

Abstract
Background
Prolonged hypothermic circulatory arrest results in neuronal cell death and neurologic injury. We have previously shown that hypothermic circulatory arrest causes both neuronal apoptosis and necrosis in a canine model. Inhibition of neuronal nitric oxide synthase reduced neuronal apoptosis, while glutamate receptor antagonism reduced necrosis in our model. This study was undertaken to determine whether glutamate receptor antagonism reduces nitric oxide formation and neuronal apoptosis after hypothermic circulatory arrest.
Methods
Sixteen hound dogs underwent 2 hours of circulatory arrest at 18°C and were sacrificed after 8 hours. Group 1 (n=8) was treated with MK-801, 0.75 mg/kg IV prior to arrest followed by 75 μg/kg/hr infusion. Group 2 dogs (n=8) received vehicle only. Intracerebral levels of excitatory amino acids and citrulline, an equal co-product of nitric oxide, were measured. Apoptosis, identified by H&E staining and confirmed by electron microscopy, was blindly scored from 0 (normal) to 100 (severe injury), while nick-end labeling demonstrated DNA fragmentation.
Results
Group 1 and 2 dogs had similar intracerebral levels of glutamate. However, MK-801 significantly reduced intracerebral glycine and citrulline levels as compared to HCA controls. MK-801 significantly inhibited apoptosis (7.92 ± 7.85 vs. 62.08 ± 6.28, Group 1 vs. 2, p<0.001).
Conclusions
Our results showed that glutamate receptor antagonism significantly reduced nitric oxide formation and neuronal apoptosis. We provide evidence that glutamate excitotoxicity mediates neuronal apoptosis in addition to necrosis after hypothermic circulatory arrest. Clinical glutamate receptor antagonists may have therapeutic benefit in ameliorating both types of neurologic injury after hypothermic circulatory arrest.
Keywords: Animal Model, Apoptosis, Brain, Hypothermic Circulatory Arrest, Nitric Oxide
Introduction
Hypothermic circulatory arrest (HCA) was initially introduced for the repair of complex congenital heart lesions, but has since been widely adopted for aortic arch and thoracic aortic procedures [1–3]. It is a technique that affords the surgeon a bloodless operative field unobstructed by vascular clamps and cannulae. However, neurological injury occurs after prolonged HCA. Morbidity and mortality increase substantially when the duration of arrest exceeds 45–60 minutes [4]. Clinical neurologic sequelae include choreoathetosis, learning and memory deficits, seizures, and impaired intellectual development. This occurs in up to 15% of children and 18% of adults [4–7].
Neuronal cell death is responsible for this HCA induced neurologic injury. There are two types of neuronal cell death, necrosis and apoptosis, or programmed cell death [8]. Selective neuronal necrosis of the neocortex, hippocampus, basal ganglia, and cerebellum has been the neuropathological hallmark of HCA. We have previously shown that neuronal apoptosis is an additional cause of neurologic damage after HCA and that inhibition of neuronal nitric oxide synthase reduces apoptosis [9]. We also demonstrated that glutamate excitotoxicity mediated neuronal necrosis after HCA [10]. We hypothesized that glutamate excitotoxicity results in increased nitric oxide (NO) production and neuronal apoptosis after HCA. The purpose of this study was to determine whether glutamate receptor antagonism reduced NO production and apoptosis in a canine model of HCA.
Material and Methods
Preparation
Our canine model of hypothermic circulatory arrest has been previously described [9–11]. Sixteen colony-bred heart-worm negative male hound dogs, 20–27 kg, 7–12 months old, were used. Anesthesia was induced with sodium thiopental (3.0 mg/kg intravenously) and maintained with 0.5% to 2.0% halothane. Bilateral tympanic membrane, nasopharyngeal, and rectal temperature probes were placed. Tympanic membrane temperature closely correlates with brain temperature. Electrocardiographic monitoring was employed. Swan-Ganz catheter through the left external jugular vein and arterial line in the left femoral artery were placed.
Cardiopulmonary Bypass (CPB) and Hypothermic Circulatory Arrest (HCA)
CPB circuit consisted of a Cobe membrane oxygenator (Cobe Laboratories, Inc., Lakewood, CO), a Sarns roller pump (Sarns Inc, Ann Arbor, MI), and a 40 μm in-line arterial filter. Circuit was primed with 1.5 L of lactated Ringer’s solution with 50 mEq of sodium bicarbonate and 10 mEq of potassium chloride. After heparinization (300U/kg intravenously), dogs were cannulated with 12F to 14F arterial cannula in the descending aorta from the right femoral artery and with 18F to 20F venous cannulae, advanced to the right atrium through the right external jugular and femoral veins.
