

Abstract
Cyclic nucleotide-sensitive ion channels, known as HCN and CNG channels, are activated by binding of ligands to a domain (CNBD) located on the cytoplasmic side of the channel. The underlying mechanisms are not well understood. To elucidate the gating mechanism, structures of both the ligand-free and -bound CNBD are required. Several crystal structures of the CNBD from HCN2 and a bacterial CNG channel (MloK1) have been solved. However, for HCN2, the cAMP-free and -bound state did not reveal substantial structural rearrangements. For MloK1, structural information for the cAMP-free state has only been gained from mutant CNBDs. Moreover, in the crystal, the CNBD molecules form an interface between dimers, proposed to be important for allosteric channel gating. Here, we have determined the solution structure by NMR spectroscopy of the cAMP-free wild-type CNBD of MloK1. A comparison of the solution structure of cAMP-free and -bound states reveals large conformational rearrangement on ligand binding. The two structures provide insights on a unique set of conformational events that accompany gating within the ligand-binding site.
Keywords: NMR solution structure, apo state, ligand removal method, potassium channel
Ion channels activated by cyclic nucleotides play key roles in neuronal excitability and sensory signaling. They belong to two subfamilies: cyclic nucleotide-gated (CNG) channels, and hyperpolarization-activated and cyclic nucleotide-gated (HCN) channels (1–4). CNG channels are voltage independent and require cyclic nucleotides to open, whereas HCN channels are activated by hyperpolarization, and their activity is modulated by cyclic nucleotides. In both channels, ligand binding to an intracellular cyclic nucleotide-binding domain (CNBD) promotes the opening of the channel by conformational changes in the CNBD that propagate via a connecting linker (C linker) to transmembrane segment S6. However, the activation mechanisms are poorly understood in part because the nature of the pore gate is not known precisely (4).
Understanding of the structural rearrangements underlying gating has been greatly advanced by several crystal structures of isolated CNBDs from HCN channels (5–8) and a prokaryotic K+-selective CNG channel, designated MloK1 (9–11). The MloK1 channel consists of four identical subunits, each encompassing six transmembrane domains (S1–S6), a “GYG” signature sequence for K+ selectivity, and a conserved CNBD. However, the C linker (approximately 20 residues) is much shorter than the C linker of mammalian CNG and HCN channels (approximately 80 residues) that is important for relaying the binding signal from the CNBD to the channel gate (12–17). In the crystal, the CNBDs from HCN channels are arranged in tetramers, where neighboring C linkers contribute most contacts between subunits (6, 8), whereas the MloK1 CNBD forms a dimer; the dimer interface between the short C linker has been proposed to be involved in channel gating (9, 11). The solution structure solved by nuclear magnetic resonance (NMR) spectroscopy of the monomeric MloK1 CNBD in complex with cAMP (18) is similar to that of the monomers in the dimer crystal (11), except for the C linker, which forms the dimer interface in the crystal structure. Therefore, the C-linker contact in the crystal might have been enforced by packing of dimers. Moreover, in a cryoelectron microscopy structure of the full-length MloK1 channel, the CNBDs appear as independent domains separated by discrete gaps, suggesting that CNBDs are not interacting with each other (19). Furthermore, ligand binding to both the tetrameric MloK1 channel and the monomeric CNBD occurs noncooperatively with similar affinity (20), suggesting that the MloK1 CNBDs are functionally and structurally independent of each other.
To elucidate the mechanism of channel gating in MloK1, knowledge of the CNBD structure in the ligand-free and -bound state is required. Because of the high affinity, cAMP copurifies with the protein and even extensive dialysis fails to remove bound ligand quantitatively (9, 11, 18, 20). For this reason, crystal structures of ligand-free CNBD were obtained from R348A and R307W mutants (9, 11). Both arginine residues are directly involved in cAMP binding and mutation of either one drastically lowers cAMP binding, and thereby allows to prepare ligand-free protein. However, considering the crucial importance of these residues for ligand binding, the mutant structure might not reflect the true wild-type structure and the conformational changes on ligand binding might be altered. We have, therefore, developed a procedure to prepare cAMP-free wild-type protein in quantities sufficient for structure determination. Here, we report the solution structure of the ligand-free CNBD and compare it with the solution structure of the cAMP-bound CNBD and with crystal structures.
Results
Solution Structure of the Wild-Type cAMP-Free CNBD.
