

Abstract
Cytochrome P450 enzymes play key roles in the metabolism of the majority of drugs. Improved models for prediction of likely metabolites will contribute to drug development. In this work, two possible metabolic routes (aromatic carbon oxidation and O-demethylation) of dextromethorphan are compared using molecular dynamics (MD) simulations and density functional theory (DFT). The DFT results on a small active site model suggest that both reactions might occur competitively. Docking and MD studies of dextromethorphan in the active site of P450 2D6 show that the dextromethorphan is located close to heme oxygen in a geometry apparently consistent with competitive metabolism. In contrast, calculations of the reaction path in a large protein model [using a hybrid quantum mechanical–molecular mechanics (QM/MM) method] show a very strong preference for O-demethylation, in accordance with experimental results. The aromatic carbon oxidation reaction is predicted to have a high activation energy, due to the active site preventing formation of a favorable transition-state structure. Hence, the QM/MM calculations demonstrate a crucial role of many active site residues in determining reactivity of dextromethorphan in P450 2D6. Beyond substrate binding orientation and reactivity of Compound I, successful metabolite predictions must take into account the detailed mechanism of oxidation in the protein. These results demonstrate the potential of QM/MM methods to investigate specificity in drug metabolism.
Cytochrome P450 enzymes (P450s) form one of the most powerful defense mechanisms of living organisms: They protect against xenobiotics via an oxidative pathway, which in most instances leads to a less harmful and more soluble product with faster excretion. The same mechanism is involved in the metabolism of most drugs and alters the pharmacological activity of many drugs. As a consequence, P450-mediated transformations of drug candidates are of crucial importance in the pharmaceutical industry. The roles of P450s are manifold. Oxidation by P450s can lead to toxic products, but, on the other hand, local activation of, e.g., anticancer prodrugs by P450s to lethal intracellular toxins at the site of the tumor is an important strategy (1). Drug compounds can induce P450 expression but can also inhibit them in various ways (2–4). The metabolic clearance of most drugs depends on P450s, and they have been implicated in a large number of drug–drug interactions (5, 6), which can result in fatalities (7, 8). Interactions of drug candidates with P450s must be taken into account during the drug discovery process if the expensive and time-consuming development of active compounds with hidden toxic effects is to be avoided.
It is increasingly recognized that methods capable of predicting P450 oxidative activity for a given substrate, and also the predominant site(s) of metabolism, could contribute significantly to drug development. There are many aspects to this problem, including isoform selectivity (i.e., which P450 isoform will contribute most to oxidation of a given substrate), chemoselectivity (which functional groups will undergo oxidation most readily), and regioselectivity (which part of a drug molecule will be oxidized). The first of these matters is determined largely by the relative abundance of the different isoforms (e.g., many drugs are oxidized by the 3A4 isoform, partly because it is very abundant), by the binding affinity of the substrate in the active site, and by distribution and P450 induction effects. Many techniques have been put forward to predict chemo- and regioselectivity, including traditional quantitative structure-activity relationship (QSAR) (9, 10) as well as three-dimensional QSAR models (10, 11), construction of pharmacophore models for individual isoforms (12), substrate docking approaches using either crystal structures or homology models of the relevant enzyme (13), ab initio or density functional theory (DFT) calculations of reaction barriers and/or electronic properties (14), and rule-based methods (15). It is likely that a combination of methods will be required in general; hybrid quantum mechanical–molecular mechanics (QM/MM) methods, which allow modeling of specific effects of the protein on reactivity, have advanced to the stage where they can potentially make an important contribution (16).
QSAR and pharmacophore approaches rely heavily on observed reactivity data and can give excellent results, provided sufficient relevant data for molecules closely related to the substrate of interest is available (9–12). However, they lack general predictive power and do not provide extensive insight into the atomic-level factors leading to selectivity. Methods based on docking are able to predict the likely binding mode of a substrate in the active site of the enzyme and to predict the likely places of metabolism based on proximity to the heme site (13), but do not themselves account for the intrinsic reactivity of the substrate. It has been recently shown that it may be necessary to dock ligands into a set of protein structures in order to obtain good results, because small differences in the enzyme structure may lead to very different docking results (17). Rule-based methods rely upon a human-annotated library of biotransformation reactions described as generic rules that are used to predict the metabolic fate of a query compound (15). They are fast, but in most cases they predict a large number of possible metabolites despite the fact that in most cases only a few are formed experimentally. Important techniques are available that combine data from docking orientation and functional group reactivity for the prediction of metabolism, with good success in many cases (18).
QM (quantum mechanical, electronic structure) methods are computationally demanding, and are thus limited to studies of relatively small molecular models, but are potentially highly accurate and can provide detailed insight into intrinsic reactivity. QM calculations on models of P450 reactions have given results that often correlate reasonably well with experimental observation (14, 19, 20). However, because the size of models that can feasibly be treated is limited, they may not account for important steric factors and binding preferences. QM/MM calculations offer the potential to do this, because larger and more extensive models can be treated, and a number of studies of issues relating to mechanisms and selectivity have started to appear (16, 21–23). However such studies remain very challenging.