Closed chest CPB was instituted; animals were surface (ice bags around head and cooling blanket) and core (CPB) cooled to a tympanic membrane temperature of 18°C within 25 to 30 minutes. Mean arterial pressures were maintained at 50–60 mmHg with pump flows of 80 to 100 ml/kg and reduced to 60 ml/kg when temperature was less than 32°C. Arterial blood gases were controlled using alpha-stat strategy. Arterial pump was turned off and venous blood was drained by gravity into the reservoir. Circulatory arrest was maintained for 2 hours followed by reinstitution of CPB and rewarming. Sodium bicarbonate (50 mEq) and lasix (20 mg) were added to the reservoir. At normothermia (37°C), animals were weaned from CPB and decannulated.
Postoperatively, dogs were survived for 8 hours after HCA. They were ventilated and anesthetized with intravenous fentanyl (10–20μg/kg) and midazolam (1 mg) as needed. Intensive care unit (ICU) monitoring was employed. Animals were then sacrificed fully anesthetized by exsanguination and perfusion with ice cold saline or 4% paraformaldehyde. Five normal dogs that did not undergo HCA had their brains harvested for normal controls.
Dizocilpine (MK-801) Protocol
Experimental dogs (group 1, n=8) received an N-methyl-D-aspartate (NMDA) receptor antagonist, MK-801, 0.75 mg/kg IV bolus prior to arrest followed by 75 μg/kg/hr IV continuous infusion. HCA control animals (group 2, n=8) received vehicle only.
Intracerebral Microdialysis
Animals, positioned in a Kopf stereotactic device, had the right side of skull exposed. Burr holes were drilled 3 mm caudal to coronal suture and 8 mm from midsagittal axis. Dura was opened and microdialysis probes (CMA 10/4; Acton, MA) stereotactically placed to depth of 20 mm in the corpus striatum. Confirmation of proper placement was determined at sacrifice. Tissue stabilized after probe placement for 180 minutes. Warmed artificial cerebrospinal fluid (CSF) (mmol/L concentration: NaCl, 131.8; NaHCO3, 24.6; CaCl2, 2.0; KCl, 3.0; MgCl2, 0.65; urea, 6.7; and dextrose, 3.7) was infused at 1 μl/min. Effluent was serially collected every 30 minutes and assayed by high performance liquid chromatography with electrochemical detection (HPLC-EC) for extracellular amino acid concentrations [12]. Citrulline concentration was used as a marker of NO production.
Histopathology
Right hemisphere of each brain was post-fixed in 10% formalin, embedded in paraffin and 8 μm sections were stained with hematoxylin and eosin (H&E) or cresyl violet (CV). Left hemisphere of saline perfused brains was frozen on dry ice for DNA nick end-labeling. Left hemisphere of perfused fixed brains was post-fixed in 4% paraformaldehyde, cryoprotected in sucrose, and frozen on dry ice for immunohistochemistry. Quantification of apoptosis was performed on H&E stained sections by a neuropathologist in a blinded fashion. Apoptosis was scored from 0 (normal) to 100 (severe injury).
Electron Microscopy
Coronal sections of perfused-fixed tissue were post-fixed in 0.1% gluteraldehyde and 4% paraformaldehyde, osmicated, dehydrated, and embedded in epoxy resin. Ultrathin sections were cut with an ultramicrotome and examined by electron microscopy. Apoptosis was confirmed by electron microscopy.
DNA Nick End-Labeling
Apoptosis activates Ca2+ and Mg2+ dependent endonucleases to cut chromatin into DNA fragments. We used a terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end-labeling (TUNEL) method (ApopTag kit, Oncor, Inc., Gaithersburg, MD) for sensitive and specific staining of DNA fragmentation and apoptotic bodies. Though not as specific as Caspase cleavage staining for apoptosis, TUNEL staining demonstrated cells with DNA fragmentation—apoptosis vs. necrosis was confirmed by H&E and electron microscopy. In addition, NMDA excitotoxicity can be capase independent via apoptosis inducing factor [13].
Statistical Analyses
All values are expressed as mean ± standard deviation of the population. Comparisons between groups were made with two way analysis of variance without replication or Student’s t-test where appropriate.
Animal Care
All experimental protocols were preapproved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions. Animals received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health (NIH Publications No. 85–23, revised 1985).
Results
Physiological Variables
Cooling times on CPB for groups 1 and 2 ranged from 25–30 minutes. No significant differences in tympanic membrane temperatures between both groups were found during cooling, arrest, rewarming, and recovery phases. Esophageal and rectal temperatures remained similar for both groups throughout the experiment. Mean arterial pressures and cardiac output were similar for both groups. No significant differences in arterial blood gases were found, with similar pH and pCO2, and bicarbonate requirements were also similar for both groups. Glucose and hemoglobin levels were examined and both groups exhibited similar hyperglycemia and hemodilution.