In this study, we have determined the solution structure of the native unliganded CNBD of the MloK1 channel using NMR spectroscopy. The isolated CNBD shows high affinity for cAMP (KD = 107 nM) (20) and copurifies with the ligand, which cannot be removed by size-exclusion chromatography or extensive dialysis (9, 11, 18, 20). For this reason, tightly bound cAMP was either removed by protein denaturation and refolding (20) or by a competition assay (9, 20). The resulting protein sample was free of cAMP, but the final yield was low. For structure determination by NMR spectroscopy or X-ray crystallography, however, large amounts of pure protein are required, preferentially prepared under nondenaturing conditions. We have therefore developed a procedure to prepare wild-type cAMP-free and -bound MloK1 CNBD in quantities sufficient for structure determination. The procedure is based on extensive washing of matrix-bound CNBD–GST fusion protein followed by ion-exchange chromatography that separated cAMP-free from cAMP-bound protein (Fig. S1). The CNBD eluted as two peaks, representing the pure monomeric forms of the cAMP-free and -bound CNBD (Fig. 1). The final yield was roughly 7 mg/l cell culture of [U-15N, 13C], or [U-15N] isotopically enriched protein. The absence of the ligand was verified by the 260/280 nm absorbance ratio (values of approximately 0.9 for cAMP-free and approximately 2 for cAMP-bound CNBD), and by NMR spectra. 1H, 15N, and 13C chemical-shift assignment of the unliganded CNBD was almost complete (97% of the backbone and 90% of the side chain CH) using multidimensional heteronuclear NMR spectroscopy with [U-15N]- or [U-15N, 13C] labeled CNBD (21). Assignments have been deposited in the BioMagResBank (accession number 16628). All spectra of the cAMP-free CNBD showed a single set of resonance signals, indicating that the protein is pure and adopts a single conformation. Moreover, the linewidths of the NMR spectra also reflect a monomeric form.
Fig. 1.

Purification and characterization of the MloK1 CNBD protein. (A) Recombinant MloK1 CNBD-GST fusion protein was expressed in Escherichia coli and purified by affinity chromatography (M: marker; lane 1: column input; lane 2–3: nonbound; lane 4–6: elution after major wash and thrombin cleavage; lane 7–9: double amount of protein as in lane 4–6). Upper and lower arrows indicate CNBD-GST fusion and cleaved CNBD protein, respectively. The sizes of the molecular weight markers (M) are indicated. The SDS-polyacrylamide gel was stained with Coomassie blue. (B) Size-exclusion chromatogram of CNBD protein desalting step (10 ml sample volume) after affinity chromatography. The CNBD protein was loaded on a HiPrep 26/10 desalting column and the desalting step was monitored at 280 nm (blue line) and 260 nm (red line). The desalting step was followed by measuring the conductivity (green dashed line). (C) Final purification of cAMP-free and -bound CNBD protein by cation-exchange chromatography. A Mono S HR 5/5 cation-exchange column was used. The absorption was monitored at 280 nm (blue line) and 260 nm (red line); the conductivity is shown in green (dashed line). cAMP-free and cAMP-bound CNBD protein were separately eluted at 6.92 mS/cm and 9.22 mS/cm, respectively.
We carried out 15N relaxation experiments to characterize the dynamics and the oligomeric state of the cAMP-free CNBD (Fig. S2). From 15N average longitudinal and transverse relaxation rates of 1.01 ± 0.05 s-1 and 13.76 ± 0.10 s-1, respectively, an isotropic rotational correlation time of 8.4 ns was derived. This value is consistent with a CNBD monomer. Nuclear Overhauser enhancement (NOE) cross-peak assignments were obtained by an iterative procedure using a combination of manual and automatic approaches. For the structure calculations, a total of 2,380 intramolecular NOE distance constraints, including 632 long-range NOEs, have been evaluated. In addition, 254 dihedral constraints were derived from chemical-shift data and used for structure calculations. A final ensemble of 15 NMR structures with the lowest CYANA target functions were used to determine the cAMP-free CNBD structure. None of the 15 structures violated NOE distances more than 0.019 nm. No dihedral-angle constraint was violated more than 4°. The vast majority of residues (89.9%) was found in the most favored regions of the Ramachandran plot. The root-mean-squared (rms) displacement of the structure ensemble compared to the average structure was 0.037 nm for backbone and 0.070 nm for all heavy atoms. These data demonstrate that the structure is well defined (Fig. 2A). Only the very N- and C-terminal residues (Q216 to R220 and A351 to A355) show higher values of rms displacement and a lower number of NOE-derived distance constraints (Fig. S3). A summary of the distance constraints and structural statistics is given in (Table S1).
Fig. 2.

Solution structure of the wild-type cAMP-free CNBD. (A) Superposition of backbone traces of a family of 15 NMR structures with the lowest CYANA target function. Backbone atoms of the amino- and carboxy-terminal ends (residues Q216-R220 and A351-A355, respectively) are not shown and were not used for least-square superposition of the structures. (B) Ribbon representation of wild-type cAMP-free CNBD. Secondary structure elements are labeled.
