

Abstract
Background
Ethanol stimulates the dopaminergic mesoaccumbal pathway, which is thought to play a role in ethanol reinforcement. Mu (μ)-opioid (MOP) receptors modulate accumbal dopamine activity, but it is not clear whether MOP receptors are involved in the mechanism of ethanol-stimulated accumbal dopamine release.
Methods
We investigated the role that MOP receptors play in ethanol (2.0 g/kg)-stimulated accumbal dopamine release by using MOP receptor knockout mice (C57BL/6J-129SvEv and congenic C57BL/6J genotypes) along with blockade of MOP receptors with a μ1 selective antagonist (naloxonazine).
Results
Both gene deletion and pharmacological antagonism of the MOP receptor decreased ethanol-stimulated accumbal dopamine release compared with controls with female mice showing a larger effect in the C57BL/6J-129SvEv genotype. However, both male and female mice showed reduced ethanol-stimulated dopamine release in the congenic MOP receptor knockout mice (C57BL/6J). No differences in the time course of dialysate ethanol concentration were found in any of the experiments.
Conclusions
The data demonstrate the existence of a novel interaction between genotype and sex in the regulation of ethanol-stimulated mesolimbic dopamine release by the MOP receptor. This implies that a more complete understanding of the epistatic influences on the MOP receptor and mesolimbic dopamine function may provide more effective pharmacotherapeutic interventions in the treatment of alcoholism.
Keywords: Dopamine, genotype, knockout mice, microdialysis, nucleus accumbens, sexual dimorphism
The mesoaccumbal dopaminergic pathway is proposed to play a role in mediating the reinforcing properties of ethanol (Doyon et al. 2003; Imperato and Di Chiara 1986; Melendez et al. 2002; Weiss et al. 1993), although the precise mechanisms by which this occurs are unclear at present. Several subtypes of opioid receptors are known to modulate dopamine activity in the nucleus accumbens (NAcc) (Borg and Taylor 1997; Spanagel et al. 1992; Yoshida et al. 1999), and these receptors are potential targets for altering ethanol-reinforced behavior. The nonselective opioid receptor antagonists naltrexone and nalmefene reduce alcohol drinking behavior in both humans and in animal models (Gonzales and Weiss 1998; Kornet et al. 1991; Mason et al. 1999; Volpicelli et al. 1992), but the subtypes of opioid receptors that mediate these effects are not yet clear.
Ethanol activation of the mesolimbic dopamine system may, in part, be mediated by stimulating the release of β-endorphin or other endogenous opioid peptides (Gianoulakis 1990; Marinelli et al. 2003; Olive et al. 2001). β-endorphin activates both delta and mu opioid receptors (MOPr) that are located either in the ventral tegmental area or the NAcc to alter accumbal dopamine release (Dilts and Kalivas 1990; Houghten et al. 1984; Mansour et al. 1987; Svingos et al. 1999). Consistent with this data, naltrexone reduced the accumbal dopamine release associated with ethanol self-administration (Gonzales and Weiss 1998). The MOPr has been implicated by data showing that systemic administration of naloxonazine, a long-lasting mu1 antagonist, inhibits ethanol-stimulated accumbal dopamine release in rats (Tanda and Di Chiara 1998). However, naloxonazine may also act on receptors other than mu1 (Dray and Nunan 1984; Ling et al. 1986), so the role of the MOPr in regulation of ethanol-stimulated dopamine release is still uncertain. Oslin et al. (2003) recently reported that a functional polymorphism of the human gene that codes for the MOPr is associated with the clinical response to naltrexone in treating alcohol dependence. Therefore, clarifying the role of the MOPr in neurochemical mechanisms that may contribute to ethanol reinforcement may lead to improvements in pharmacotherapy for treatment of alcohol dependence.
A decrease in both ethanol self-administration and/or conditioned place preference has been shown in three MOPr knockout mouse models, further suggesting a key role for the MOPr in ethanol reward and reinforcement (Becker et al. 2002; Hall et al. 2001; Roberts et al. 2000). In this study, we investigated the role of the MOPr in ethanol stimulation of mesolimbic dopamine activity using both null mutants and pharmacological blockade. The importance of sex influences in neuroscience studies is well known (Cahill 2006), and several sex differences in opioid responsiveness have been detected (Baamonde et al. 1989; Bartok and Craft, 1997; Cicero et al. 1996; Cicero et al. 2000). We therefore included both sexes in our studies and tested a secondary hypothesis that MOPr-mediated alcohol responsiveness included sexually dimorphic influences.
Methods and Materials
Subjects
Mice (male and female, 4–16 months old, 21–38 g) were obtained from the National Institute on Drug Abuse (NIDA, Baltimore, Maryland) or bred at the University of Texas at Austin. The mice were housed in a humidity and temperature controlled room with a 12:12-hour light/dark cycle (lights on 7 am), and animals had free access to food and water. All animal experiments were conducted under protocols approved by the Institutional Animal Care and Use Committee at the University of Texas, following National Institute of Heath (NIH) and United States Department of Agriculture (USDA) guidelines and regulations.