In Vivo Microdialysis
Extracellular amino acid concentrations of glutamate, glutamine, glycine, arginine, and citrulline were measured over time. Peak levels of each amino acid were examined over 3 time periods, arrest, reperfusion CPB, and recovery (2–8 hours after HCA), and compared to baseline. Intracerebral glutamate levels, which increased during HCA and throughout the experiment, did not significantly change with administration of MK-801 (Figure 1A). Glycine, a co-agonist with glutamate on the NMDA receptor, increased during reperfusion CPB and recovery; glycine levels were significantly reduced by MK-801 treatment (Figure 1B). No significant changes in glutamine occurred over time during HCA, with or without MK-801 treatment.
Figure 1.

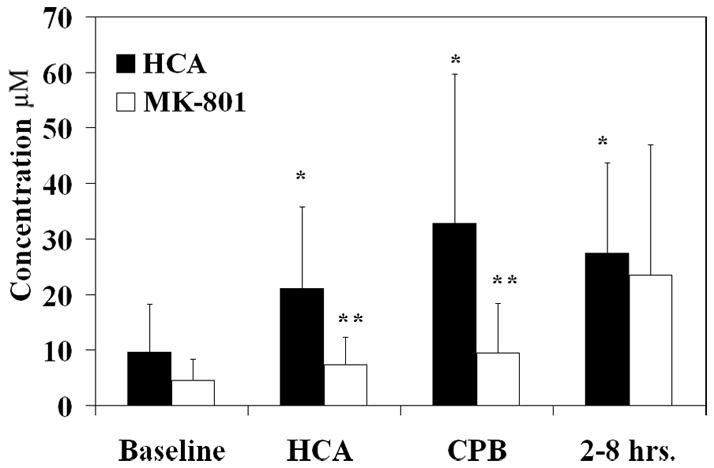
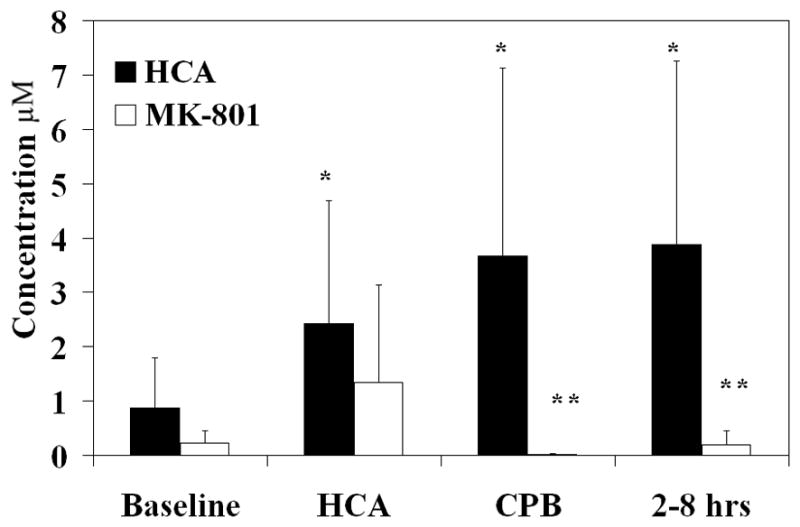
Intracerebral glutamate levels (μM) over time with and without MK-801 treatment (figure 1A). Intracerebral glycine levels (μM) (figure 1B) and intracerebral citrulline levels (μM) (figure 1C) over time with and without MK-801 treatment. *, p<0.05 compared to baseline. **, p<0.05 compared to HCA without MK-801 treatment.
NOS converts arginine and oxygen to citrulline and nitric oxide in a 1:1 ratio. Since ornithine transcarbamylase in the liver is the only other enzyme that performs this reaction, citrulline production in the brain can be used as a marker for NO production by NOS activity. NO production increased significantly during reperfusion and recovery and was completely inhibited by MK-801 (Figure 1C). NO production at the end of the experiment was found to be reduced as compared to baseline. No significant changes in arginine concentration were found with or without MK-801 during arrest.
Identification of Apoptosis
Apoptosis was identified morphologically by electron microscopy (EM) and light microscopy, and biochemically DNA fragmentation was identified by the TUNEL assay. Morphological criteria for identifying apoptosis were cytoplasmic and cellular shrinkage, nuclear chromatin condensation and aggregation, fragmentation of the cell with or without nuclear fragments to produce apoptotic bodies, nuclear pyknosis with a peripheral distribution in the nucleus, plasma membrane blebbing with no loss of integrity, and the lack of an inflammatory response. Similarly, as previously demonstrated for neuronal nitric oxide synthase inhibition [9], electron microscopy confirmed apoptotic cells in contrast to normal cells after 2 hours of HCA. Ultrastructurally, the integrity of the plasma membrane and organelles of the apoptotic cell was preserved and apoptotic bodies were present. By light microscopy, H&E stained sections also demonstrated apoptosis in HCA treated animals with or without MK-801 treatment (Figure 2B–2C) versus controls (Figure 2A).