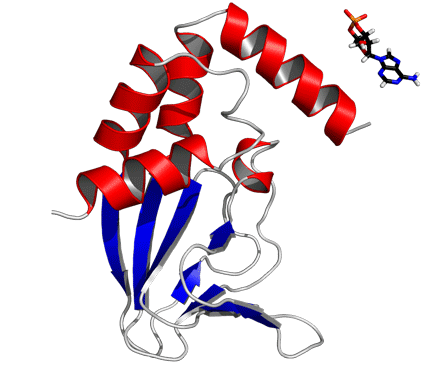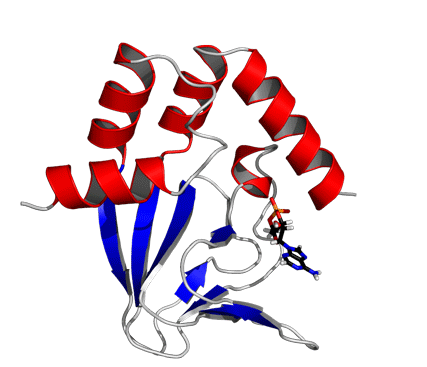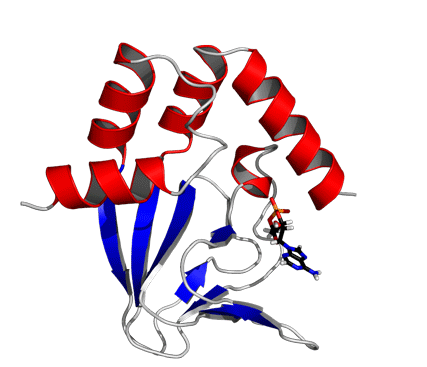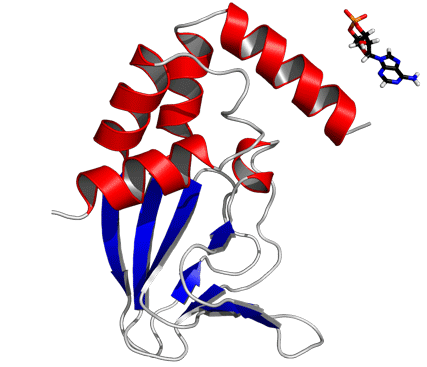
The cAMP-free CNBD structure (Fig. 2B) features an antiparallel β roll with a short internal α helix, known as phosphate-binding cassette (PBC) similar to other CNBDs from HCN channels (6–8), the cAMP-dependent protein kinase A (PKA) (22, 23), the exchange protein activated by cAMP (Epac) (24), and the catabolite activator protein (CAP) (25), which lacks, however, the internal α helix. This core element consists of eight antiparallel β strands (β1: R252–V256, β2: V261–C263, β3: R271–E277, β4: V280–V282, β5: V288–L290, β6: F295–F296, β7: V311–A313, β8: V317–H323) and one α helix (PBC helix: E298–I302). The core element is topped by a region of four α helices (αA’: F223–A231, αA: P241–A250, αB: S324–S333, αC: P335–R349).
In the cAMP-bound CNBD structure, the binding site is formed by the β roll and the PBC helix that provide interactions with the phosphate and ribose moieties of cAMP. The C-terminal helix αC is placed like a “lid” above the binding pocket and thereby stabilizes the complex. In the cAMP-bound CNBD structure, 25 intermolecular NOE distance constraints between the CNBD and cAMP have been derived. In the cAMP-free form, significant changes in chemical shifts and NOE pattern for atoms located in the binding site and in contact with cAMP have been observed (Fig. S4).
Titration of the Ligand-Free CNBD with cAMP.
To verify that the cAMP-free CNBD is functional, cAMP was stepwise added to cAMP-free CNBD and (15N-1H)-HSQC spectra were recorded (Fig. 3A). During the initial steps of titration, a second set of NMR signals appeared originating from cAMP-bound CNBD (Fig. 3B). At an equimolar ratio of CNBD and cAMP, NMR signals from the cAMP-free protein disappeared, demonstrating that the protein is homogenous with respect to binding of cAMP. Moreover, the HSQC spectrum recorded after cAMP titration is indistinguishable from HSQC spectra of CNBD samples that copurified with cAMP (Fig. 3C) (18).
Fig. 3.

Titration of cAMP-free CNBD with cAMP. (A) 2D (15N - 1H)-HSQC spectrum of 0.2 mM cAMP-free [U - 15N]-labeled CNBD (T = 25 °C, 10 mM Tris/HCl, pH 7, 100 mM sodium chloride, 0.2 mM EDTA, 0.02% (w/v) sodium azide, 5% (v/v) 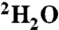 ). (B) 2D (15N - 1H)-HSQC spectrum recorded in the presence of 50 μM cAMP. A second set of NMR signals emerged that originate from CNBD that has bound to cAMP. Two resonance signals characteristic for some amides are highlighted by brackets. Resonance signals of the cAMP-free CNBD spectrum are shown in red. (C) 2D (15N - 1H)-HSQC spectrum recorded in the presence of 250 μM cAMP. NMR signals originating from cAMP-free CNBD disappeared, proving that all CNBD protein has bound to cAMP. The spectrum is indistinguishable from spectra that were recorded using samples where cAMP copurified with CNBD (shown in blue) and that were reported previously (18). (D) Chemical-shift differences of resonance signals between cAMP-free and -bound CNBD; the most pronounced perturbations are highlighted by blue arrows. The chemical-shift differences were mapped on ribbon diagrams of the cAMP-free (E) and cAMP-bound (F) solution structures. cAMP-induced changes in both spectrum dimensions (
). (B) 2D (15N - 1H)-HSQC spectrum recorded in the presence of 50 μM cAMP. A second set of NMR signals emerged that originate from CNBD that has bound to cAMP. Two resonance signals characteristic for some amides are highlighted by brackets. Resonance signals of the cAMP-free CNBD spectrum are shown in red. (C) 2D (15N - 1H)-HSQC spectrum recorded in the presence of 250 μM cAMP. NMR signals originating from cAMP-free CNBD disappeared, proving that all CNBD protein has bound to cAMP. The spectrum is indistinguishable from spectra that were recorded using samples where cAMP copurified with CNBD (shown in blue) and that were reported previously (18). (D) Chemical-shift differences of resonance signals between cAMP-free and -bound CNBD; the most pronounced perturbations are highlighted by blue arrows. The chemical-shift differences were mapped on ribbon diagrams of the cAMP-free (E) and cAMP-bound (F) solution structures. cAMP-induced changes in both spectrum dimensions (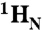 and 15N) were classified according to the following equation:
and 15N) were classified according to the following equation: 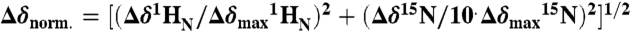 . The cAMP molecule is shown in stick representation.