Here we report a QM/MM study of the factors accounting for selectivity in oxidation of a drug molecule by a human cytochrome P450 enzyme. These calculations are time-consuming, so it is not yet possible to study large numbers of species in a large range of isoforms. The system we have focused on is oxidation of the cough suppressant drug dextromethorphan by P450 2D6. This isoform of P450s has attracted much interest in recent years because it is the second most important drug-metabolizing enzyme in man and is highly polymorphic. The home page of the Human Cytochrome P450 Allele Nomenclature Committee (http://www.imm.ki.se/cypalleles/cyp2d6.htm) lists more than 70 alleles of P450 2D6 with activities for substrate metabolism ranging from several times that of the wild-type enzyme to almost zero (24, 25). It has been implicated in many drug–drug interactions, and therefore there is a significant effort in preclinical drug discovery to ensure that drug candidates are neither metabolites nor inhibitors of P450 2D6 (26). Dextromethorphan has frequently been used as a probe drug in clinical studies of metabolism, and there is abundant data available on its metabolites. It is O-demethylated by P450 2D6 (27), although N-demethylation can also occur at high substrate concentration or with other isoforms, and with mutation of active site residues other products have also been observed (28) Aromatic oxidation of dextromethorphan in P450s is never observed, although this is a major metabolic route for other methoxyaromatic rings (e.g., anisole) (29–31) (Scheme 1).
Scheme 1.

Metabolic routes of aromatic ethers. (A) Metabolic products of anisole in rat29 (plain arrow) and rabbit31 (dotted arrow). (B) Major metabolite of dextromethorphan by human P450 2D6. Bold arrows indicate major products.
According to experimental and computational evidence, the major part of the large variety of reactions catalyzed by P450 enzymes all involve the same active species, a high-valent iron-oxo derivative of the active site heme group, known as Compound I (14, 32). Its ground state has three unpaired electrons, two in Fe–O π∗ orbitals, and one in a π-orbital of the porphyrin. Due to the weak coupling between the Fe–O based and porphyrin orbitals, the energy difference between the resulting quartet (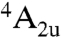 ) and open-shell doublet (
) and open-shell doublet (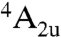 ) states of Compound I is very small, giving rise to two-state reactivity of P450 enzymes (14).
) states of Compound I is very small, giving rise to two-state reactivity of P450 enzymes (14).
The aim of the present work was to investigate the reasons why P450 aromatic carbon oxidation of dextromethorphan is not observed, despite the fact that it occurs with other anisole derivatives. Previous QM/MM studies of cytochrome P450s have concentrated on the metabolism of model compounds such as propene (22, 33), cyclohexene (22), or benzene (21), or on camphor metabolism by the bacterial P450cam enzyme (34). These studies gave important insights into the effect of enzyme environment on the mechanism of metabolism, but due to the small size of the ligands, specific interactions between the ligands and protein residues are not crucial determinants of reactivity. Here, we have studied and compared the mechanism of O-demethylation and aromatic carbon oxidation of dextromethorphan.
The O-demethylation reaction is a stepwise process, with the highest barrier due to hydrogen atom abstraction from the methyl group to the oxo group of Compound I. This is followed by fast rebound, in which the -OCH2 radical species forms a carbon–oxygen bond with the iron-hydroxo species (35, 36). The resulting ferric complex of the hemiacetal derivative of the substrate dissociates from the enzyme and is hydrolyzed to form formaldehyde and a phenolic product. We focus here only on the key hydrogen atom abstraction step.
Aromatic carbon oxidation is another typical reaction catalyzed by P450s, which in a multistep process may lead to various products, including phenols, ketones, or epoxides. Although several mechanisms have been proposed, including direct formation of arene epoxides (37), recent experimental (38, 39) and computational (40) evidence favors the addition-rearrangement mechanism. According to this mechanism, the first and rate-determining step of the reaction is the addition of the oxo group of Compound I to the aromatic ring forming a tetrahedral intermediate (σ-complex), which can undergo rearrangement reactions to yield phenols, ketones, or epoxides.
Computational Details
QM Calculations.
All calculations were carried out using the B3LYP functional with the Jaguar 6.0 program package (41). Geometry optimizations and frequency calculations in the gas phase were carried out using an unrestricted ansatz with the LACVP** basis set. Barrier heights were calculated from single point calculations on the optimized geometries performed using a larger basis set, referred to here as BS I, which includes the LACV3P basis on iron and the 6–311++G(d,p) basis on other atoms. The barrier heights were corrected for zero-point energy.
Molecular Dynamics (MD) Simulations.
The initial structure used in the simulations was constructed on the basis of the crystallographic structure of P450 2D6 (42) obtained from the Protein Data Bank (PDB code 2F9Q). The CHARMM27 force field (43) and the CHARMM package version c27b2 (44) were used in all MM minimizations and MD simulations, with Compound I parameters for the heme group (45). For dextromethorphan, standard CHARMM27 parameters were used, extended with parameters for the ether group (SI Appendix) (46). All MM minimizations were performed by using 1,500 steps of steepest descent and 1,500 steps of adopted basis Newton–Raphson. A 13-Å cutoff for nonbonded interactions was used for all MM energy minimizations and MD simulations. A time step of 1 fs was used in all MD simulations, and SHAKE restraints were applied for X–H bonds. For stochastic boundary MD simulations, the system was divided into a buffer region (atoms between 21 and 25 Å from the heme iron) treated by Langevin dynamics and a reaction region (atoms in a 21-Å sphere centered on the heme iron) treated by full Newtonian MD. Friction coefficients of 62 and 250 ps-1, respectively, were applied to water oxygen atoms and protein heavy atoms in the buffer region. Protein atoms within the buffer region were also restrained harmonically to their minimized positions. The buffer restraints were dependent on the distance from the center of the system, increasing toward the boundary according to a protocol used previously (47).