Figure 2.
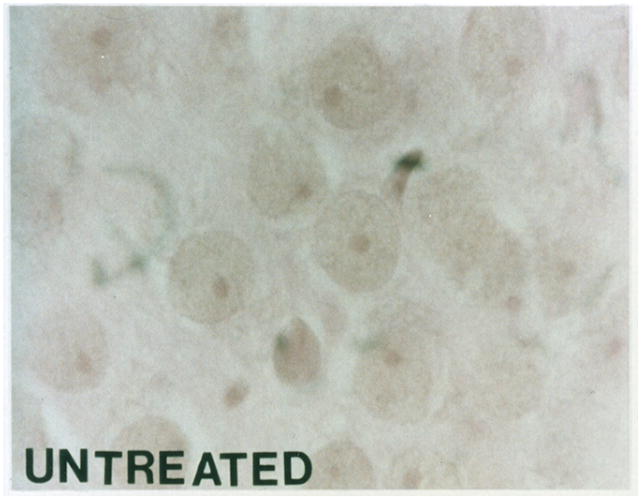
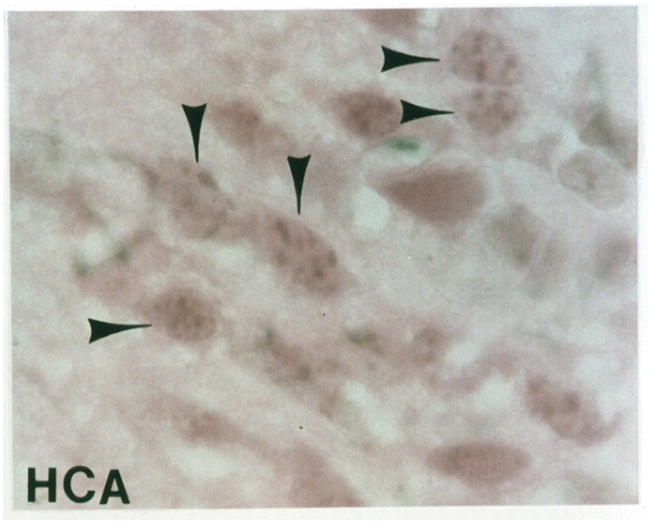
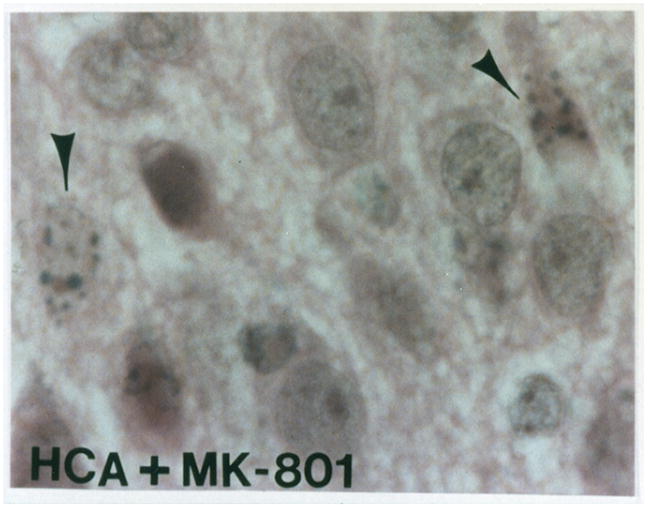
Light microscopy of H&E stained sections for a normal dog (figure 2A). Apoptosis is seen in an HCA treated dog (figure 2B). Reduction in apoptosis seen in an MK-801 treated HCA dog shown by light microscopy of H&E stained sections (figure 2C).
Biochemically, apoptosis results in internucleosomal fragmentation of DNA into multiples of single nucleosome length of DNA, approximately 180 bp. The TUNEL assay, an in situ method for staining of DNA fragments, is nonspecific marker for apoptosis. TUNEL showed cells with DNA fragmentation after HCA with or without MK-801 treatment (Figure 3B–3C) compared with controls (Figure 3A). Confirmation of apoptosis vs. necrosis was performed by H&E and electron microscopy.
Figure 3.