Heterozygous MOPr knockout mice were derived from the colony originally developed by Sora et al. (1997) at NIDA. Two colonies of MOPr knockout mice were established by nonsibling heterozygous breeding at the Animal Resources Center at the University of Texas at Austin. The first colony was on a mixed genetic background (C57BL/6J-129SvEv; 1:1) that had been maintained for approximately 30 generations, and breeding pairs were transferred to the University of Texas. The inbreeding coefficient as described by Falconer and Mackay (1996) was calculated at .926 indicating that by the 30th generation, 89% of the C57BL/6J-129SvEv genome was homozygous with a certainty of less than 11% possible segregation of B6 and 129 alleles. The second colony was from a congenic line created at NIDA in which the mixed background knockout mice were backcrossed for 10 generations onto the C57BL/6J strain. The C57BL/6J model was chosen for the backcross because these mice are well known to have high levels of drinking relative to other inbred strains (Belknap et al. 1993).
Genotyping
Genotyping was determined by polymerase chain reaction of tissue extracts. Details are provided in Supplement 1 online.
Surgery, Recovery, and Handling
Surgical preparation of the mice for microdialysis was conducted as previously described (Tang et al. 2003). We anesthetized the mice with 2.5% Avertin (2,2,2-tribromoethanol; .016 ml/g) or isoflurane (2%). The stainless steel guide cannula (8 mm long, .8 mm 20–21 gauge; Small Parts Inc., Miami Lakes, Florida) was positioned above the right NAcc (anterior (AP) +1.5–1.7, lateral (LAT) +.8, ventral (DV) −2.0–2.5 from bregma; Paxinos and Franklin 2001) and secured with a skull screw and dental cement. An obturator was placed into the guide cannula to prevent blockage. After the surgery, each animal was singly housed and given at least 4 days for recovery. During recovery, each mouse was injected daily with .1 ml saline for at least 3 days immediately before the microdialysis experiment in order to habituate the mouse to the intraperitoneal (IP) injection.
Microdialysis
Dialysis probes with 1 mm active dialyzing area were constructed as previously described (Tang et al. 2003). A probe was inserted into the guide cannula under halothane or isoflurane anesthesia (2.0% in air, flow rate at 2.0 liter/min) the night before the experiment, and the mouse was placed in a dialysis chamber. The probe was perfused with artificial cerebrospinal fluid (ACSF: 145 mM NaCl, 2.8 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, .25 mM ascorbic acid, and 5.4 mM D-glucose) at .2 μl/min overnight. The flow rate was increased to 1.0 μl/min the next morning, and after one hour, sample collection (15–17 min intervals) commenced. After the collection of four basal samples, we injected each animal with either saline or 2.0 g/kg ethanol (15% w/v, in saline). Sample collection continued for 3–4 hours. Calcium-dependency (percent decrease in dopamine concentration obtained during perfusion with calcium-free ACSF from regular ACSF) was determined by perfusing calcium-free ACSF for one hour at the end of the experiment. Dialysate samples were handled and analyzed by gas chromatography (for ethanol) and high performance liquid chromatography with electrochemical detection (for dopamine) as described in Tang et al. (2003) and Doyon et al. (2003). The detection limit for dopamine was 2 fmol.
Experimental Design
The first experiment used wildtype and MOPr knockout mice with a mixed genetic background (C57BL/6J – 129SvEv) to investigate the effect of the MOPr on ethanol-evoked (2.0 g/kg, IP) dopamine release in the ventral striatum. This dose was chosen because we and others previously showed that it reliably stimulates dopamine release (Olive et al. 2000; Tang et al. 2003). In addition, doses of ethanol in this range produce conditioned place preference in a variety of mouse strains indicating that this route and dose induce ethanol reward (Boyce-Rustay and Risinger 2003; Cunningham et al. 1996; Nocjar et al. 1999). The second experiment used mixed background wildtype animals to determine the effect of pharmacological antagonism of the mu1 opioid receptor with naloxonazine on the dopamine response stimulated by ethanol. Mice were pretreated with saline or naloxonazine (15 mg/kg, IP) 19 – 20 hours prior to microdialysis sampling. The third experiment was similar to the first except that the mice used were from the congenic line with a C57BL/6J background. Sex was an independent variable in the first three experiments. In addition, we performed a separate control experiment to examine the potential effect of saline injection on dopamine release in wildtype and MOPr knockout mice (congenic line).
Histology
After each experiment the mouse was given an intraperitoneal injection of chloral hydrate (60 mg/mouse) or pentobarbital (500 mg/kg) and was perfused intracardially with a 10% (v/v) formalin solution. Analysis of probe placement was carried out as previously described (Tang et al. 2003).
Data Analysis
Statistical significance was determined using a random block analysis of variance (ANOVA) with two between-subject variables (genotype or pretreatment and sex) and one within-subject variable (time before and after the injection). The basal response was defined as the dopamine level in the last two samples prior to the ethanol injection. ANOVA was used to determine the differences in the dialysate ethanol peak concentration and the rate of decline between either the genotypes or the pretreatments across sexes. The rate of decline was calculated as the slope of the linear part of the time-concentration curve. The criterion for the type I error was set to p < .05. Simple effect tests with Bonferroni correction factors were used to distinguish the influence of one variable on another variable when a significant interaction was detected. During the HPLC analysis there were a few samples lost due to technical reasons (either unidentified contaminants co-eluting with dopamine or the recorder was off-scale during dopamine elution) and these were estimated by averaging the dopamine responses before and after the missing point for statistical analysis. The degrees of freedom were corrected for each ANOVA based on the number of estimated points in each specific data set. Overall there were six missing points in the knockout experiment with the mixed background, three missing points in the naloxonazine experiment, and one missing point in the congenic experiment. Values are reported as mean ± SEM.
