

Abstract
We present two novel allyl-based terminating agents that can be used to end-functionalize living polymer chains obtained by ring-opening metathesis polymerization (ROMP) using Grubbs’ third generation catalyst. Both terminating agents can be easily synthesized and yield ROMP polymers with stable, storable activated ester groups at the chain-end. These end-functionalized ROMP polymers are attractive building blocks for advanced polymeric materials, especially in the biomedical field. Dye-labeling and surface-coupling of antimicrobially active polymers using these end-groups were demonstrated.
Keywords: Dye-labeling, End-functionalization, Ring-opening metathesis polymerization (ROMP), Surface modification, Terminating agent
Introduction
Polymers with defined reactive end-groups are of great interest for a variety of applications, such as attaching fluorescent or radioactive labels, building polymers with defined molecular architecture, or functionalizing surfaces and nanoparticles.[1–5] Reactive end-groups allow precise incorporation of one entity at a defined position on the macromolecule, and thus provide superior control and specificity compared to other methods, such as using partially functionalized macromolecules, or copolymers of functionalized and non-functionalized repeat units. End-group incorporation techniques are well established for polymerization methods such as anionic and controlled radical polymerizations. However, there have been few efficient methods to obtain end-functionalized polymers by ring-opening metathesis polymerization (ROMP) using Grubbs’ catalyst. This is due to the nature of the Grubbs’ catalyst - it is extremely tolerant of many functional groups including alcohols, aldehydes, esters, amides, ketones, carboxylic acids and even water. Thus, it can yield polymers with extremely rich functionalities imparting water solubility,[6–8] antimicrobial[9–13] and antibiofouling[14] activity, and even metal-containing functional groups.[15] However, with this high functional group tolerance, finding a suitable terminating agent that efficiently removes the Grubbs’ catalyst at the chain-end is not trivial. While the less functional-group tolerant metathesis catalysts containing titanium, tungsten and molybdenum can be quenched by carboxylic acids, alcohols, and aldehydes, the ruthenium-containing Grubbs’ catalysts can only undergo metathesis with olefins and molecular oxygen. The latter yields aldehyde end groups, however this reaction is rather slow.[16]
The end-functionalization methods for ROMP polymers obtained from titanium, tungsten, molybdenum and ruthenium-containing metathesis catalysts have been summarized recently in an excellent review.[17] Several methods for end-functionalizing polymers obtained using Grubbs’ catalysts have been reported, which we will quickly summarize. As ruthenium carbenes only react with olefins in a metathesis reaction, the end-functionalization agent must contain an olefinic group and form a Ru=C species that is inactive as a metathesis catalyst. In the ‘lactone terminating approach’, this is an unsaturated lactone or cyclic carbonate, which yields a carboxylic acid group or an aldehyde, respectively, at the chain-end.[18] In the ‘sacrificial synthesis route’, block copolymers of the desired monomer and an acid-labile monomer are prepared. This yields hydroxy end groups after hydrolysis of the block with the labile repeat units.[19] Heterotelechelic polymers, i.e. polymers with two different functional groups at each chain-end, were obtained by combination of the ‘sacrificial’ and the ‘lactone’ approach.[20]
The most widely used approach to end-functionalize the living ROMP polymer is to terminate the chain with substituted vinyl ethers. These molecules transfer a substituted methylene group onto the polymer and simultaneously remove the catalyst from the polymer chain-end.[17] By this ‘vinyl ether type terminating’ approach, a variety of functional groups were appended to the chain-end of ROMP polymers, such as trimethylsilyl groups, carboxylate groups, trimethylsilyl esters, masked acids, ketones, masked amines, and azides.[21–23] However, in some cases, these functionalization reactions were not quantitative. Allyl ethers can also be used to end-functionalize ROMP polymers, yielding acid or amine end-groups.[24] By a similar approach using an allyl ether, ester end-groups have been incorporated, which could be hydrolyzed to OH groups.[25] Only very recently, Grubbs reported terminating agents that yielded groups able to initiate atom transfer radical polymerization, as well as an N-hydroxysuccinimide ester end-group.[26]
For our research on biomedical polymers, we desired a stable activated ester end-group for dye-labeling and surface-modification. This group should be reactive in solution, but inactive in bulk so that the functionalized polymer would have a reasonably long shelf life and a number of materials could be synthesized from one master batch. For this purpose, we chose a pentafluorophenol ester as the activated ester group, which had been previously employed in combination with other polymerization techniques,[27] and designed two allyl type terminating agents containing the pentafluorophenol group (Figure 1). These terminating agents should be easy to synthesize, yield quantitatively end-functionalized polymers, and allow coupling of the end-functionalized polymers to small molecules, fluorescent dyes and polymer surfaces. Our novel pentafluorophenol ester-based terminating agents 1 and 2 were shown to fulfil all of these criteria, as will be detailed below, and thus were valuable coupling tools for a variety of new materials.