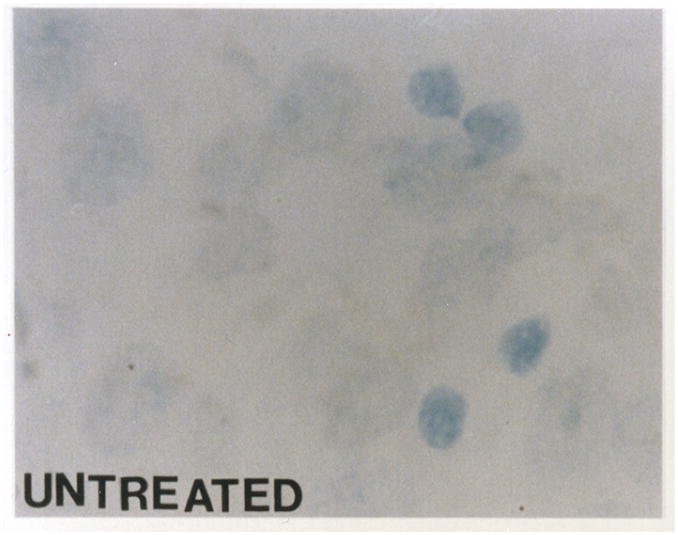

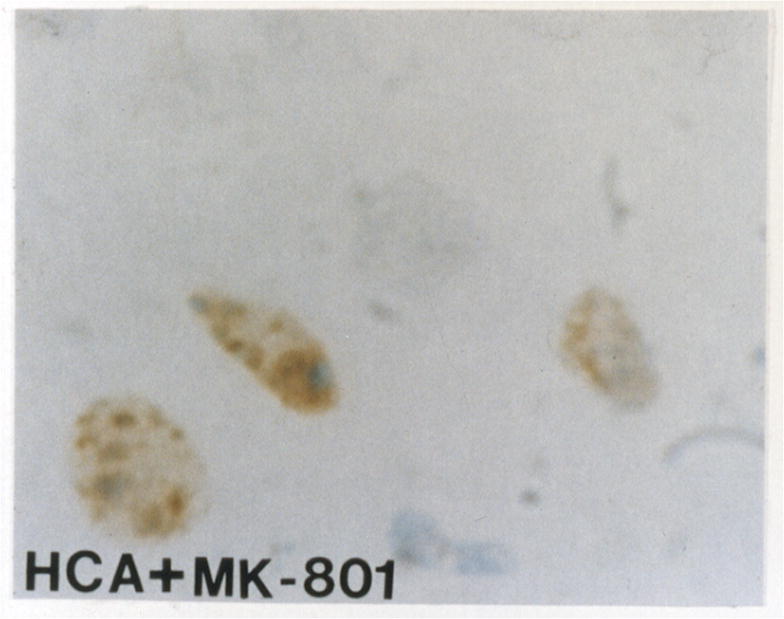
TUNEL assay shows no DNA fragmentation in a normal dog (figure 3A). TUNEL demonstrates cells with DNA fragmentation in an HCA dog (figure 3B). Reduction in cells with DNA fragmentation is seen in an HCA dog treated with MK-801 (figure 3C).
Inhibition of Apoptosis
As we have previously described, apoptosis was distributed in selective neuronal populations, including layer 2 of the neocortex, layers 2, 3, and 5 of the entorhinal cortex, and dentate gyrus of the hippocampus [9]. Notably, apoptosis did not occur in the CA1-CA4 regions of the hippocampus. Of all the regions, however, apoptosis most extensively targetted the granule cell layer of the dentate gyrus of the hippocampus. With such severity in a well-defined region, apoptosis was most amenable to quantification in the dentate gyrus.
Since apoptosis occurred maximally in the dentate gyrus of the hippocampus, that region was closely examined. Investigation of H&E stained sections revealed reduction in apoptosis after HCA in dogs treated with MK-801 (Figure 2C). Quantitatively, apoptosis was scored using the H&E stained sections in a blinded fashion by a neuropathologist from 0 (normal) to 100 (severe injury), based on the percentage of apoptotic cells. MK-801 significantly reduced apoptosis in dogs that underwent 2 hours of HCA (7.92 ± 7.85 vs. 62.08 ± 6.28, p<0.001). TUNEL staining also demonstrated a reduction in the number of cells with DNA fragmentation (Figure 3C).
Comment
HCA is an important technique for repair of complex congenital heart lesions and operations on the aortic arch and thoracic aorta [1–3]. It provides a bloodless operative field, unobstructed by vascular clamps and cannulae. However, the central nervous system is exquisitely sensitive to ischemia, limiting the duration of safe arrest. Neurological injury occurs when the duration of arrest exceeds 45–60 minutes [4, 7]. Clinical sequelae include seizures, choreoathetosis, learning and memory deficits, and impaired intellectual development [4–7, 14]. Clinically apparent neurologic damage occurs in up to 12% of children and 15% of adults [4–7, 14].