MD Simulations: System Setup.
Different starting structures for the MD simulations were generated manually by placing the dextromethorphan molecule in the active site of the enzyme in five different positions. Then, hydrogen atoms were added to the initial enzyme/substrate complexes, and their positions were optimized. Histidine protonation states were assigned according to the local environment, as in our previous work (45). The protein was truncated to a 25-Å sphere centered on the heme iron. Charged residues at the surface were neutralized to avoid unrealistic effects due to protein truncation and insufficient screening of charges by solvent. For glutamate, aspartate, lysine, and arginine residues near the surface, neutral “patch” residues, with the same geometry but modified atomic charges, were used. This resulted in a net charge of +1 for the P450 2D6 model. The structures were solvated within a 30-Å box with 8,000 preequilibrated water molecules, represented by the TIP3P model [as implemented in CHARMM (44)], centered on the heme iron. The added water was then equilibrated by stochastic boundary MD at 300 K over 20 ps with respect to the substrate-bound enzyme structure and minimized. Water molecules more than 25 Å from the heme iron were removed. Then, all atoms were minimized, followed by stochastic boundary MD simulation of the whole system. All systems were heated to 300 K over 60 ps. Subsequent MD equilibrations at 300 K were carried out over 2 ns.
QM/MM Calculations.
Starting structures were taken from the MD simulations described above and were first extensively MM minimized. For hydrogen abstraction, starting structures were chosen that could serve as a favorable reactant complex for the reaction to occur; i.e., with a short hydrogen ferryl-oxygen distance, and favorable Fe–O–H angle (between 110–130°) and almost linear O–H–C angle. For aromatic carbon oxidation, based upon our previous results for benzene (21), the starting structures were chosen with short C–O distances and favorable Fe–O–C angles (between 110–130°). The QM region comprised the heme group without substituents on the porphyrin ring, the SCH2 group of the cysteinyl ligand, and the benzene molecule. The Jaguar 6.0 software (41) was used with the standard UB3LYP density functional. In the case of the hydrogen abstraction reaction, most of the calculations were carried out in the quartet state (one profile was calculated in the doublet state), because the QM calculations indicated that the energetics in the two spin states are similar. Conversely, based on previous work (48) and our QM calculations here, only the doublet state was used for aromatic carbon oxidation. The QM/MM adiabatic mapping energy profiles used the LACVP* basis set (and the Los Alamos effective core potential on Fe). Single point QM/MM energy calculations were then carried out at the B3LYP/BSI-CHARMM27 QM/MM level. The results include a correction for zero-point energy derived from the QM calculations above. The reaction coordinate used in adiabatic mapping was the distance between the ferryl oxygen and the abstracted hydrogen atom or the aromatic carbon. Reactant complexes were minimized without restraints.
The TINKER MM code (49) with the CHARMM27 all-atom force field was used for the treatment of the MM part of the system. All atoms within a sphere of 20 Å centered on the heme iron were free to move. Atoms beyond this distance were fixed. Input and output from the Jaguar and Tinker codes were coupled by our own set of routines (QoMMMa) (50), which was also used to optimize the positions of the QM atoms. The MM charges are included in the QM Hamiltonian. The valences of the covalent bonds at the QM/MM boundary were satisfied by using hydrogen “link” atoms (51). MM charges on the atom replaced by the link atom, as well as those on atoms directly bonded to this atom, were set to zero to avoid nonphysical effects, while retaining overall integer charge.
Topology file and (additional) CHARMM22 parameters for dextromethorphan, Cartesian coordinates and total energies of all stationary points located in the QM-only calculations, and Cartesian coordinates of QM atoms from QM/MM minima and TSs are provided in the SI Appendix.
Results and Discussions
QM Calculations.
The relative barrier heights for Compound I hydrogen atom abstraction and addition to the aromatic ring will determine product selectivity for oxidation of anisole derivatives. We started by locating the corresponding transition states (TSs) using B3LYP calculations for a small model system (Table 1), in which anisole was used to represent dextromethorphan, and an unsubstituted heme ring with a ferryl oxygen and a hydrogenosulfide ligand represented P450 Compound I. The TSs for hydrogen abstraction from the methoxy group of anisole, and for addition to the ortho-position, were optimized on both the quartet and doublet potential energy surfaces (PESs) (Fig. 1 and Table 1). Hydrogen abstraction has a similar barrier on the quartet and doublet PESs (ΔE‡ = 11.1 and 9.8 kcal/mol), whereas aromatic carbon oxidation has a lower barrier on the doublet surface (ΔE‡ = 14.2 kcal/mol). These results are in line with previous B3LYP studies on hydrogen abstraction from a methoxy group (19) and on aromatic hydroxylation of anisole in the para-position (48). The barrier found here for ortho-oxidation of anisole is very similar to the published result for the para-position (12.6 kcal/mol). The key conclusion here is that, although the QM calculations predict hydrogen abstraction to be slightly favored, they also show both pathways to have roughly similar barrier heights, consistent with experimental observations whereby both oxidation processes are observed in P450 oxidation of anisole derivatives (29–31). The QM calculations on small models therefore suggest that dextromethorphan could undergo aromatic oxidation, yet this is not observed in P450 2D6.