Results
Histological Analysis and Ca2+-Dependence of Dopamine Release
The microdialysis probe placements are shown in Figure 1. They were distributed either in the medial NAcc and passed through both the shell and core regions, or exclusively in the shell. The calcium-dependency, a functional measure of the physiological state of the dopamine nerve terminals during microdialysis sampling, was determined for all the animals used in the study. The criterion for acceptable calcium-dependency was the attainment of at least a 40% decrease in dialysate dopamine concentrations during the calcium-free ACSF perfusion compared with regular ACSF perfusion. Mean calcium-dependency for all groups was between 47–84%.
Figure 1.
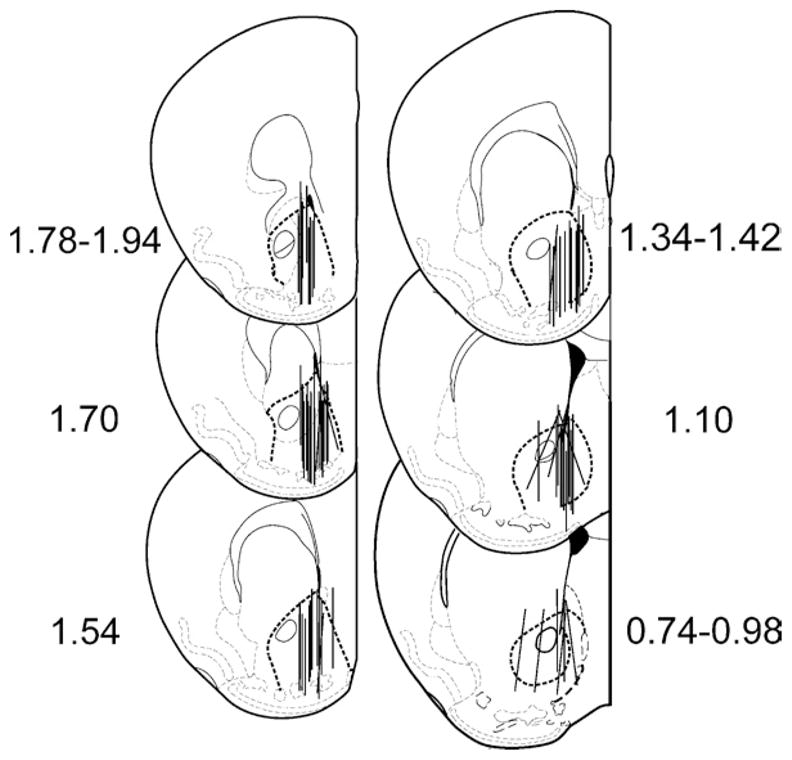
Microdialysis probe placements in the ventral striatum in MOPr knockout mice and their wildtype controls. Numbers on either side of the figure represent the position of the slice (in millimeters) relative to bregma. The border of the NAcc is highlighted with a heavy dashed line. Drawings were from Paxinos and Franklin (2001). MOPr, mu opioid receptors; NAcc, nucleus accumbens.
Effect of MOPr Knockout on Ethanol-Evoked Dopamine Release in the Ventral Striatum: Mixed Background
The increase in dialysate dopamine concentration stimulated by 2.0 g/kg ethanol (IP) was dependent on the MOPr and sex with female knockouts showing a greater reduction in the ethanol stimulation compared with males (Figure 2A, 2B). ANOVA on dialysate dopamine concentration revealed a significant three-way interaction, sex × genotype × time (F(8, 218) = 2.9, p < .05). Simple effects tests for the female mice revealed that deletion of the MOPr virtually abolished ethanol-stimulated dopamine release (F(8, 218) = 5.7, p < .05, for the genotype × time interaction). In control mice the maximal response was observed at the first point after the injection (F(1, 218) = 79, p < .05; simple effects within the female wildtype group), and the dopamine concentration was significantly elevated for 102 min compared with basal levels. In contrast, ethanol did not significantly stimulate dopamine release in the female knockout mice. Instead, there was a gradual decrease in accumbal dopamine levels relative to basal that was significant at 85 min after the injection (F(1, 218) > 10.9, p < .05; simple effects within the female knockout group).
Figure 2.