Neuronal cell death causes HCA induced neurologic injury. Necrosis is one neuropathologic hallmark of HCA induced injury; however, we have previously shown that neuronal apoptosis is an additional cause of neurologic injury after HCA [9]. We demonstrated that neuronal apoptosis is a process occurring early, peaking 8 hours, diminishing by 20 hours, and disappearing by 72 hours after HCA. This time frame may be different in other animal models, such as neonatal pigs, and the acute nature of this time frame may also be influenced by the degree and severity of ischemia, where shorter durations of HCA may show a longer time till onset of apoptosis [15].
Previous work in our laboratory has demonstrated the utility of glutamate receptor antagonists in ameliorating necrosis associated with HCA at 72 hours [16, 17]. In contrast, one study showed no neuroprotective effect of memantine, an NMDA receptor antagonist in a porcine model of 75 minutes of HCA at 20°C after 7 days [18]. They found no significant changes in histology or behavioral scoring; however, since they did not investigate changes in nitric oxide, it is uncertain whether the dose response curve of memantine was sufficient to reduce nitric oxide production. In this study, we investigated the mechanism of apoptosis which occurs more acutely in our HCA model using an NMDA receptor antagonist, MK-801. We used the same dose of MK-801 as Dr. Redmond’s chronic study examining necrosis [10], and demonstrated not only inhibition of apoptosis after HCA but also near complete suppression of nitric oxide production.
In our prior microdialysis study, we examined changes in extracellular amino acid concentrations in the brain associated with neuronal apoptosis after hypothermic circulatory arrest [19]. Glutamate levels increased significantly during arrest, reperfusion CPB, and recovery, supporting the hypothesis of glutamate excitotoxicity. In addition, citrulline levels, reflective of NO production, increased significantly during reperfusion CPB and recovery [19]. In this study, MK-801, a noncompetitive antagonist, did not change glutamate levels; however, it did significantly reduce NO production, supporting the upstream actions of glutamate. MK-801 also reduced glycine levels, a requisite co-agonist of glutamate on the NMDA receptor.
We have demonstrated that glutamate excitotoxicity mediates neuronal apoptosis in this model of HCA. Apoptosis was pathologically defined by cellular shrinkage and nuclear chromatin condensation and aggregation on H&E and electron microscopy. Apoptosis was the morphologic representation of regularly cut nucleosome length DNA, and the TUNEL assay of nick-end labeling demonstrated cells with DNA fragmentation. Subsequent work in Dr. Johnston’s laboratory has demonstrated good correlation of TUNEL positive staining with activated caspase staining in the dentate gyrus for apoptosis. Increased glutamate via its effects on NO production may result in DNA strand breaks by base deamination to result in apoptosis.
Glutamate Excitotoxicity and Cell Death Mechanism
Neuronal injury may be caused by overstimulation of excitatory amino acid receptors, including glutamate and aspartate [20, 21]. This excitotoxicity is predominantly mediated by calcium influx through ionic channels of activated glutamate receptors. Hypoxia and ischemia result in overaccumulation of the excitatory amino acid, glutamate. Glutamate, the principal neurotransmitter of the brain, is responsible for many physiologic functions, including cognition, memory, movement, and sensation. Pathophysiologically, excessive glutamate activates N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA), and kainate glutamate receptors. Glutamate along with co-agonist glycine stimulates NMDA receptors to increase intracellular calcium, which triggers a cascade of intracellular reactions, activating phospholipases, proteases, protein kinases, phosphatases, and nitric oxide synthase (NOS). NOS causes increased nitric oxide production, which may damage DNA by base deamination to result in DNA strand breaks. Damaged DNA activates poly(adenosine 5′-diphosphoribose) polymerase (PARP) to add dATP to the ends of nicked DNA, resulting in depletion of energy sources from the cell. These processes ultimately lead to cell death, which can be necrotic or apoptotic in nature [22, 23].
Apoptosis, or programmed cell death, is a tightly regulated process, requiring energy, macromolecular synthesis, and gene transcription [21, 24]. It results in nonrandom oligonucleosomal length DNA fragmentation. A number of genes have been implicated in the programmed cell death process, including antiapoptotic genes, i.e. bcl-2, and proapoptotic genes, i.e. bax and interleukin converting enzyme (ICE) family of cysteine proteases [21, 24]. However, what shifts the balance of antiapoptosis to proapoptosis is unknown. Furthermore, why cells die via apoptosis vs. necrosis is unknown. Evidence suggests that any insult below the threshold to cause necrosis may result in apoptosis, so that mild and intense insults result in apoptosis and necrosis, respectively [21, 24, 25]. Initiation of the apoptotic pathways appears to involve increasing intracellular calcium, acidosis, reactive oxygen species, and Fas receptor activation [21]. Reduction in neuronal apoptosis has primarily focused on calpain and caspase inhibition and antioxidants [26].