Table 1.
QM barriers (kcal/mol, including correction for zero-point energy) to hydrogen abstraction and C2 aromatic carbon oxidation in the doublet and quartet spin states at the B3LYP/LACVP** and B3LYP/BSI levels of theory
| Hydrogen abstraction | Aromatic carbon oxidation | |||
| LACVP** | BSI | LACVP** | BSI | |
| Quartet | 12.4 | 11.1 | 17.2 | 15.9 |
| Doublet | 12.0 | 9.8 | 15.3 | 14.2 |
Fig. 1.

Calculated TS structures and activation energies (at the B3LYP/BSI//B3LYP/LACVP** level, including ZPE) in QM and QM/MM calculations for quartet state hydrogen abstraction from the O-methyl group of dextromethorphan and doublet state C2 aromatic hydroxylation. The angle between the best-fit planes for the aromatic ring and the heme group is shown for the aromatic oxidation TS structures.
MD Simulations and QM/MM Calculations
We used the protonated form of dextromethorphan in our simulations, because the presence of a basic nitrogen is one of the most important pharmacophores of P450 2D6 substrates (25). The choice of initial structure for the MD is important, because the 2-ns timescale is sufficient to sample a representative sample of structures surrounding the initial docking pose, but it is not long enough to sample radically different substrate binding modes such as those that would be needed to allow N-demethylation. It has been shown that interaction between the Glu216 or Asp301 residues and the protonated amine is very important for many substrates, as mutation of these residues alters the regioselectivity of the enzyme (52). There are two stereoisomers of the N-protonated compound, both of which can easily be docked in the active site. The pose found here for isomer A involves a salt bridge between the protonated nitrogen and Glu216, whereas for the other isomer (B), the pose involves interaction with Asp301 (see Fig. S1). In both cases, the aromatic ring of dextromethorphan is oriented to form π–π interactions with Phe120. Other favorable docking poses without the salt bridge with Glu216 or Asp301 were found and may play a role in accounting for selectivity when these residues are mutated (28), but they were not considered further here. Note that because the MD simulations are not long enough to sample the binding mode with the N-methyl group close to the heme ferryl, it is not practical in this study to address the selectivity for O- vs. N-demethylation (53).
In the MD simulations starting from the favored pose of isomer A, both the methoxy group and the C2 aromatic carbon remained close to the ferryl oxygen, and the salt bridge with Glu216 was preserved. In the MD simulation starting from the favored pose of stereoisomer B, the proximity to the ferryl group was less well maintained (see Fig. S2). Because gas-phase calculations at the B3LYP/6-31G* level indicate that stereoisomer A is more stable by 3 kcal/mol, further calculations considered this form only. The close interaction between the ferryl oxygen and the methoxy group and the C2 aromatic carbon, combined with the QM calculations showing similar barriers to oxidation of both groups, suggest that oxidation of both positions might be expected.
QM/MM calculations provide an efficient way to model the reaction while including in the calculations a large number of solvent and protein atoms. Although similar results for barrier heights might well be obtained by carrying out QM-only calculations with a much expanded QM model, such calculations would be significantly more computationally expensive, and it is not feasible to study systems remotely as large as our QM/MM models here. We have shown previously (33) that it is important to preselect starting structures for QM/MM calculations in order to obtain reasonable reaction barriers, especially when fairly loosely bound substrates are involved, as in this case. The different structures may be in rapid equilibrium, as shown by the MD simulations, but some of them may not be connected to a low energy barrier through a continuous reaction path. We therefore extracted from the MD simulations three snapshots, each corresponding to a priori favorable structures for hydrogen abstraction and aromatic carbon oxidation, based on similarity to the known TS structures derived from QM calculations. Many such suitable structures for both reactions were sampled throughout the MD simulation, showing that selecting suitable geometries does not create a significant bias to the free energy of activation. In the case of hydrogen abstraction, we selected structures within the MD simulation with a short ferryl oxygen to hydrogen distance, and a near-linear ferryl oxygen—hydrogen—carbon arrangement, and an Fe–O–H angle near 120°. For aromatic oxidation, the key parameter is the O–C distance, and this was found to be between 3 and 4 Å throughout the MD (see Fig. S3); we picked starting structures toward the lower end of this range.
The QM/MM energy profiles for hydrogen abstraction and aromatic carbon oxidation are shown in Fig. S4. Considering O-demethylation first, the TS energies for the hydrogen abstraction step calculated starting from the three snapshots chosen are all similar, at 18.2, 19.0, and 19.7 kcal/mol (including a correction for zero-point energy), at the B3LYP/BS I QM/MM level. A single doublet QM/MM profile at the same level of theory was also calculated, yielding, as in the QM calculations, a similar value (17.0 kcal/mol) to the quartet barrier. The basis set effect is small (as in the QM-only calculations): The values obtained with the smaller basis set (used for optimization), LACVP*, are quite similar at 20.8, 21.8, and 21.1 kcal/mol.