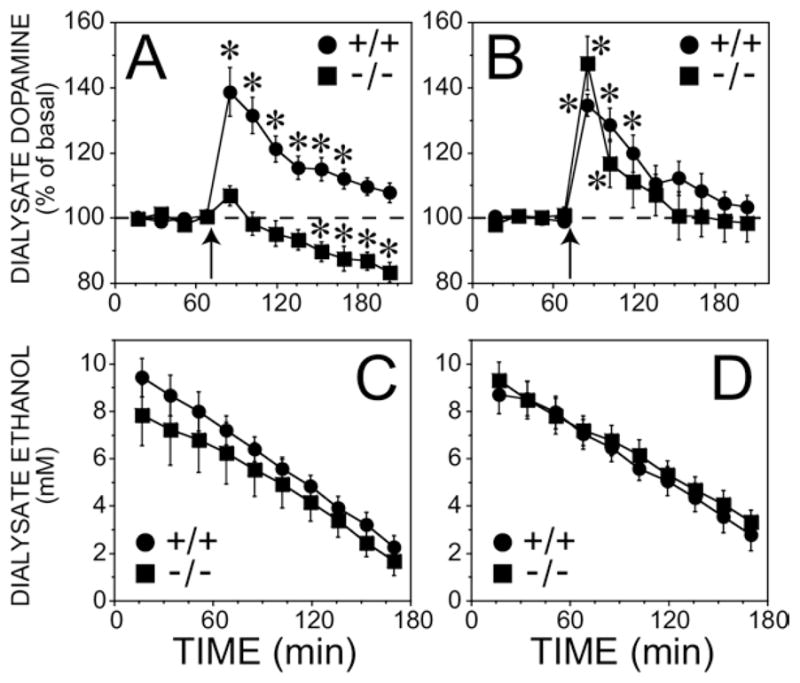
Dopamine response and dialysate ethanol in the ventral striatum after intraperitoneal administration of ethanol (2 g/kg) in MOPr knockout mice on a mixed genetic background (C57BL/6J-129SvEv) and their wild-type controls. (A) Female mice. The basal dopamine concentrations were 1.1 ± .2 nM and .4 ± .1 nM for wildtype and knockout groups, respectively (n = 8).(B) Male mice. The basal dopamine concentrations were 1.3 ± .1 nM and 1.7 ± .3 nM for wildtype and knockout groups, respectively (n = 8). The arrow indicates when the ethanol injection was given. *indicates significant different from basal dopamine concentration by simple effects post hoc analysis (p < .05) after a significant three-way interaction in the overall ANOVA. The ethanol content in samples taken after the injection is shown in (C) female mice and (D) male mice. Note that the x-axis starts at the point of the ethanol injection which is different from that shown in panels A and B. MOPr, mu opioid receptors; NAcc, nucleus accumbens; ANOVA, analysis of variance.
In male mice the effect of the null mutation of the MOPr was more subtle than in the females (Figure 2B). The peak ethanol stimulated dopamine response was similar in both the knockout and wildtype mice, but the knockout mice showed a more rapid return to baseline compared with the controls (F(8, 218) = 2.84, p < .05).
Basal dopamine levels were lower in the female knockout mice compared with the other groups (F(1, 28) = 5.10, p < .05 for the sex × genotype interaction, see caption Figure 2). Although the low basal level in female knockout mice tends to magnify changes in dopamine after ethanol injection when expressed as percentage of basal, this cannot account for the significant three-way interaction (sex × genotype × time) found in the ethanol-induced accumbal dopamine response because this group had the least response to ethanol.
Dialysate ethanol concentrations were maximal in the first sample, and this was followed by a linear decline in ethanol concentrations (Figure 2C, 2D). Neither sex nor genotype significantly affected peak concentration of ethanol (F(1, 21) = 1.12, p > .05) or the rate of decline (F(1, 21) = .01, p > .05).
Effect of Pharmacological Blockade of the mu1 Receptor on Ethanol-Evoked Dopamine Release in the Ventral Striatum: Mixed Background
Antagonism of the mu1 receptor with naloxonazine (15 mg/kg, IP) caused a sex dependent attenuation of the ethanol-stimulated dopamine response similar to our findings with the MOPr knockout mice (Figure 3A, 3B; p < .05 for the sex × pretreatment × time interaction by ANOVA, F(8, 165) = 2.3). Naloxonazine pretreatment abolished the stimulation of dialysate dopamine concentrations in response to the 2.0 g/kg ethanol challenge in the females (F (8, 165) = 2.9, p < .05). In males ethanol produced a similar increase in dialysate dopamine concentrations in the naloxonazine and saline groups (F (8, 165) = .39, p > .05 for the interaction between pretreatment and time).
Figure 3.
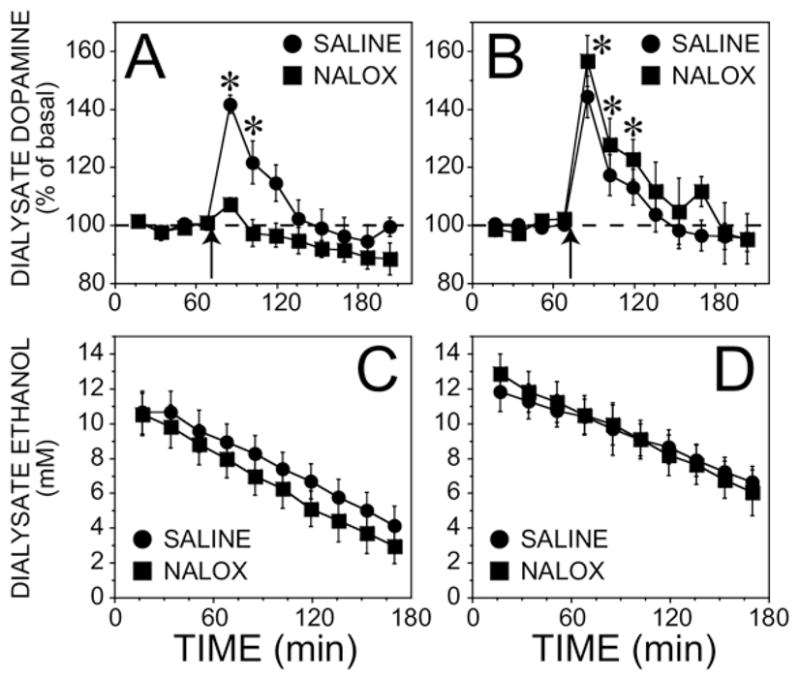
Effect of naloxonazine on ethanol-stimulated (2 g/kg, IP) dialysate dopamine and ethanol concentrations in the ventral striatum in wildtype mice with a mixed genetic background (C57BL/6J-129SvEv). Naloxonazine (NALOX) (15 mg/kg), IP, or saline was given 19 –20 hours prior to the experiment. (A) Female mice. The basal dopamine concentrations were 1.7 ± .4 nM and 1.9 ± .6 nM for saline and naloxonazine groups, respectively (n = 7). (B) Male mice. The basal dopamine concentrations were 1.1 ± .2 nM (n = 6) and .8 ± .1 nM (n = 5) for saline and naloxonazine groups, respectively. The arrow indicates when the ethanol injection was given. *Significantly different from basal dopamine concentration by simple effects post hoc analysis (p < .05) after a significant three-way interaction in the overall ANOVA. The ethanol content in samples taken after the injection is shown in (C) female mice and (D) male mice. Note that the x-axis starts at the point of the ethanol injection which is different from that shown in panels A and B. ANOVA, analysis of variance; IP, intraperitoneally.