Neuronal Cell Death after HCA
In hypothermic circulatory arrest, glutamate excitotoxicity mediates both apoptosis and necrosis [27]. How glutamate triggers one cell to undergo apoptosis, while another necrosis is not known. Severe ischemia with large increases in glutamate may result in necrosis, characterized by cellular swelling and lysis, while milder insults cause apoptosis. Greater insults may generate more superoxide anion to convert nitric oxide to peroxynitrite, which leads to lipid peroxidation and cellular lysis. Milder insults may generate nitric oxide, but less peroxynitrite, to alter the oxidative state of the cell and shift the balance from antiapoptotic to proapoptotic forces.
In this study, we have demonstrated that NMDA receptor antagonism with MK-801 significantly reduced downstream nitric oxide production with no effect on glutamate concentrations. This reduction in nitric oxide resulted in significant reduction in neuronal apoptosis after HCA. Apoptotic cell death is significant not only for its role in neurologic injury after HCA, but also for its role in hypoxia, ischemia, Huntington’s, Alzheimer’s, and physiological neural development. Strategies for cerebral protection after HCA require amelioration of both forms of neuronal cell death, necrosis and apoptosis. Glutamate receptor antagonists in HCA are efficacious in reducing both necrosis and apoptosis; however, long-term efficacy and outcomes are uncertain due to the complicating factors of their own neurologic side effects, including problems with memory and cognition. Clinically safe glutamate receptor antagonists with minimal side effects are challenging to develop, but would be beneficial in reducing neurologic complications after HCA.
Acknowledgments
This work was supported by The Dana and Albert Broccoli Center for Aortic Diseases and grant 2RO1NS31238-05 from the National Institutes of Health. Elaine Tseng was supported by the Nina Braunwald Research Fellowship Award from the Thoracic Surgery Foundation for Research and Education.
Footnotes
Paper was presented in part at the 51st Annual Sessions of the Owen H. Wangensteen Surgical Forum, at the 82nd Annual Clinical Congress of the American College of Surgeons, San Francisco, CA, October 6-11, 1996.
References
- 1.Kouchoukos NT, Masetti P, Rokkas CK, Murphy SF. Hypothermic cardiopulmonary bypass and circulatory arrest for operations on the descending thoracic and thoracoabdominal aorta. Ann Thorac Surg. 2002;74:S1885–7. doi: 10.1016/s0003-4975(02)04153-x. discussion S1892–8. [DOI] [PubMed] [Google Scholar]
- 2.LeMaire SA, Carter SA, Coselli JS. The elephant trunk technique for staged repair of complex aneurysms of the entire thoracic aorta. Ann Thorac Surg. 2006;81:1561–9. doi: 10.1016/j.athoracsur.2005.11.038. discussion 1569. [DOI] [PubMed] [Google Scholar]
- 3.Niazi SA, Lewis FJ. Profound hypothermia in man; report of a case. Ann Surg. 1958;147:264–6. doi: 10.1097/00000658-195802000-00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kouchoukos NTBE, Doty D, Hanley F, Karp R. Cardiac Surgery. In: Kirklin JW, B-BBG, editors. Hypothermic, Circulatory Arrest, and Cardiopulmonary Bypass. 3. Vol. 1. Philadelphia: Churchill Livingstone, Inc; 2003. pp. 66–130. 2128. [Google Scholar]
- 5.Bellinger DC, Jonas RA, Rappaport LA, Wypij D, et al. Developmental and neurologic status of children after heart surgery with hypothermic circulatory arrest or low-flow cardiopulmonary bypass. N Engl J Med. 1995;332:549–55. doi: 10.1056/NEJM199503023320901. [DOI] [PubMed] [Google Scholar]
- 6.Bellinger DC, Wypij D, duDuplessis AJ, Rappaport LA, et al. Neurodevelopmental status at eight years in children with dextro-transposition of the great arteries: the Boston Circulatory Arrest Trial. J Thorac Cardiovasc Surg. 2003;126:1385–96. doi: 10.1016/s0022-5223(03)00711-6. [DOI] [PubMed] [Google Scholar]
- 7.Wypij D, Newburger JW, Rappaport LA, duPlessis AJ, et al. The effect of duration of deep hypothermic circulatory arrest in infant heart surgery on late neurodevelopment: the Boston Circulatory Arrest Trial. J Thorac Cardiovasc Surg. 2003;126:1397–403. doi: 10.1016/s0022-5223(03)00940-1. [DOI] [PubMed] [Google Scholar]