. The cAMP molecule is shown in stick representation.
Comparison of the (15N-1H)-HSQC spectra of cAMP-free and -bound CNBD shows that chemical shifts of more than half of the resonances changed on binding (Fig. 3D). The chemical-shift differences were mapped on ribbon diagrams of the cAMP-free and -bound structures (Fig. 3 E and F). Although differences are not localized to a particular region, changes are most prominent in the helical parts rather than the β roll. In particular, a cluster of residues in the cAMP-binding site making contact with cAMP and regions nearby display pronounced differences of their chemical shifts. These differences primarily reflect changes in the chemical environment induced by cAMP. Furthermore, a multitude of resonances from residues located further away from the cAMP-binding site undergo significant changes in their chemical shifts (Fig. 3D; e.g., R249, A231, L235, L301, L330, and E336).
Superposition of cAMP-bound and -free CNBD shows that the β roll core is almost identical with an rms displacement of only 0.084 nm (Fig. 4). In contrast, the helical parts show substantial rearrangements between the two forms (rms displacement of 0.271 nm). The orientation of all helices, in particular αA’, αB, and αC, with respect to the β roll differs remarkably in the two structures, whereas the position of αB and αC relative to each other is similar: The angle between the two helices in the cAMP-free and -bound state is 65.5° and 60.5°, respectively (rms displacement of 0.053 nm for αB-turn-αC). This almost static behavior is likely mediated by hydrophobic interactions between residues F327 and F341 in αB and αC, respectively (Fig. S5). In the cAMP-free state, a network of mainly hydrophobic interactions is observed within the helical region and those parts of the β roll that form the interface with the helical region [including residues in αA’ (W227, L229, V230, A231), loop region between αA’ and αA (L235, F236, L239), αA (V243, L244, I247, V248), PBC helix (L301), αB (F327, L330), αC (F341, I340, I337), β3 (M272, F274, V276), and β8 (L322)]. These interactions are also present in the cAMP-bound state, in addition to the hydrophobic interactions of residues in the 310-helix (V233), αA (L251), PBC helix (I302), αB (M329), αC (A345), β1 (A253), β6 (F295), and β8 (L320).
Fig. 4.

Structure comparison of cAMP-free and -bound CNBD. Stereoview of the cAMP-free and -bound CNBD structure overlay. The helical portion of the cAMP-bound and cAMP-free CNBD is shown in white and red, respectively. β strands in both structures are shown in blue, and the N-terminal loop region in the cAMP-free structure is highlighted in yellow. Secondary structure elements are labeled (label “0000C” stands for “cAMP-bound state”; label “0000F” stands for “cAMP-free state”).
The movement of αC is accompanied by a substantial displacement of other helical parts right up to the N-terminal helix αA’, and residues in the helical parts undergo a significant change in their chemical shift (e.g., A231, L235, K238, R249, G269, M299, L301, L330, E336, and A338; Fig. 3 D–F). For example, the position of two residues D222 and P234 located in the αA’ and loop region, respectively, shifted by 0.8 nm relative to their position in the cAMP-bound structure. In the cAMP-bound state, a short 310-helix formed by residues P234 to K238 is located between αA’ and αA. This 310-helix is adjacent to αC. In particular, L235 and K238 of the 310-helix undergo hydrophobic interactions with I337, I340, and F341 in αC. This short 310-helix is absent in the cAMP-free state. However, residues in this loop region show heteronuclear NOE values close to 0.8, indicating a rather rigid region. Furthermore, residues L235, F236, K238, and L239 in this loop region show hydrophobic interactions similar to residues I337, I340, and F341 located in αC. These interactions might be responsible for the fixed orientation of αC.
The cAMP-Free CNBD Is Rigid.
Hydrogen exchange of protons can provide valuable information on a protein’s local flexibility, structure, and function (26–28). The proton-deuterium exchange rates provide information on slow internal motions (slower than minutes) that permit breaking of hydrogen bonds whereby labile protons can exchange with the solvent (29). The exchange rates of the cAMP-free and -bound state have been determined by a set of sequentially recorded (15N-1H)-HSQC spectra (Fig. S6).