Basal concentrations of dopamine were not significantly affected by naloxonazine, and male and female mice also had similar basal values (F(1, 21) = 2.19, p > .05). Neither blockade of the mu1 receptor nor sex affected the time course of dialysate ethanol concentrations in terms of peak concentration (F (1, 15) = .36, p > .05) and the rate of decline (Figure 3C, 3D; F (1, 15) = .01, p > .05).
Effect of MOPr Knockout on Ethanol-Evoked Dopamine Release in the Ventral Striatum: Congenic C57BL/6J Background
Deletion of the MOPr gene strongly inhibited the dopamine response due to ethanol administration in mice that were back-crossed onto the C57BL/6J strain (Figure 4A, 4B, F(8, 160) = 9.4, p < .05, genotype by time interaction in the overall ANOVA). Although ANOVA revealed that the effect of genotype was significantly different between males and females (F(8, 160) = 2.3, p < .05), the MOPr knockout in both sexes clearly showed a dramatic reduction in the stimulation of dopamine release immediately after the injection. Instead, the sex-dependence of the effect of MOPr deletion was due to a difference between males and females in the delayed reduction of dialysate dopamine concentrations below basal levels after the ethanol injection. Specifically, for both the male and female wildtype mice, the maximal dopamine response occurred immediately after the injection (F(1, 160) > 40.1, p < .05; simple effects test within the group). In contrast, in the male and female knockout mice, no changes were observed immediately after the ethanol injection. However, there was a slowly developing decrease in accumbal dopamine levels relative to basal that reached significance 45 min after the injection in the females (F(1, 160) = 9.3, p < .05; simple effects test within the knockouts), and 2 hr after the injection in males (F(1,160) = 9.1, p < .05).
Figure 4.
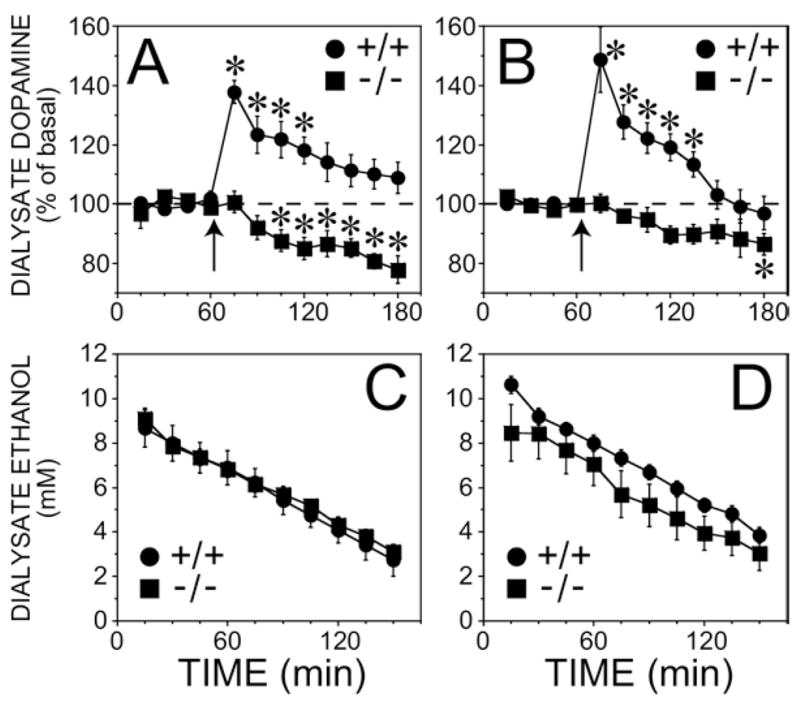
Dopamine response and dialysate ethanol in the ventral striatum after intraperitoneal administration of ethanol (2 g/kg) in MOPr knockout mice and their wildtype controls with a C57BL/6J background. Dialysate samples were collected every 15 min. (A) Female mice. The basal dopamine concentrations were .7 ± .1 nM (n = 5) and .7 ± .2 nM (n = 7) for wildtype and knockout groups, respectively. (B) Male mice. The basal dopamine concentrations were .9 ± .1 nM (n = 6) and 1.1 ± .3 nM (n = 6) for wildtype and knockout groups, respectively. The arrow indicates when the ethanol injection was given. *Significantly different from basal values by simple effects post hoc analyses (p <.05) after a significant three-way interaction overall ANOVA. The ethanol content in samples taken after the injection is shown in (C) female mice and (D) male mice. Note that the x-axis starts at the point of the ethanol injection which is different from that shown in panels A and B. MOPr, mu opioid receptors; ANOVA, analysis of variance.