These QM/MM barriers for hydrogen abstraction are significantly higher than that obtained in the gas phase at the same QM level (11.1 kcal/mol), while still consistent with reasonably efficient turnover. In the gas phase we calculated the barrier with respect to the separated fragments, whereas in the QM/MM calculations the barrier is obtained relative to the reactant complex, in which there is a weak attractive interaction between the substrate and the heme group. This probably accounts for a small part of the difference. More significant, however is the fact that the structures are more constrained in the QM/MM calculations (because of interactions at the active site). This means that the ideal TS geometry cannot be achieved within the enzyme, leading to a higher barrier. Similar effects have been found in previous comparisons between QM and QM/MM studies of P450 oxidation (21, 33).
The structures of the TSs in the three QM/MM profiles for hydrogen abstraction were very similar. Important features of the lowest energy abstraction TS are shown in Fig. 1. Comparison with the gas-phase calculations for hydrogen abstraction shows that the benzene rings of anisole and dextromethorphan are differently oriented in the QM and QM/MM TSs. In the QM TS, the most favorable orientation for reaction can be attained because there is no steric hindrance for the movement of any of the atoms. In the enzyme environment, which is included in the QM/MM calculations, the topology of the active site cavity, hydrogen bond, and steric interactions limit the movement of the substrate in the active site; these prevent it from attaining the most favorable position for reaction. Apart from the orientation of the aromatic ring with respect to the porphyrin ring, the structure of the TSs are very similar in the QM and QM/MM calculations (see Table S1).
We now consider the aromatic oxidation TSs. Previous QM and QM/MM studies on the aromatic hydroxylation of benzene by P450 2C9 indicated that there are two favorable pathways for the initial oxygen addition step corresponding to two different orientations of the benzene ring with respect to the heme group (21, 40, 48). The benzene ring can approach in a “side-on” way, in which the plane of the benzene ring is approximately at right angles to the plane of the heme group, or “face-on,” in which the two planes are roughly parallel. The QM results here correspond to the face-on mode. For dextromethorphan, the docking and MD here showed that only the side-on approach of the methoxy aromatic ring is feasible, at least if one wishes to maintain the hydrogen-bond interaction between the protonated amine group and the acidic Glu216 or Asp301 residues. From the three starting geometries picked from the MD simulation, the QM/MM barriers for sigma complex formation are similar to one another, at 32.9, 33.5, and 36.7 kcal/mol, and are much larger than both the QM value (14.2 kcal/mol) for the same reaction and the QM/MM values for methoxy hydrogen atom abstraction. These results depend only weakly on the basis set.
These results indicate that in cytochrome P450 2D6, aromatic carbon oxidation of dextromethorphan is strongly disfavored. This agrees with experiment, in that the corresponding metabolite is not observed (27). In order to explain the large difference between the QM and QM/MM activation energies, we studied in detail the QM and QM/MM TS structures. The three QM/MM TS structures are very similar, but significantly different from the QM (gas-phase) TS (Fig. 1 and Table S1). The typical QM/MM TS structure shown in Fig. 1 has a shorter O–C and longer Fe–O distance than the QM TS and is hence much more product-like. Another significant difference is the orientation of the phenyl ring of dextromethorphan with respect to the porphyrin ring. The angles between the best-fit planes through the heavy atoms of the porphyrin ring and the heavy atoms of the phenyl ring for both cases are shown in Fig. 1. The QM value is slightly under 60°, indicating that the substrate “bends over” the ferryl group, and is similar to the values obtained previously for TSs for side-on addition of Compound I to benzene (21). This structure presumably optimizes electronic and steric interactions in the TS. The QM/MM TS structures are very different: The corresponding angle is close to 95°, so that the aromatic ring bends away from the ferryl group. This is energetically much less favorable, probably both for steric and electronic reasons. This will destabilize the resulting tetrahedral adduct also, thereby accounting both for the high energy of the TS, and its product-like character.
Fig. 2. shows the orientation of dextromethorphan in the active site cavity of P450 2D6 as derived from MD simulations. This orientation is favored by hydrogen bonding to Glu216 and steric interactions with the amino acid residues around the cavity. Dextromethorphan interacts with Phe120 on one side of the cavity, and Ser304, Ala305, Val308, and Thr 309 on the other side. Although these interactions do not much constrain the motion of the methoxy group, and hence do not impede O-demethylation, they do prevent adoption of the favored orientation for addition to the aromatic ring, hence the very high energies for the aromatic ring addition TSs.
Fig. 2.

Binding orientation of dextromethorphan in the active site of P450 2D6 as derived from QM/MM minimization. The protonated amine in dextromethorphan interacts with Glu216, whereas residues Ser304, Ala305, Val308, and Thr 309 border the cavity from the right.