In both states, residues in the β roll showed slow exchange rates, indicating that the β roll is structurally rigid (Fig. S6). Calculated protection factors indicate that parts of β1, β3, and β8 are even more protected in the cAMP-free compared to the cAMP-bound state (Fig. S6 E and G). This might be due to the spatial rearrangement of αA’, which covers more of the β sheet in the cAMP-free state. However, exchange rates of the helical parts revealed major differences. In the cAMP-free state, only residues in the αA helix exhibited slow exchange rates (Fig. S6 A, C, and G), perhaps reflecting a specific interaction between αA’ and αA, whereby αA becomes shielded from the solvent. Furthermore, αA and the β roll present the structurally most invariant parts of the CNBD. Amide protons of all other helices were already exchanged before recording of the first HSQC spectrum (600 s after dissolving protein in D2O). In contrast, in the cAMP-bound state, residues forming the cAMP-binding pocket displayed slower exchange rates (e.g., E298, A301, R307, V282, V288, L290, and V311; Fig. S6 B, D, and F). Additionally, amide protons of a cluster of residues in the hinge region (αB), in αC, and in the PBC helix exhibited slow exchange rates (e.g., F327, M329 to C331 and A338 to R342). However, helical propensities calculated from secondary 13C chemical shifts do not differ substantially for both states (Fig. S5C).
Furthermore, we carried out heteronuclear steady-state {1H}-15N-NOE experiments to characterize the dynamic behavior of the cAMP-free CNBD on a subnanosecond time scale (30, 31). Heteronuclear NOE values are sensitive to dynamics of the local environment. Positive values close to 0.8 are expected in the absence of rapid internal motions of protein backbone N-H bond vectors, whereas values < 0.8 reveal fast internal motions; for highly mobile residues, NOE values even might become negative.
Most residues of the cAMP-free CNBD show heteronuclear NOE values close to 0.8 (with an average value of 0.75 for residues G221 to G350), indicating a rigid fold (Fig. S2). However, residues of the very N- and C-terminal regions (Q216 to R219 and G350 to A355) exhibit higher values of average local displacement and a lower number of NOE-derived distance constraints (Fig. S3). The small heteronuclear NOE values for these residues support the idea that the N- and C-terminal ends (five residues each) are more flexible.
Discussion
Previously, crystal structures were reported from two CNBD mutants R307W and R348A. These two Arg residues undergo key interactions with the ligand and their replacement lowers the cAMP affinity by > 100-fold (9, 11, 20). Arg 307 makes Coulomb contact with the charged phosphoryl group of cAMP, which perhaps represents the first step of binding. During or after seating of the ligand in the β roll, the αC helix repositions and stacks on top of the cAMP purine ring to cap the mouth of the cavity, and the side chain of R348 undergoes an ion-pair interaction with E298 in the PBC. In a total of six crystal structures from these two mutants, the αC position varies considerably over large distances and is even disordered or unwound (9). The different structures might either reflect unique crystal environments or a conformational space that is explored by αC movements, or might represent functional intermediates during repositioning of αC. Moreover, all crystal structures of the CNBDs in the cAMP-free and -bound state (9, 11) reveal a dimer in the asymmetric unit. In solution, however, the CNBD exists as a monomer (18). The interactions within the dimer interface might also induce changes in the β roll or the αB and αC helices. Given the functional importance of the two Arg residues, the position of the αC helix, and the potential functional implications of the C-linker interface, it seems necessary to resolve the structure of the wild-type CNBD in either state by an independent technique.
The overall fold of the monomeric, cAMP-free state reported here is similar to the monomer structure in the dimer crystal (Fig. 5). The backbone coordinates for residues in the β roll are virtually identical (rms displacement value of approximately 0.07 nm). However, three major differences in αA’ and αC helices and the β4/β5 loop come to light when comparing the solution and crystal coordinates. First, in the solution structure, the N-terminal αA’ helix is straight rather than bent in the crystal structures. Consequently, the rms displacement values are largest for this segment of the superimposed solution and crystal structures (0.17 and 0.22 nm). A similar observation has been made for the cAMP-bound state (18). Second, the position of αC differs remarkably (rms displacement values for αC backbone coordinates of 0.24 to 0.57 nm between solution and all crystal structures). The ensemble of αC positions has been attributed to a looser conformation of the cAMP-free state and the enhanced flexibility might allow αC to sample a conformational space during binding. In contrast to the various crystal structures, the αC helix in the solution structure is rigid and adopts a single orientation. This conclusion is supported by various NMR spectroscopic parameters, including 15N relaxation measurements (Fig. S2), a large number of long-range NOEs between αC and other parts of the molecule (Fig. S5B), and a high helical propensity as calculated from secondary 13C chemical shifts (Fig. S5C).
Fig. 5.

Comparison of the monomeric cAMP-free wild-type solution structure with structures of mutant dimers in the crystal. (A) Superposition of backbone traces for all cAMP-free structures [wild-type PDB ID code 2KXL, R348A mutant dimer (11), molecules A and B, PDB ID code 1U12, R307W mutant dimer (9), molecules A–D, PDB ID code 3CO2]. (B) Superposition of ribbon representations for all cAMP-free structures. The least-square superposition of the structures was done using the backbone atoms of the β roll strands.