Basal dopamine concentrations were not affected by the deletion of the MOPr knockout in the congenic strain in either males or females (F(1, 19) = 4.28, p > .05 for the interaction between genotype and sex). Additionally, peak dialysate ethanol concentrations (F(1,20) = 3.11, p > .05) and the rate of decline in ethanol concentration (Figure 4C, 4D, F(1,20) = .03, p > .05) were not different among the groups.
Effect of MOPr Knockout on Dopamine Release in the Ventral Striatum after Saline Injection: Congenic C57BL/6J Background
To determine whether the stimulation of dialysate dopamine concentrations may be affected by the IP injection procedure, we performed a separate experiment using the congenic strain of MOPr knockouts and their wildtype controls. Basal concentrations of dopamine were 2.0 ± .5 (n = 6), 1.2 ± .4 (n = 4), 1.8 ± .6 (n = 6), and 1.8 ± .5 nM (n = 4) for the male wildtype, male knockout, female wildtype, and female knockout mice, respectively. Saline injection did not significantly alter dialysate dopamine concentrations compared with basal concentrations in any of the groups (no effect of genotype × time, sex × time, or genotype × sex × time, F(4, 64) < 1.4, p > .05, ANOVA). In addition, neither the MOPr knockout nor sex significantly affected basal dopamine concentrations (F(1, 16) < .8, p > .05, ANOVA for main effects and interaction).
Discussion
Our study is the first to report diminished ethanol-stimulated dopamine release in any MOPr knockout model. A secondary finding from the present study is the novel interaction between sex and genotype in the MOPr-dependence of ethanol-stimulated mesolimbic dopamine release. Female, but not male, 129SvEv-C57BL/6J mice were found to require the MOPr for the expression of the neurochemical response to ethanol. Similar results were observed following pharmacological blockade of the mu1 receptor. In contrast, both male and female C57BL/6J mice required the MOPr for the ethanol-stimulated dopamine response. Previous work in rats has shown that the mu1 receptor is involved in ethanol-stimulated accumbal dopamine release (Tanda and Di Chiara 1998), but our study is the first to show that this mechanism is influenced by sex and genotype. Together, the results strongly support the hypothesis that activation of MOPr is a necessary step, under specific conditions, in the mesoaccumbal dopamine pathway that is activated by ethanol. This pathway is likely to mediate, at least in part, the reinforcing or rewarding properties of ethanol. The variation in the mechanism of the effect of ethanol on mesolimbic dopamine activity has important implications for understanding the development of alcohol abuse as well as for its treatment.
Our conclusion that the MOPr regulates ethanol-stimulated dopamine release in a sex and genotype dependent manner rests primarily on the results with the 129SvEv-C57BL/6J mice. On the basis of the breeding strategy used to maintain this model, the level of gene fixation is approximately 90%. Because we are the first to report this epistatic interaction between the MOPr and mesolimbic dopamine function, it is not known whether this interaction occurs in MOPr knockout mice generated by other investigators (Loh et al. 1998; Matthes et al. 1996; Schuller et al. 1999; Tian et al. 1997). However, one previous study using the same MOP knockout strain as the present experiments also reported a sexual dimorphism in ethanol reward (Hall et al. 2001). The correspondence between our neurochemical data and the previous behavioral studies further strengthens the idea that the MOPr mediation of mesolimbic dopamine activity contributes to the expression of the rewarding and reinforcing properties of ethanol. Taken together, these findings suggest that the influence of sex should be included in future studies of the interaction between the MOPr and ethanol.
A large body of evidence from behavioral and clinical studies has suggested that ethanol reward is influenced by endogenous opioid peptide dependent mechanisms (for review see Gianoulakis 2001). However, there is a controversy regarding the opioid receptor subtypes that mediate these effects of ethanol. Both MOP and delta opioid receptor antagonists decrease ethanol consumption (Honkanen et al. 1996; Hyytia 1993; Krishnan-Sarin et al. 1995; Krishnan-Sarin et al. 1998; Le et al. 1993), but clinical studies that investigate the effectiveness of opioid receptor antagonists for reducing relapse in alcoholism have mainly utilized nonselective antagonists (Mason et al. 1999; Volpicelli et al. 1992). Furthermore, the clinical efficacy of naltrexone, the most widely studied opioid receptor antagonist used for treatment of alcohol dependence, is low and inconsistent according to some authors (Kranzler and Van Kirk 2001). However, meta-analyses have demonstrated significant effects of naltrexone on relapse rate (Bouza et al. 2004). It has been suggested that genetic contributions to therapeutic response may underlie the somewhat disappointing effects of opiate antagonists in broader populations of alcoholics, but that specific, genetically distinct, subpopulations might exhibit better responses to naltrexone (Edenberg and Kranzler 2005). One possible explanation for the variability in clinical responses may be that there is underlying variability of the alcoholic population in the possible mechanisms that contribute to the rewarding and reinforcing properties of ethanol in the first place. This may include the regulation by opioid versus nonopioid mechanisms of actions of ethanol on the mesoaccumbal dopamine system.