Figure 1.

Synthesis of terminating agent molecules 1 and 2.
Results and Discussion
The terminating agent synthesis is illustrated in Figure 1. The first terminating agent molecule (1) was obtained from 2-butene-1,4-diol and succinic anhydride, followed by esterification of the acid groups with pentafluorophenol, using EDC as a coupling agent (Figure 1a).
The synthesis of the second terminating agent (2) is shown in b. It is obtained from the Michael addition of 2-butene-1,4-diol to tert-butyl acrylate, followed by deprotection using trifluoroacetic acid, and esterification of the resulting acid groups with pentafluorophenol. Both terminating agents are symmetric molecules and can undergo metathesis with a living polymer chain-end carrying Grubbs’ catalyst (Figure 2), analogous to the traditional terminating agent, ethyl vinyl ether, which is commonly used to obtain non-functionalized ROMP polymers.
Figure 2.

End-functionalization of living ROMP polymers with terminating agents 1 (pentafluoro-allyl ester) and 2 (pentafluoro-allyl ether).
In order to investigate the efficiency of the termination reaction using MALDI-TOF mass spectrometry, we used the non-polar polymer 3 (Figure 3) with a molecular weight of 5000 g mol−1 as a model system. Due to its non-polar nature, this polymer can be more easily investigated by MALDI-TOF MS than our more polar biologically active polymers. The living polymer chain 3 was quenched with two equivalents of terminating agent 1, which yielded the end-functionalized polymer 4. As can be seen from its MALDI spectrum in Figure 4a, the inter-peak distance corresponds to the mass of the repeat unit, and only one molar mass distribution is present, which indicates that the quenching of polymer 3 with 1 was quantitative within the accuracy of the method. Polymer 4 was subsequently reacted with an amine-bearing siloxane and with tert-butyl amine to demonstrate the reactivity of the new end-group, yielding polymers 6 and 5, respectively (Figure 3). The siloxane was chosen because of its distinct MALDI shift as compared to the parent polymer, while tert-butyl amine was used to demonstrate that the activated ester was also capable of reacting with bulkier primary amine groups.
Figure 3.

End-functionalization and polymer-analogous reactions of model polymers 3 and 4.
Figure 4.

a) MALDI-TOF mass spectrum of polymer 4, b) MALDI-TOF mass spectrum of polymer 6, c) overlay of the calculated and observed MALDI-TOF MS peaks. Open symbols refer to the calculated, solid symbols to the observed peak positions; triangles – polymer 4; diamonds – polymer 5; squares – polymer 6; d) Mn, calc/Mn, obs versus M/Z for polymers 4, 5, and 6.
Representative MALDI spectra of polymers 4 and 6 are shown in Figure 4a and b, together with an overlay of the calculated and observed MALDI peaks for polymers 4, 5 and 6 in Figure 4c. The calculated and measured peak positions coincide for all polymers. Like polymer 4, the spectra for both polymer 5 (MALDI not shown) and 6 (Figure 4b) also contain only one molar mass distribution, indicating a unique and well-defined end-group. The ratio of the calculated molecular weight over the molecular weight observed by MALDI (Mn.calc/Mn,obs) is shown in Fig 4d. This data demonstrates that these polymer-analogous reactions proceed quantitatively and emphasizes the versatility of terminating agent 1.