In a similar vein, the β4/β5 hairpin in the crystal adopts a single conformation in the cAMP-bound state but adopts multiple orientations in the absence of the ligand (9). The β4/β5 hairpin is situated near the mouth of the binding pocket. On binding, the hairpin closes around the ligand. Thus, cAMP is sandwiched between R348 and three hydrophobic residues in the hairpin that can undergo van der Waals interaction with the purine ring of cAMP. However, in the solution structure of the cAMP-free state, the hairpin adopts a single conformation that is not significantly different from the conformation in the liganded state (Figs. 4 and 5).
We tentatively conclude that the conformations of some exposed regions are particularly sensitive to packing in the crystal and, therefore, might reflect different crystal environments rather than a functionally important conformational ensemble that becomes “frozen” in the crystal. A case in point is the HCN2 channel. The structures of the CNBD in the free and bound state of HCN2 (7, 8) are almost identical except for a coil-to-helix transition (αF’). This transition resembles the formation of the 310-helix in MloK1 CNBD, which occupies a similar position as αF’. However, rearrangements of the helical parts were not observed (Fig. S7). This surprising finding might result from crystal contacts in the αC helix region that lock the helix in a nonphysiological conformation (7). From experiments using transition metal ion FRET, it was argued that the αC helix is partially unstructured in the cAMP-free state and adopts a more α helical structure on ligand binding (7). In CAP, on cAMP binding, a coil-to-helix transition occurs that extends αC by three turns. (32). For the MloK1 channel, there is no evidence that the αC segment undergoes major changes either in secondary structure or length.
The solution structures of the cAMP-free and -bound CNBD, allow to reconstruct, unbiased by mutations or crystal contacts, the sequence of dynamical events that propagate through the binding fold on cAMP binding. There are commonalities and differences among different CNBDs. All CNBDs share a common fold consisting of a β roll and an α helical bundle. A common feature is also a lid region that caps the binding cavity of the β roll. In all CNBDs, except Epac, the αC helix forms the lid. Epac is lacking an αC helix; instead three β strands form the lid. Another commonality might be the structural rearrangements that move the hinge-lid region closer to the binding pocket. The PBC, αB, and αC put up a cavity that is filled with hydrophobic side chains that probably fix the relative position of these three helical elements (Fig. S5A). These hydrophobic residues (L301, F327, and F341) are highly conserved in almost all CNBDs. On ligand binding, the PBC is displaced, allowing the hydrophobic residues to enter the void, which gives rise to a tilting movement of the hinge-lid region. This mechanism has been first proposed for Epac (24, 33, 34). These structural rearrangements, however, are differently relayed to the effector domains that are located either N- or C-terminally of the CNBD. In ion channels, the signal is transferred via an N-terminal helix bundle to the transmembrane segment S6, involving the body movement of αB/αC toward the β roll, the formation of a short 310-helix in the loop between αA’ and αA, a sliding movement of αA’ across the surface of the β roll, and displacement of αA relative to αA’ (Movie S1). Although these structural rearrangements may serve as guideline, the “gating movements” in the CNBDs of other CNG or HCN channels might be different. Crucial amino acids like R348 in αC or the F327/F341 pair are absent in classic CNG channels. Moreover, classic CNG and HCN channels are cooperatively activated by ligand or voltage, respectively. The allosteric mechanisms involve intersubunit contacts between long C linkers (4); the C-linker interface seems to be less important in MloK1 or CNGK channels that are activated noncooperatively (4, 20, 35). In contrast, in CAP, Epac, and PKA the signal is relayed to the DNA-binding domain or catalytic sites, respectively, that are located in the C terminus.
To delineate the sequence of events, conformational movements need to be resolved with high spatio-temporal accuracy. One way to provide such structural information is the use of double electron-electron resonance (DEER) technique (36, 37). If combined with rapid-mixing techniques, DEER could provide a distance map of selected residues along the reaction pathway and, thereby, reveal the pattern of conformational movements. Thus CNG channels hold great promise to further our understanding of the interplay between ligand binding and gating.
Materials and Methods
Protein Preparation.
Isotopically labeled MloK1 CNBD samples for NMR studies were prepared as detailed described in SI Text. NMR samples contained 0.5 mM [U-15N] or [U-15N, 13C] labeled CNBD in aqueous solution.
NMR Spectroscopy, Data Evaluation, and Structure Calculation.
A detailed description about NMR experiments, data evaluation and structure calculation is provided in SI Materials and Methods. In brief, the procedures were as follows. All NMR experiments were performed at 298 K on Varian UnityINOVA and VNMRS instruments, equipped with a cryogenic Z-axis pulse-field-gradient (PFG) triple resonance probe at proton frequencies of 800 and 600 MHz. Backbone and side chain assignments of cAMP-free and -bound CNBD were obtained using heteronuclear standard experiments as reported previously (21, 38). All NMR spectra were processed using NMRPipe (39) and evaluated in CARA (40). Nuclear Overhauser Effect (NOE) distance constraints for structure calculations were derived from three- and two-dimensional NOESY experiments. NOE cross-peak assignments were obtained by an iterative procedure using a combination of manual and automatic steps. CARA was used to evaluate NOE spectra and to manually assign nearly all of the apparently unambiguous NOE distance constraints. NOE cross-peak intensities were classified as strong, medium, and weak, corresponding to upper limit distance constraints of 2.7, 3.8, and 5.5 Å, respectively. For NOE cross peaks involving methyl groups upper limit distance constraints of 2.9, 4.0, and 5.7 Å were used. Structure calculation was done with CYANA (41) using the standard protocol for simulated annealing. Using manually assigned NOEs an initial fold of the protein was calculated. Additional NOEs were automatically assigned in an iterative approach using ATNOS/CANDID (42, 43) algorithms in combination with CYANA. Dihedral-angle constraints and helical propensities were determined by TALOS (44, 45). Geometry of the resulting structures, structural parameters, and secondary structure elements were analyzed and visualized using the following programs: MOLMOL (46), PyMOL (47), MolProbity (48), and PROCHECK (49).