Conclusions
QM/MM calculations demonstrate a crucial role of the protein in determining reactivity of dextromethorphan in P450 2D6, and (in contrast to MD simulations, docking, and QM model calculations) explain the experimentally observed lack of aromatic hydroxylation. The QM (small model) results on a model P450 Compound I species and the model substrate anisole suggest that both reactions might occur competitively, and indeed some anisole derivatives undergo both types of oxidation with P450 enzymes. The puzzle has been why aromatic carbon oxidation is not observed for dextromethorphan in P450 2D6. Docking of dextromethorphan into the active site of P450 2D6, and MD simulations, suggest that both reactions could be viable, based on the fact that both of the putative reacting sites in the substrate can approach the Compound I ferryl oxygen atom quite readily, and with approximately the correct orientation. In contrast, QM/MM calculations show a very strong preference for O-demethylation, with the aromatic carbon oxidation reaction predicted to have a high activation energy, consistent with experiment. Aromatic oxidation is strongly disfavored in the protein because interactions at the active site prevent the aromatic ring of dextromethorphan from attaining the preferred TS structure. This has the effect of significantly increasing the barrier relative to the ideal gas-phase geometry. The barrier for hydrogen abstraction (the first step of O-demethylation) is also increased from that in the gas phase, for similar reasons, but is significantly lower and is consistent with the experimentally observed reactivity. These QM/MM calculations, exploring oxidation selectivity for a drug compound in a human drug-processing isoform of P450, show that the active site environment plays an important role not only in controlling access of the substrate to the active site, but also in modulating its reactivity.
It is worth noting that sampling of additional QM/MM reaction paths, or calculation of a potential of mean force (54) (e.g., by umbrella sampling MD simulations, which are not feasible at present with DFT QM/MM methods for P450 enzymes) might change the results slightly. In particular, the very large aromatic oxidation barrier obtained here would probably be somewhat reduced. However, the structural argument based on Figs. 1 and 2 provides strong support for a high aromatic oxidation barrier, and the conclusion whereby the latter is significantly higher than the barrier for hydrogen abstraction should not change. Hence, the present observations should be sound in qualitative terms.
These results show that some important selectivity effects are unlikely to be captured by current simple methods to predict metabolites in P450 oxidation. Many methods explicitly or implicitly account for the following two important factors: (i) relative intrinsic reactivity of different sites in the compound [which can be probed computationally by QM model studies (14)] and (ii) proximity of the different sites to the heme iron in the preferred docking/binding mode of the compound in the active site of a given P450 isoform (which can be assessed using docking methods or MD simulations). This reactivity/accessibility paradigm, although often successful, neglects the fact that while a site may be close to the heme iron, active site constraints may prevent it from reacting easily with Compound I. This factor is rarely a major consideration for enzyme catalysis with a natural substrate, because evolution of efficient reaction will ensure that substrate in the active site is able to reach the TS. Prevention of formation of a favorable TS structure by active site constraints does, however, appear to be crucial in understanding of reactivity of dextromethorphan in P450 2D6. It may also be a significant factor in metabolism of other xenobiotics (e.g., drugs) and could be important in understanding reactivity of designed or engineered P450 catalysts. We propose that models for metabolite predictions could be improved by including a third criterion aimed at assessing likely reactivity of the docked structure. One way to achieve this might be to seek to identify a low energy docking pose for the relevant oxidation TS, based on knowledge of TS structure. Models of reactive TSs could be based on QM/MM studies of P450 reactivity (14), and should be very useful for this purpose.
Supplementary Material
Acknowledgments.
We thank Richard Lonsdale for assistance with the QM/MM calculations. J.O. acknowledges receipt of a European Union Marie Curie Fellowship (Project “Modelling CYPs”). A.J.M. is an Engineering and Physical Sciences Research Council Leadership Fellow.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1010194108/-/DCSupplemental.
References
- 1.McFadyen MCE, Melvin WT, Murray GI. Cytochrome P450 enzymes: Novel options for cancer therapeutics. Mol Cancer Ther. 2004;3:363–371. [PubMed] [Google Scholar]
- 2.Zhou SF, Liu JP, Lai XS. Substrate specificity, inhibitors and regulation of human cytochrome P450 2D6 and implications in drug development. Curr Med Chem. 2009;16:2661–2805. doi: 10.2174/092986709788681985. [DOI] [PubMed] [Google Scholar]