While a slight excess of terminating agent 1 yielded completely end-functionalized polymer (after stirring over night at room temperature), similar conditions did not yield end-functionalized polymer with terminating agent 2. Therefore, several functionalization conditions were studied for this molecule. It was found that 10 equivalents (relative to Grubbs catalyst) were needed to get complete conversion, while 1, 2 and even 5 equivalents did not shift the equilibrium to quantitative termination (23%, 47% and 70% conversion, respectively, were obtained). Longer reaction times (48 h and 72 h at room temperature) and higher temperatures (40°C and 60°C) did not improve the conversion. Since terminating agent 2 is easy to synthesize and the excess is removable by simple filtration through silica gel, the higher excess needed is not a big disadvantage. The classical terminating agent, ethyl vinyl ether, is typically added in much larger excess. Also, the advantage of terminating agent 2 over terminating agent 1 is that it connects the polymer to the functional group by an ether bond, rather than an ester bond, resulting in polymers with better long term stability, as the ether bond is not cleavable by simple hydrolysis. However, there might be cases where hydrolyzability is a desired property, in which cases terminating agent 1 may be preferable. Like terminating agent 2, its excess is also removable by filtration through silica gel (see SI).
The above described chemistry was developed for biomedical applications the including dye-labeling of ROMP polymers as well as their attachment to surfaces. We therefore will now focus on terminating agent 2 when discussing these applications, as they require a non-hydrolysable linkage. After our successful studies with model polymer 4, we used this approach both for dye incorporation onto and surface functionalization of bioactive polymers, as will be detailed below.
Dye incorporation
Dye incorporation onto polymers is an important tool for tracking them in an organism. We were interested in studying the interaction of previously published synthetic mimics of antimicrobial peptides (SMAMPs) with E. coli cell membranes.[12] With our end-functionalization approach, it was possible to position exactly one dye molecule per polymer chain at the chain-end. Following the above described procedure, but this time using a monomer that imparts antimicrobial activity, the terminating agent 2 was used to obtain the end-functionalized SMAMP precursor 7. Unfortunately, as 7 did not give meaningful MALDI-TOF MS data, we had to demonstrate the successful incorporation of the end group into the polymer by 1H-NMR spectroscopy (Figure 5). Figure 5 is an overlay of the 1H-NMR spectrum of the terminating agent 2 (Figure 5, top) and the SMAMP precursor 7 (Figure 5, bottom). The SMAMP precursor 7 had been fractioned previously by preparative GPC to remove any terminating agent. As shown in Fig. 5, the signals of the two methylene groups in the new end-group became broad triplets when attached to the polymer chain (black arrows). The integration of these signals versus peaks from the polymer indicates, within the accuracy of the method, near quantitative end group incorporation.
Figure 5.
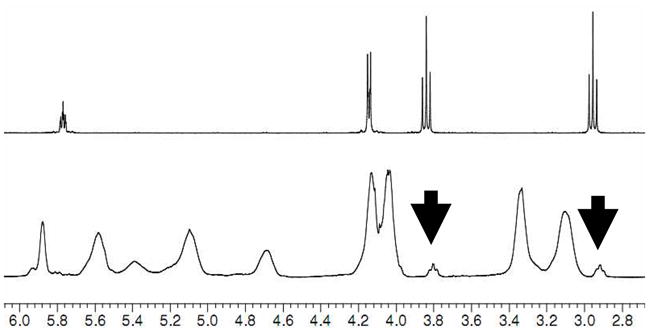
1H-NMR spectra of terminating agent 2 (top) and SMAMP 7 (bottom).
When reacted with the fluorescent nitro benzoxadiazole derived dye 8 that had been synthesized as described in the literature[28] (Figure 6a), the fluorescently labeled polymer 9 was obtained. Polymer 9 was purified by column chromatography to remove excess dye. After removal of the Boc protective group with trifluoroacetic acid, followed by dialysis, the active SMAMP 10 was obtained (Figure 6a). This molecule was then incubated with E. coli cells and imaged using confocal microscopy. The results are shown in Figure 6b. As seen, especially for the two bacteria cells highlighted by arrows, the fluorescent SMAMP tends to accumulate in the cell membrane region rather than inside the cytosol. This is in line with current theories on SMAMP-cell interaction, which state that the antimicrobial activity of the SMAMP is due to interaction with the cell membrane, leading either to fatal membrane pore formation or membrane disruption.[9, 29]
Figure 6.

Dye incorporation. a) reaction scheme, b) confocal micrograph of E. coli bacteria incubated with the SMAMP 10. The right image illustrates the position of the bacteria cells, which are mostly viewed side on, except for the central one, which is viewed end-on.