15N longitudinal (R1) and transverse (R2) relaxation rates, together with the steady-state {1H}-15N-NOE, were recorded at 298 K on a [U-15N] labeled protein sample using standard methods (30) at 600 MHz and 800 MHz proton frequency. The correlation time was determined for an isotropic tumbling model using the TENSOR2 (50) package. NMR titration experiments were carried out at 298 K using [U-15N] labeled unliganded MloK1 CNBD sample. A set of (15N-1H)-HSQC spectra were recorded with increasing ligand concentrations. Proton-deuterium exchange experiments were performed at 298 K. Slowly exchanging amide protons were identified by sequently recording a set of two-dimensional SOFAST (15N-1H)-HMQC (51) spectra after dissolving the freeze-dried CNBD samples in D2O. The last spectrum was recorded after 14 h for the cAMP-free and 24 h for the cAMP-bound CNBD.
Supplementary Material
Acknowledgments.
This study was supported by a fellowship from the International Research School BioStruct to S.S. and a research grant from the Helmholtz–Gemeinschaft (Virtual Institute of Structural Biology) to D.W.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: Resonance assignments and the atomic coordinates for the resulting 15 NMR structures with the lowest target function have been deposited in the BioMagResBank (accession code 16628) and the Protein Data Bank, www.pdb.org (PDB ID code 2KXL), respectively.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1015890108/-/DCSupplemental.
References
- 1.Kaupp UB, Seifert R. Molecular diversity of pacemaker ion channels. Annu Rev Physiol. 2001;63:235–257. doi: 10.1146/annurev.physiol.63.1.235. [DOI] [PubMed] [Google Scholar]
- 2.Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82:769–824. doi: 10.1152/physrev.00008.2002. [DOI] [PubMed] [Google Scholar]
- 3.Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: From molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- 4.Cukkemane A, Seifert R, Kaupp UB. Cooperative and uncooperative cyclic-nucleotide-gated ion channels. Trends Biochem Sci. 2011;36:55–64. doi: 10.1016/j.tibs.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Craven KB, Zagotta WN. CNG and HCN channels: Two peas, one pod. Annu Rev Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. [DOI] [PubMed] [Google Scholar]
- 6.Flynn GE, Black KD, Islas LD, Sankaran B, Zagotta WN. Structure and rearrangements in the carboxy-terminal region of SpIH channels. Structure. 2007;15:671–682. doi: 10.1016/j.str.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taraska JW, Puljung MC, Olivier NB, Flynn GE, Zagotta WN. Mapping the structure and conformational movements of proteins with transition metal ion FRET. Nat Methods. 2009;6:532–537. doi: 10.1038/nmeth.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zagotta WN, et al. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- 9.Altieri SL, et al. Structural and energetic analysis of activation by a cyclic nucleotide binding domain. J Mol Biol. 2008;381:655–669. doi: 10.1016/j.jmb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clayton GM, Altieri S, Heginbotham L, Unger VM, Morais-Cabral JH. Structure of the transmembrane regions of a bacterial cyclic nucleotide-regulated channel. Proc Natl Acad Sci USA. 2008;105:1511–1515. doi: 10.1073/pnas.0711533105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clayton GM, Silverman WR, Heginbotham L, Morais-Cabral JH. Structural basis of ligand activation in a cyclic nucleotide regulated potassium channel. Cell. 2004;119:615–627. doi: 10.1016/j.cell.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 12.Gordon SE, Zagotta WN. Localization of regions affecting an allosteric transition in cyclic nucleotide-activated channels. Neuron. 1995;14:857–864. doi: 10.1016/0896-6273(95)90229-5. [DOI] [PubMed] [Google Scholar]
- 13.Johnson JP, Jr, Zagotta WN. Rotational movement during cyclic nucleotide-gated channel opening. Nature. 2001;412:917–921. doi: 10.1038/35091089. [DOI] [PubMed] [Google Scholar]
- 14.Paoletti P, Young EC, Siegelbaum SA. C-Linker of cyclic nucleotide-gated channels controls coupling of ligand binding to channel gating. J Gen Physiol. 1999;113:17–34. doi: 10.1085/jgp.113.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Chen S, Siegelbaum SA. Regulation of hyperpolarization-activated HCN channel gating and cAMP modulation due to interactions of COOH terminus and core transmembrane regions. J Gen Physiol. 2001;118:237–250. doi: 10.1085/jgp.118.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou L, Siegelbaum SA. Gating of HCN channels by cyclic nucleotides: Residue contacts that underlie ligand binding, selectivity, and efficacy. Structure. 2007;15:655–670. doi: 10.1016/j.str.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zong X, Zucker H, Hofmann F, Biel M. Three amino acids in the C-linker are major determinants of gating in cyclic nucleotide-gated channels. EMBO J. 1998;17:353–362. doi: 10.1093/emboj/17.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schünke S, Stoldt M, Novak K, Kaupp UB, Willbold D. Solution structure of the Mesorhizobium loti K1 channel cyclic nucleotide-binding domain in complex with cAMP. EMBO Rep. 2009;10:729–735. doi: 10.1038/embor.