha in BCR-ABL-expressing cells. J Biol Chem. 2001;276:28570–28577. doi: 10.1074/jbc.M011685200. [DOI] [PubMed] [Google Scholar]
- 55.Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, Zink MC. Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol. 2004;164:355–362. doi: 10.1016/S0002-9440(10)63125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18:443–451. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- 57.Chatterjee-Kishore M, Kishore R, Hicklin DJ, Marincola FM, Ferrone S. Different requirements for signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 in the regulation of low molecular mass polypeptide 2 and transporter associated with antigen processing 1 gene expression. J Biol Chem. 1998;273:16177–16183. doi: 10.1074/jbc.273.26.16177. [DOI] [PubMed] [Google Scholar]
- 58.Chatterjee-Kishore M, Wright KL, Ting JP, Stark GR. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000;19:4111–4122. doi: 10.1093/emboj/19.15.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR. Defective TNF-alpha-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science. 1997;278:1630–1632. doi: 10.1126/science.278.5343.1630. [DOI] [PubMed] [Google Scholar]
- 60.Stephanou A, Scarabelli TM, Brar BK, Nakanishi Y, Matsumura M, Knight RA, Latchman DS. Induction of apoptosis and Fas receptor/Fas ligand expression by ischemia/reperfusion in cardiac myocytes requires serine 727 of the STAT-1 transcription factor but not tyrosine 701. J Biol Chem. 2001;276:28340–28347. doi: 10.1074/jbc.M101177200. [DOI] [PubMed] [Google Scholar]
- 61.Nguyen H, Chatterjee-Kishore M, Jiang Z, Qing Y, Ramana CV, Bayes J, Commane M, Li X, Stark GR. IRAK-dependent phosphorylation of Stat1 on serine 727 in response to interleukin-1 and effects on gene expression. J Interferon Cytokine Res. 2003;23:183–192. doi: 10.1089/107999003765027384. [DOI] [PubMed] [Google Scholar]
- 62.Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol. 41:242–247. doi: 10.1007/s12035-010-8105-9. [DOI] [PubMed] [Google Scholar]
- 63.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 64.Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol. 2000;74:3366–3378. doi: 10.1128/jvi.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Heteren JT, Rozenberg F, Aronica E, Troost D, Lebon P, Kuijpers TW. Astrocytes produce interferon-alpha and CXCL10, but not IL-6 or CXCL8, in Aicardi-Goutieres syndrome. Glia. 2008;56:568–578. doi: 10.1002/glia.20639. [DOI] [PubMed] [Google Scholar]
- 66.Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, Michaud J, Roberts E, Stephenson JB, Woods CG, Lebon P. Cree encephalitis is allelic with Aicardi-Goutieres syndrome: implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. 2003;40:183–187. doi: 10.1136/jmg.40.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shakil AO, Di Bisceglie AM, Hoofnagle JH. Seizures during alpha interferon therapy. J Hepatol. 1996;24:48–51. doi: 10.1016/s0168-8278(96)80185-1. [DOI] [PubMed] [Google Scholar]
- 68.Schaefer M, Engelbrecht MA, Gut O, Fiebich BL, Bauer J, Schmidt F, Grunze H, Lieb K. Interferon alpha (IFNalpha) and psychiatric syndromes: a review. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:731–746. doi: 10.1016/s0278-5846(01)00324-4. [DOI] [PubMed] [Google Scholar]
- 69.Dieperink E, Willenbring M, Ho SB. Neuropsychiatric symptoms associated with hepatitis C and interferon alpha: A review. Am J Psychiatry. 2000;157:867–876. doi: 10.1176/appi.ajp.157.6.867. [DOI] [PubMed] [Google Scholar]
- 70.Amodio P, De Toni EN, Cavalletto L, Mapelli D, Bernardinello E, Del Piccolo F, Bergamelli C, Costanzo R, Bergamaschi F, Poma SZ, Chemello L, Gatta A, Perini G. Mood, cognition and EEG changes during interferon alpha (alpha-IFN) treatment for chronic hepatitis C. J Affect Disord. 2005;84:93–98. doi: 10.1016/j.jad.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 71.Ban M, Goris A, Lorentzen AR, Baker A, Mihalova T, Ingram G, Booth DR, Heard RN, Stewart GJ, Bogaert E, Dubois B, Harbo HF, Celius EG, Spurkland A, Strange R, Hawkins C, Robertson NP, Dudbridge F, Wason J, De Jager PL, Hafler D, Rioux JD, Ivinson AJ, McCauley JL, Pericak-Vance M, Oksenberg JR, Hauser SL, Sexton D, Haines J, Sawcer S, Compston A. Replication analysis identifies TYK2 as a multiple sclerosis susceptibility factor. Eur J Hum Genet. 2009;17:1309–1313. doi: 10.1038/ejhg.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mero IL, Lorentzen AR, Ban M, Smestad C, Celius EG, Aarseth JH, Myhr KM, Link J, Hillert J, Olsson T, Kockum I, Masterman T, Oturai AB, Sondergaard HB, Sellebjerg F, Saarela J, Kemppinen A, Elovaara I, Spurkland A, Dudbridge F, Lie BA, Harbo HF. A rare variant of the TYK2 gene is confirmed to be associated with multiple sclerosis. Eur J Hum Genet. 18:502–504. doi: 10.1038/ejhg.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bieber AJ, Suwansrinon K, Kerkvliet J, Zhang W, Pease LR, Rodriguez M. Allelic variation in the Tyk2 and EGF genes as potential genetic determinants of CNS repair. Proc Natl Acad Sci U S A. 107:792–797. doi: 10.1073/pnas.0906589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boutros T, Croze E, Yong VW. Interferon-beta is a potent promoter of nerve growth factor production by astrocytes. J Neurochem. 1997;69:939–946. doi: 10.1046/j.1471-4159.1997.69030939.x. [DOI] [PubMed] [Google Scholar]
- 75.Chabot S, V, Yong W. Interferon beta-1b increases interleukin-10 in a model of T cell-microglia interaction: relevance to MS. Neurology. 2000;55:1497–1505. doi: 10.1212/wnl.55.10.1497. [DOI] [PubMed] [Google Scholar]
- 76.Paul S, Ricour C, Sommereyns C, Sorgeloos F, Michiels T. Type I interferon response in the central nervous system. Biochimie. 2007;89:770–778. doi: 10.1016/j.biochi.2007.02.009. [DOI] [PubMed] [Google Scholar]