Surface functionalization
The SMAMP precursor 7 was further used to functionalize a silica surface with a thin layer of antimicrobial polymers. This was achieved by an amine-functionalized silicon wafer to which the SMAMP precursor 7, with a molecular weight of a 10 000 g mol−1, was grafted. Preparation details are given in Figure 7a and the Supporting Information. The resulting surface was characterized by ellipsometry and atomic force microscopy (AFM). According to ellipsometry data, a 5 to 7 nm thick layer of polymer was grafted to the silicon wafer (Δ = 155.2/Ψ = 11.00). This was confirmed by AFM images (Figure 7b). Both the height and the phase image indicate the presence of a thin, roughly 4 nm thick, diffuse polymer layer on the silica wafer surface. The finding that our polymer with an activated ester end group can be attached to a surface is consistent with literature, in which an in-situ created activated ester was used to attach a polymer to a surface.[30] The antimicrobial activity of these surfaces is currently under investigation and will be reported in due course.
Figure 7.

a) surface functionalization steps. b) AFM images of functionalized surface (left: height image, right: phase image), arrow 1 indicates a zone on the Si-wafer that is not covered by polymer, arrow 2 points at a diffuse surface feature that represents a polymer coil.
Conclusions
In this paper, we presented the synthesis of two terminating agents and their possible uses to end- functionalize living ROMP polymers. By subsequent polymer analogous chemistry, it was shown that small molecules, such as a fluorescent dye and amine-substituted surfaces, could be easily coupled to the end-functionalized polymer. We will take advantage of this method in future research, including surface functionalization studies and dye-labeling of antimicrobial and cell-penetrating peptides. Additionally, due to the ease of synthesis of the terminating molecules, as well as the stable yet reactive nature of the resulting activated ester end-group, we expect that this approach will find application in areas such as polymer-polymer coupling and bioconjugation reactions, and prove to be a versatile nanoparticle functionalization technique.
Supplementary Material
Acknowledgments
Funding by the German Research Foundation (DFG-Forschungsstipendium to K.L.), the Ludcke Foundation (fellowship to K.L.), German National Academic Foundation (Studienstiftung des deutschen Volkes, travel grant to A.H.R.K.), NIH (AI-074866) and ONR (N000140710520) is gratefully acknowledged. This work utilized facilities supported in part by the National Science Foundation Materials Research Science and Engineering Center (DMR 0820506) and Center for Hierarchical Manufacturing (CMMI 0531171).
Footnotes
Experimental details and more characterization data can be found in the Supporting Information. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Aoshima S, Kanaoka S. Chemical Reviews. 2009;109:5245. doi: 10.1021/cr900225g. [DOI] [PubMed] [Google Scholar]
- 2.Tomczak N, Janczewski D, Han M, Vancso GJ. Progress in Polymer Science. 2009;34:393. [Google Scholar]
- 3.Barner L, Perrier S. Handbook of RAFT Polymerization. 2008:455. [Google Scholar]
- 4.Tsarevsky NV, Golas PL, Matyjaszewski K. Polymer Preprints. 2008;49:141. [Google Scholar]
- 5.Lo Verso F, Likos CN. Polymer. 2008;49:1425. [Google Scholar]
- 6.Alfred Sterling F, Lienkamp Karen, Madkour Ahmad E, Tew Gregory N. J Polymer Sci A - Polymer Chem. 2008;46:6672. [Google Scholar]
- 7.Alfred Sterling F, Al-Badri Zoha M, Madkour Ahmad E, Lienkamp Karen, Tew Gregory N. Journal of Polymer Science Part A-Polymer Chemistry. 2008;46:2640. [Google Scholar]