- 3.Zhou SF, Zhou ZW, Yang LP, Cai JP. Substrates, inducers, inhibitors and structure-activity relationships of human cytochrome P450 2C9 and implications in drug development. Curr Med Chem. 2009;16:3480–3675. doi: 10.2174/092986709789057635. [DOI] [PubMed] [Google Scholar]
- 4.Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- 5.Bjornsson TD, et al. The conduct of in vitro and in vivo drug-drug interaction studies: A Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos. 2003;31:815–832. doi: 10.1124/dmd.31.7.815. [DOI] [PubMed] [Google Scholar]
- 6.Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34:17–35. doi: 10.1081/dmr-120001387. [DOI] [PubMed] [Google Scholar]
- 7.Kohler GI, et al. Drug-drug interactions in medical patients: Effects of in-hospital treatment and relation to multiple drug use. Int J Clin Pharmacol Ther. 2000;38:504–513. doi: 10.5414/cpp38504. [DOI] [PubMed] [Google Scholar]
- 8.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: A meta-analysis of prospective studies. JAMA J Am Med Assoc. 1998;279:1200–1205. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- 9.Hansch C, Zhang L. Quantitative structure-activity-relationships of cytochrome P450. Drug Metab Rev. 1993;25:1–48. doi: 10.3109/03602539308993972. [DOI] [PubMed] [Google Scholar]
- 10.Roy K, Roy PP. QSAR of cytochrome inhibitors. Expert Opin Drug Metab Toxicol. 2009;5:1245–1266. doi: 10.1517/17425250903158940. [DOI] [PubMed] [Google Scholar]
- 11.Locuson CW, Wahlstrom JL. Three-dimensional quantitative structure-activity relationship analysis of cytochromes P450: Effect of incorporating higher-affinity ligands and potential new applications. Drug Metab Dispos. 2005;33:873–878. doi: 10.1124/dmd.105.004325. [DOI] [PubMed] [Google Scholar]
- 12.de Groot MJ, Ekins S. Pharmacophore modeling of cytochromes P450. Adv Drug Deliver Rev. 2002;54:367–383. doi: 10.1016/s0169-409x(02)00009-1. [DOI] [PubMed] [Google Scholar]
- 13.Maréchal J-D, et al. Insights into drug metabolism by cytochromes P450 from modelling studies of CYP2D6-drug interactions. Brit J Pharmacol. 2008;153:S82–S89. doi: 10.1038/sj.bjp.0707570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 15.Testa B, Balmat A-L, Long A, Judson P. Predicting drug metabolism—An evaluation of the expert system METEOR. Chem Biodiversity. 2005;2:872–885. doi: 10.1002/cbdv.200590064. [DOI] [PubMed] [Google Scholar]
- 16.Shaik S, et al. P450 enzymes: Their structure, reactivity, and selectivity—Modeled by QM/MM calculations. Chem Rev. 2010;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 17.Hritz J, de Ruiter A, Oostenbrink C. Impact of plasticity and flexibility on docking results for cytochrome P450 2D6: A combined approach of molecular dynamics and ligand docking. J Med Chem. 2008;51:7469–7477. doi: 10.1021/jm801005m. [DOI] [PubMed] [Google Scholar]
- 18.Cruciani G, et al. MetaSite: Understanding metabolism in human cytochromes from the perspective of the chemist. J Med Chem. 2005;48:6970–6979. doi: 10.1021/jm050529c. [DOI] [PubMed] [Google Scholar]
- 19.Olsen L, Rydberg P, Rod TH, Ryde U. Prediction of activation energies for hydrogen abstraction by cytochrome P450. J Med Chem. 2006;49:6489–6499. doi: 10.1021/jm060551l. [DOI] [PubMed] [Google Scholar]
- 20.Lonsdale R, Harvey JN, Mulholland AJ. Inclusion of dispersion effects significantly improves accuracy of calculated reaction barriers for cytochrome P450 catalysed reactions. J Phys Chem Lett. 2010;1:3232–3237. [Google Scholar]
- 21.Bathelt CM, Mulholland AJ, Harvey JN. QM/MM modeling of benzene hydroxylation in human cytochrome P450 2C9. J Phys Chem A. 2008;112:13149–13156. doi: 10.1021/jp8016908. [DOI] [PubMed] [Google Scholar]
- 22.Cohen S, Kozuch S, Hazan C, Shaik S. Does substrate oxidation determine the regioselectivity of cyclohexene and propene oxidation by cytochrome P450? J Am Chem Soc. 2006;128:11028–11029. doi: 10.1021/ja063269c. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, et al. Theoretical and experimental studies of the conversion of chromopyrrolic acid to an antitumor derivative by cytochrome P450 StaP: The catalytic role of water molecules. J Am Chem Soc. 2009;131:6748–6762. doi: 10.1021/ja9003365. [DOI] [PubMed] [Google Scholar]
- 24.Ekhart C, Rodenhuis S, Smits PHM, Beijnen JH, Huitema ADR. An overview of the relations between polymorphisms in drug metabolising enzymes and drug transporters and survival after cancer drug treatment. Cancer Treat Rev. 2009;35:18–31. doi: 10.1016/j.ctrv.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5:6–13. doi: 10.1038/sj.tpj.6500285. [DOI] [PubMed] [Google Scholar]
- 26.de Groot MJ, Wakenhut F, Whitlock G, Hyland R. Understanding CYP2D6 interactions. Drug Discovery Today. 2009;14:964–972. doi: 10.1016/j.drudis.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Schmider J, Greenblatt DJ, Fogelman SM, von Moltke LL, Shader RI. Metabolism of dextromethorphan in vitro: Involvement of cytochromes P450 2D6 and 3A3/4, with a possible role of 2E1. Biopharm Drug Dispos. 1997;18:227–40. doi: 10.1002/(sici)1099-081x(199704)18:3<227::aid-bdd18>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 28.Flanagan JU, et al. Phe120 contributes to the regiospecificity of cytochrome P450 2D6: Mutation leads to the formation of a novel dextromethorphan. Biochem J. 2004;380:353–360. doi: 10.1042/BJ20040062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daly J. Metabolism of acetanilides and anisoles with rat liver microsomes. Biochem Pharmacol. 1970;19:2979–2993. doi: 10.1016/0006-2952(70)90084-5. [DOI] [PubMed] [Google Scholar]