Our finding that genotype dependent epistasis influences the mechanism by which the MOPr regulates the mesolimbic dopamine system has important implications for the pharmacological treatment of alcoholism. First, alterations in MOPr function through functional polymorphisms may affect the role of mesolimbic dopamine in ethanol reward. Alcohol dependent patients who possess a functional polymorphism in the MOPr gene have been shown to have lower rates of relapse in response to naltrexone (Oslin et al. 2003). These results are consistent with the findings that individuals with the polymorphism report enhanced positive feelings after ethanol administration (Ray and Hutchison 2004). Taken together, the results support the idea that genetic variation in the MOPr system, or in systems that interact with the MOPr, may influence the rewarding and reinforcing properties of ethanol. This implies that genetic variability in the MOPr system will also influence the ability of MOPr antagonists to reduce these effects. It is unclear whether the MOPr regulation of ethanol-stimulated dopamine release in the ventral striatum plays a role in human alcohol addiction, but our results suggest that genetic null variants of the MOPr could influence the vulnerability to ethanol addiction. It should be noted, however, that a null mutation in the MOPr gene will likely produce a much larger decrease in MOPr signaling compared with the Asn40Asp polymorphism that has been studied in human alcoholism.
Second, our finding that sex is an important factor that interacts with genotype in the modulation of ethanol-stimulated dopamine release by the MOPr suggests that sexual dimorphism may exist for the clinical response to MOPr antagonists used for the treatment of alcoholism. Recent reports suggest a sexually dimorphic response to naltrexone in the treatment of alcoholism (Garbutt et al. 2005; Kiefer et al. 2005), further pointing to the potential significance of the present findings. Our results imply that a more complete understanding of the influence of sex and genotype on ethanol responses may eventually lead to more effective pharmacological treatments for alcoholism.
The mechanisms that underlie MOPr regulation of ethanol-stimulated accumbal dopaminergic transmission are not clear. However, several lines of evidence show 1) the existence of endogenous opioid peptides in the VTA (Greenwell et al. 2002; Khachaturian et al. 1983), 2) that ethanol increases the release of a variety of endogenous opioid peptides from brain tissue in vitro and in vivo (Gianoulakis 1990; Olive et al. 2001), and 3) the stimulation of VTA dopamine neuron firing rate by activation of MOPr on GABA interneurons in the VTA (Johnson and North 1992). Our studies specifically implicate the MOPr as a regulator of the stimulation of VTA dopamine neurons by ethanol, but the present studies do not allow us to make conclusions regarding the anatomical sites of action of the MOPr that mediate these effects.
Ethanol concentrations in NAcc dialysates were monitored concurrently with the dopamine concentrations. No differences in the ethanol peak concentration and the rate of decline were observed in either the gene deletion or antagonist experiments. This confirms that the decrease in ethanol-stimulated accumbal dopamine we observed is the result of dysfunction of central mechanisms mediated by MOP receptors and potential epistatic interactions rather than an effect on ethanol pharmacokinetics. In agreement with our previous findings in C57BL/6 mice (Tang et al. 2003), we confirmed that the stimulation of dopamine release by ethanol administration is not due to the IP injection procedure. The results from our saline control experiments suggest that the ethanol-evoked dopamine response in the ventral striatum was due to a central pharmacological effect rather than a stress related phenomenon in the congenic MOPr knockouts of either sex. Therefore, we believe it is unlikely that the attenuation of ethanol-stimulated dopamine response in the female 129SvEv-C57BL/6J MOPr knockouts is due to an attenuation of the response due to the injection alone, although we cannot totally exclude this possibility.
Another interesting finding from our study is that females, but not males, from the 129SvEv-C57BL/6J MOPr knockout mice exhibited an apparent decrease in basal dopamine concentration, an effect not found in the naloxonazine experiment. This suggests that the apparent decrease in basal dopamine was due to developmental or compensatory mechanisms due to the lifelong deletion of the MOPr and not specifically due to MOPr mediated mechanisms. Male MOPr null mutants on a C57BL/6 background were previously reported to have decreased dopamine uptake and reduced dopamine release (Chefer et al. 2003) compared with wildtype controls, but females were not studied. It is possible that similar mechanisms occur in the female 129SvEv-C57BL/6J mice in the present study, and this could contribute to the apparent reduction in basal dopamine we observed.
Several pharmacological effects of morphine previously have been shown to differ between males and females, for example, antinociception (Baamonde et al. 1989; Bartok and Craft 1997; Cicero et al. 1996) and reinforcement (Cicero et al. 2000). In addition, an interaction between sex and genotype in MOPr mediated analgesia has previously been reported (Kest et al. 1999). Furthermore, sex differences in behavior have been reported in null mutant mice on a mixed genetic background (Hall et al. 2001; Walther et al. 2000). We now report an interaction between sex and genotype in the role of the MOPr in neurochemical effects of ethanol. The mechanism of the sex dependence of the various phenotypes cited above, including our data, is unclear at present. For our studies it is apparent that the 129SvEv strain contributes the gene(s) that is/are responsible for the epistatic interaction(s) we observed since there was no difference in the C57BL/6J congenic strain. Additional genetic studies are needed to determine the specific gene or genes that produce the sexual dimorphism of the MOPr response that we observed. In particular, it would be useful to replicate these experiments using the MOPr knockout backcrossed onto the 129SvEv strain. However, this model is not yet available.