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu PL, et al. The structure of the prokaryotic cyclic nucleotide-modulated potassium channel MloK1 at 16 A resolution. Structure. 2007;15:1053–1064. doi: 10.1016/j.str.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cukkemane A, et al. Subunits act independently in a cyclic nucleotide-activated K(+) channel. EMBO Rep. 2007;8:749–755. doi: 10.1038/sj.embor.7401025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schünke S, Lecher J, Stoldt M, Kaupp UB, Willbold D. Resonance assignments of the nucleotide-free wildtype MloK1 cyclic nucleotide-binding domain. Biomol NMR Assign. 2010;4:147–150. doi: 10.1007/s12104-010-9231-z. [DOI] [PubMed] [Google Scholar]
- 22.Diller TC, Madhusudan, Xuong NH, Taylor SS. Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: Crystal structure of the type II beta regulatory subunit. Structure. 2001;9:73–82. doi: 10.1016/s0969-2126(00)00556-6. [DOI] [PubMed] [Google Scholar]
- 23.Su Y, et al. Regulatory subunit of protein kinase A: Structure of deletion mutant with cAMP binding domains. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- 24.Rehmann H, et al. Structure and regulation of the cAMP-binding domains of Epac2. Nat Struct Biol. 2003;10:26–32. doi: 10.1038/nsb878. [DOI] [PubMed] [Google Scholar]
- 25.Weber IT, Steitz TA. Structure of a complex of catabolite gene activator protein and cyclic AMP refined at 2.5 A resolution. J Mol Biol. 1987;198:311–326. doi: 10.1016/0022-2836(87)90315-9. [DOI] [PubMed] [Google Scholar]
- 26.Hvidt A, Nielsen SO. Hydrogen exchange in proteins. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 27.Jeng MF, Dyson HJ. Comparison of the hydrogen-exchange behavior of reduced and oxidized Escherichia coli thioredoxin. Biochemistry. 1995;34:611–619. doi: 10.1021/bi00002a028. [DOI] [PubMed] [Google Scholar]
- 28.Taylor SS, et al. PKA: A portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259–269. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 29.Wagner G, Wüthrich K. Amide protein exchange and surface conformation of the basic pancreatic trypsin inhibitor in solution. Studies with two-dimensional nuclear magnetic resonance. J Mol Biol. 1982;160:343–361. doi: 10.1016/0022-2836(82)90180-2. [DOI] [PubMed] [Google Scholar]
- 30.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: Application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 31.Yao J, Chung J, Eliezer D, Wright PE, Dyson HJ. NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into the early events in protein folding. Biochemistry. 2001;40:3561–3571. doi: 10.1021/bi002776i. [DOI] [PubMed] [Google Scholar]
- 32.Popovych N, Tzeng SR, Tonelli M, Ebright RH, Kalodimos CG. Structural basis for cAMP-mediated allosteric control of the catabolite activator protein. Proc Natl Acad Sci USA. 2009;106:6927–6932. doi: 10.1073/pnas.0900595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rehmann H, Wittinghofer A, Bos JL. Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat Rev Mol Cell Biol. 2007;8:63–73. doi: 10.1038/nrm2082. [DOI] [PubMed] [Google Scholar]
- 34.Rehmann H, et al. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature. 2008;455:124–127. doi: 10.1038/nature07187. [DOI] [PubMed] [Google Scholar]
- 35.Bönigk W, et al. An atypical CNG channel activated by a single cGMP molecule controls sperm chemotaxis. Sci Signal. 2009;2:ra68. doi: 10.1126/scisignal.2000516. [DOI] [PubMed] [Google Scholar]
- 36.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci USA. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fleissner MR, et al. Site-directed spin labeling of a genetically encoded unnatural amino acid. Proc Natl Acad Sci USA. 2009;106:21637–21642. doi: 10.1073/pnas.0912009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schünke S, Novak K, Stoldt M, Kaupp UB, Willbold D. Resonance assignment of the cyclic nucleotide binding domain from a cyclic nucleotide-gated K+ channel in complex with cAMP. Biomol NMR Assign. 2007;1:179–181. doi: 10.1007/s12104-007-9049-5. [DOI] [PubMed] [Google Scholar]
- 39.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 40.Keller R. The Computer Aided Resonance Assignment Tutorial. Zurich: Cantina; 2004. [Google Scholar]
- 41.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 42.Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR. 2002;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- 43.Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 44.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 45.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 47.DeLano W. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 48.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 50.Dosset P, Hus JC, Blackledge M, Marion D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J Biomol NMR. 2000;16:23–28. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- 51.Schanda P, Brutscher B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J Am Chem Soc. 2005;127:8014–8015. doi: 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}