- 30.Ohi H, Takahara E, Ohta S, Hirobe M. Effects of oxygen concentration on the metabolism of anisole homologs by rat-liver microsomes. Xenobiotica. 1992;22:1329–1337. doi: 10.3109/00498259209053161. [DOI] [PubMed] [Google Scholar]
- 31.Bray HG, James SP, Thorpe WV, Wasdell MR. The metabolism of ethers in the rabbit. 1. Anisole and diphenyl ether. Biochem J. 1953;54:547–551. [PMC free article] [PubMed] [Google Scholar]
- 32.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 33.Lonsdale R, Harvey JN, Mulholland AJ. Compound I reactivity defines alkene oxidation selectivity in cytochrome P450cam. J Phys Chem B. 2010;114:1156–1162. doi: 10.1021/jp910127j. [DOI] [PubMed] [Google Scholar]
- 34.Altun A, Guallar V, Friesner RA, Shaik S, Thiel W. The effect of heme environment on the hydrogen abstraction reaction of camphor in P450cam catalysis: A QM/MM study. J Am Chem Soc. 2006;128:3924–3925. doi: 10.1021/ja058196w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groves JT, McClusky GA. Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J Am Chem Soc. 1976;98:859–861. [Google Scholar]
- 36.Ogliaro F, et al. A model “rebound” mechanism of hydroxylation by cytochrome P450: Stepwise and effectively concerted pathways, and their reactivity patterns. J Am Chem Soc. 2000;122:8977–8989. [Google Scholar]
- 37.Jerina DM, Daly JW, Witkop B, Zaltzman-Nirenberg P, Udenfriend S. The role of arene oxide-oxepin systems in the metabolism of aromatic substrates. III. Formation of 1,2-naphthalene oxide from naphthalene by liver microsomes. J Am Chem Soc. 1968;90:6525–6527. doi: 10.1021/ja01025a058. [DOI] [PubMed] [Google Scholar]
- 38.Korzekwa KR, Swinney DC, Trager WF. Isotopically labeled chlorobenzenes as probes for the mechanism of cytochrome P-450 catalyzed aromatic hydroxylation. Biochemistry. 1989;28:9019–9027. doi: 10.1021/bi00449a010. [DOI] [PubMed] [Google Scholar]
- 39.Rietjens IMCM, Soffers AE, Veeger C, Vervoort J. Regioselectivity of cytochrome P-450 catalyzed hydroxylation of fluorobenzenes predicted by calculated frontier orbital substrate characteristics. Biochemistry. 1993;32:4801–4812. doi: 10.1021/bi00069a015. [DOI] [PubMed] [Google Scholar]
- 40.de Visser SP, Shaik S. A proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome P450 enzymes. J Am Chem Soc. 2003;125:7413–7424. doi: 10.1021/ja034142f. [DOI] [PubMed] [Google Scholar]
- 41.New York: Schrödinger, LLC; 2005. Jaguar. Version 6.0. [Google Scholar]
- 42.Rowland P, et al. Crystal structure of human cytochrome P450 2D6. J Biol Chem. 2006;281:7614–7622. doi: 10.1074/jbc.M511232200. [DOI] [PubMed] [Google Scholar]
- 43.MacKerell AD, Jr, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 44.Brooks BR, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bathelt CM, Zurek J, Mulholland AJ, Harvey JN. Electronic structureof Compound I in human isoforms of cytochrome P450 from QM/MM modeling. J Am Chem Soc. 2005;127:12900–12908. doi: 10.1021/ja0520924. [DOI] [PubMed] [Google Scholar]
- 46.Vorobyov I, et al. Additive and classical drude polarizable force fields for linear and cyclic ethers. J Chem Theory Comput. 2007;3:1120–1133. doi: 10.1021/ct600350s. [DOI] [PubMed] [Google Scholar]
- 47.Zurek J, Bowman AL, Sokalski WA, Mulholland AJ. MM and QM/MM modeling of threonyl-tRNA synthetase: Model testing and simulations. Struct Chem. 2004;15:405–414. [Google Scholar]
- 48.Bathelt CM, Ridder L, Mulholland AJ, Harvey JN. Mechanism and structure-reactivity relationships for aromatic hydroxylation by cytochrome P450. Org Biomol Chem. 2004;2:2998–3005. doi: 10.1039/B410729B. [DOI] [PubMed] [Google Scholar]
- 49.Ponder JW. TINKER: Software Tools for Molecular Design. Saint Louis, MO: Ponder Lab; 2003. Version 4.0. [Google Scholar]
- 50.Harvey JN. Spin-forbidden CO ligand recombination in myoglobin. Faraday Discuss. 2004;127:165–177. doi: 10.1039/b314768a. [DOI] [PubMed] [Google Scholar]
- 51.Field MJ, Albe M, Bret C, Proust-De Martin F, Thomas A. The dynamo library for molecular simulations using hybrid quantum mechanical and molecular mechanical potentials. J Comput Chem. 2003;21:1088–1100. [Google Scholar]
- 52.Paine MJI, et al. Residues glutamate 216 and aspartate 301 are key determinants of substrate specificity and product regioselectivity in cytochrome P450 2D6. J Biol Chem. 2003;278:4021–4027. doi: 10.1074/jbc.M209519200. [DOI] [PubMed] [Google Scholar]
- 53.Schyman P, Usharani D, Wang Y, Shaik S. Brain chemistry: How does P450 catalyze the O-demethylation reaction of 5-methoxytryptamine to yield serotonin? J Phys Chem B. 2010;114:7078–7089. doi: 10.1021/jp1008994. [DOI] [PubMed] [Google Scholar]
- 54.Sharma PK, Chu ZT, Olsson MHM, Warshel A. A new paradigm for electrostatic catalysis of radical reactions in vitamin B12 enzymes. Proc Natl Acad Sci USA. 2007;104:9661–9666. doi: 10.1073/pnas.0702238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.