- 8.Lienkamp Karen, Kins Christoph F, Alfred Sterling F, Madkour Ahmad E, Tew Gregory N. J Polym Sci, Part A: Polym Chem. 2009;47:1266. [Google Scholar]
- 9.Lienkamp Karen, Tew Gregory N. Chemistry--A European Journal. 2009;15:11784. doi: 10.1002/chem.200900049. [DOI] [PubMed] [Google Scholar]
- 10.Lienkamp Karen, Kumar Kushi-Nidhi, Som Abhigyan, Nuesslein Klaus, Tew Gregory N. Chemistry--A European Journal. 2009;15:11710. doi: 10.1002/chem.200802558. [DOI] [PubMed] [Google Scholar]
- 11.Lienkamp Karen, Madkour Ahmad E, Kumar Kushi-Nidhi, Nuesslein Klaus, Tew Gregory N. Chemistry--A European Journal. 2009;15:11715. doi: 10.1002/chem.200900606. [DOI] [PubMed] [Google Scholar]
- 12.Lienkamp Karen, Madkour Ahmad E, Musante Ashlan, Nelson Christopher F, Nusslein Klaus, Tew Gregory N. J Am Chem Soc. 2008;130:9836. doi: 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabriel Gregory J, Maegerlein Janet A, Nelson Christopher F, Dabkowski Jeffrey M, Eren Tarik, Nüsslein Klaus, Tew Gregory N. Chemistry - A European Journal. 2009;15:433. doi: 10.1002/chem.200801233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semra Colak, Tew Gregory N. Biomacromolecules. 2009;10:353. doi: 10.1021/bm801129y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Badri Zoha M, Tew Gregory N. Macromolecules. 2008;41:4173. [Google Scholar]
- 16.Biagini SCG, Gareth Davies R, Gibson VC, Giles MR, Marshall EL, North M. Polymer. 2001;42:6669. [Google Scholar]
- 17.Hilf Stefan, Kilbinger Andreas FM. Nature Chemistry. 2009;1:537. doi: 10.1038/nchem.347. [DOI] [PubMed] [Google Scholar]
- 18.Hilf Stefan, Grubbs Robert H, Kilbinger Andreas FM. Journal of the American Chemical Society. 2008;130:11040. doi: 10.1021/ja8022863. [DOI] [PubMed] [Google Scholar]
- 19.Hilf Stefan, Berger-Nicoletti Elena, Grubbs Robert H, Kilbinger Andreas FM. Angewandte Chemie, International Edition. 2006;45:8045. doi: 10.1002/anie.200602323. [DOI] [PubMed] [Google Scholar]
- 20.Hilf Stefan, Kilbinger Andreas FM. Macromolecules. 2010;43:208. [Google Scholar]
- 21.Gordon Eva J, Gestwicki Jason E, Strong Laura E, Kiessling Laura L. Chemistry & Biology. 2000;7:9. doi: 10.1016/s1074-5521(00)00060-0. [DOI] [PubMed] [Google Scholar]
- 22.Gestwicki Jason E, Cairo Christopher W, Mann David A, Owen Robert M, Kiessling Laura L. Analytical Biochemistry. 2002;305:149. doi: 10.1006/abio.2002.5652. [DOI] [PubMed] [Google Scholar]
- 23.Mangold Shane L, Carpenter Rachael T, Kiessling Laura L. Organic Letters. 2008;10:2997. doi: 10.1021/ol800932w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morita Takeharu, Maughon Bob R, Bielawski Christopher W, Grubbs Robert H. Macromolecules. 2000;33:6621. [Google Scholar]
- 25.Vernon C, Gibson, Toshiyuki Okada. Macromolecules. 2000;33:655. [Google Scholar]
- 26.Matson John B, Grubbs RH. Macromolecules. 2010;43:213. doi: 10.1021/ma9019366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Theato P. Journal of Polymer Science, Part A: Polymer Chemistry. 2008;46:6677. [Google Scholar]
- 28.Ohba Y, Kanao Y, Takatsuji M, Ito M, Yabuta N, Nojima H, Kita Y. Tetrahedron. 2007;63:3754. [Google Scholar]
- 29.Som Abhigyan, Vemparala Satyavani, Ivanov Ivaylo, Tew Gregory N. Biopolymers (Peptide Science) 2008;90:83. doi: 10.1002/bip.20970. [DOI] [PubMed] [Google Scholar]
- 30.Owen Robert M, Gestwicki Jason E, Young Travis, Kiessling Laura L. Organic Letters. 2002;4:2293. doi: 10.1021/ol0259239. [DOI] [PubMed] [Google Scholar]
- 31.Jennifer A Love, John P Morgan, Tina M Trnka, Robert H Grubbs. Angewandte Chemie International Edition. Vol. 41. 2002. p. 4035. [Google Scholar]
- 32.Kevin Sill, Todd Emrick. Journal of Polymer Science, Polymer Chemistry. 2005;43:5429. [Google Scholar]
- 33.Fadeev Alexander Y, McCarthy Thomas J. Langmuir. 1999;15:3759. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.