- 8.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tseng EE, Brock MV, Lange MS, Blue ME, et al. Neuronal nitric oxide synthase inhibition reduces neuronal apoptosis after hypothermic circulatory arrest. Ann Thorac Surg. 1997;64:1639–47. doi: 10.1016/s0003-4975(97)01110-7. [DOI] [PubMed] [Google Scholar]
- 10.Redmond JM, Gillinov AM, Zehr KJ, Blue ME, et al. Glutamate excitotoxicity: a mechanism of neurologic injury associated with hypothermic circulatory arrest. J Thorac Cardiovasc Surg. 1994;107:776–86. discussion 786–7. [PubMed] [Google Scholar]
- 11.Brock MV, Blue ME, Lowenstein CJ, Northington FA, et al. Induction of neuronal nitric oxide after hypothermic circulatory arrest. Ann Thorac Surg. 1996;62:1313–20. doi: 10.1016/0003-4975(96)00775-8. [DOI] [PubMed] [Google Scholar]
- 12.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A. 1990;87:682–5. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Yu SW, Koh DW, Lew J, et al. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–73. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newburger JW, Jonas RA, Wernovsky G, Wypij D, et al. A comparison of the perioperative neurologic effects of hypothermic circulatory arrest versus low-flow cardiopulmonary bypass in infant heart surgery. N Engl J Med. 1993;329:1057–64. doi: 10.1056/NEJM199310073291501. [DOI] [PubMed] [Google Scholar]
- 15.Mennander A, Paakko P, Hirvonen J, Anttila V, et al. Apoptotic activity is increased in brain cortex infarct after hypothermic circulatory arrest in a porcine model. Scand Cardiovasc J. 2002;36:247–9. doi: 10.1080/14017430260180427. [DOI] [PubMed] [Google Scholar]
- 16.Redmond JM, Gillinov AM, Blue ME, Zehr KJ, et al. The monosialoganglioside, GM1, reduces neurologic injury associated with hypothermic circulatory arrest. Surgery. 1993;114:324–32. discussion 332–3. [PubMed] [Google Scholar]
- 17.Redmond JM, Zehr KJ, Blue ME, Lange MS, et al. AMPA glutamate receptor antagonism reduces neurologic injury after hypothermic circulatory arrest. Ann Thorac Surg. 1995;59:579–84. doi: 10.1016/0003-4975(94)01047-1. [DOI] [PubMed] [Google Scholar]
- 18.Rimpilainen J, Pokela M, Kiviluoma K, Vainionpaa V, et al. The N-methyl-D-aspartate antagonist memantine has no neuroprotective effect during hypothermic circulatory arrest: a study in the chronic porcine model. J Thorac Cardiovasc Surg. 2001;121:957–68. doi: 10.1067/mtc.2001.112934. discussion 968–70. [DOI] [PubMed] [Google Scholar]
- 19.Tseng EE, Brock MV, Kwon CC, Annanata M, et al. Increased intracerebral excitatory amino acids and nitric oxide after hypothermic circulatory arrest. Ann Thorac Surg. 1999;67:371–6. doi: 10.1016/s0003-4975(99)00033-8. [DOI] [PubMed] [Google Scholar]
- 20.Amir G, Ramamoorthy C, Riemer RK, Reddy VM, Hanley FL. Neonatal brain protection and deep hypothermic circulatory arrest: pathophysiology of ischemic neuronal injury and protective strategies. Ann Thorac Surg. 2005;80:1955–64. doi: 10.1016/j.athoracsur.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 21.Won SJ, Kim DY, Gwag BJ. Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol. 2002;35:67–86. doi: 10.5483/bmbrep.2002.35.1.067. [DOI] [PubMed] [Google Scholar]
- 22.Chan PH. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann N Y Acad Sci. 2005;1042:203–9. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- 23.Ha HC, Snyder SH. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol Dis. 2000;7:225–39. doi: 10.1006/nbdi.2000.0324. [DOI] [PubMed] [Google Scholar]
- 24.Banasiak KJ, Xia Y, Haddad GG. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol. 2000;62:215–49. doi: 10.1016/s0301-0082(00)00011-3. [DOI] [PubMed] [Google Scholar]
- 25.Snider BJ, Gottron FJ, Choi DW. Apoptosis and necrosis in cerebrovascular disease. Ann N Y Acad Sci. 1999;893:243–53. doi: 10.1111/j.1749-6632.1999.tb07829.x. [DOI] [PubMed] [Google Scholar]
- 26.Ray SK. Currently evaluated calpain and caspase inhibitors for neuroprotection in experimental brain ischemia. Curr Med Chem. 2006;13:3425–40. doi: 10.2174/092986706779010342. [DOI] [PubMed] [Google Scholar]
- 27.Baumgartner WA, Walinsky PL, Salazar JD, Tseng EE, et al. Assessing the impact of cerebral injury after cardiac surgery: will determining the mechanism reduce this injury? Ann Thorac Surg. 1999;67:1871–3. doi: 10.1016/s0003-4975(99)00445-2. discussion 1891–4. [DOI] [PubMed] [Google Scholar]