A small but significant decrease in accumbal dopamine release occurred approximately 60–120 min after ethanol administration in the MOPr knockouts, but not in the naloxonazine experiment. This delayed inhibitory response in the MOPr knockouts may reflect the activation of kappa opioid (KOP) receptors in the NAcc by dynorphin-like peptide release produced by ethanol (Marinelli et al. 2006), which would reduce dopamine release (Spanagel et al. 1992). This effect would not be observed in wildtype mice because of the predominant effect of MOPr-mediated stimulation of dopamine release. Alternatively, the small delayed reduction in dopamine could reflect a general baseline drift that could be due, in part, to circadian variation. We think this is unlikely because it did not occur in every experiment, and we did not previously observe this phenomenon in C57BL/6 mice (Tang et al. 2003). Furthermore, this is supported by previously published data that showed minimal variation of accumbal extracellular dopamine during the light cycle (Paulson and Robinson 1994). The lack of delayed inhibition of dopamine release produced after ethanol administration in naloxonazine-pretreated mice suggests that dysfunction of the MOPr system at the gene level or at the receptor level may unmask the inhibitory tone to varying degrees.
The precise role that ethanol-stimulated dopamine release plays in the rewarding or reinforcing effects of ethanol is not completely clear at present. Accumbal dopamine signals may contribute to ethanol reward (Weiss et al. 1993), incentive-salience of ethanol (Robinson and Berridge 1993), or reward prediction (Schultz et al. 1997). In the present study the mice were ethanol naive, so the incentive and rewarding properties of ethanol were not established. Regardless of the role or roles that dopamine plays in the development and maintenance of ethanol reinforcement, our data suggests that the MOPr may be part of the neurochemical mechanisms that contribute to the behavioral consequences of ethanol administration, at least in specific populations.
In summary, our data demonstrate a novel interaction between genotype and sex in MOPr mediation of the stimulation of mesolimbic dopamine activity by ethanol. This interaction was not due to compensatory mechanisms in the MOPr knockout mice because it was observed after pharmacological blockade by naloxonazine. The results suggest that both sex and genotype specific epistasis are major factors that determine the role of the MOPr in mechanisms that contribute to ethanol reinforcement. Our results may help explain some of the variation in the effectiveness of MOPr antagonists in the treatment of alcohol dependence.
Acknowledgments
This work was supported by grants AA11852 and AA13486 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA), the Texas Commission on Alcohol and Drug Abuse, and by intramural funding at the National Institute on Drug Abuse, National Institute of Health (NIH)/Department of Health and Human Services (DHHS) (GRU, FSH, IS). Martin Job and Amanda Tang were supported by a training grant T32 AA07471 from NIAAA and fellowships from the Waggoner Center for Alcohol and Addiction Research. We thank Judith A. Randall for excellent technical assistance, and Julie Wu, Julie Owen, and Christina Schier for assistance with genotyping.
Footnotes
Supplementary material cited in this article is available online.
Both MOJ and AT should be considered first authors based on their respective contributions.
References
- Baamonde AI, Hidalgo A, Andres-Trelles F. Sex-related differences in the effects of morphine and stress on visceral pain. Neuropharmacology. 1989;28:967–970. doi: 10.1016/0028-3908(89)90197-4. [DOI] [PubMed] [Google Scholar]
- Bartok RE, Craft RM. Sex differences in opioid antinociception. J Pharmacol Exp Ther. 1997;282:769–778. [PubMed] [Google Scholar]
- Becker A, Grecksch G, Kraus J, Loh HH, Schroeder H, Hollt V. Rewarding effects of ethanol and cocaine in mu opioid receptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:296–302. doi: 10.1007/s00210-002-0533-2. [DOI] [PubMed] [Google Scholar]
- Belknap JK, Crabbe JC, Young ER. Voluntary consumption of ethanol in 15 inbred mouse strains. Psychopharmacology (Berl) 1993;112:503–510. doi: 10.1007/BF02244901. [DOI] [PubMed] [Google Scholar]
- Borg PJ, Taylor DA. Involvement of mu- and delta-opioid receptors in the effects of systemic and locally perfused morphine on extracellular levels of dopamine, DOPAC and HVA in the nucleus accumbens of the halothane-anaesthetized rat. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:582–588. doi: 10.1007/pl00004987. [DOI] [PubMed] [Google Scholar]
- Bouza C, Angeles M, Munoz A, Amate JM. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction. 2004;99:811–828. doi: 10.1111/j.1360-0443.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Risinger FO. Dopamine D3 receptor knockout mice and the motivational effects of ethanol. Pharmacol Biochem Behav. 2003;75:373–379. doi: 10.1016/s0091-3057(03)00091-1. [DOI] [PubMed] [Google Scholar]
- Cahill L. Why sex matters for neuroscience. Nat Rev Neurosci. 2006;7:477–484. doi: 10.1038/nrn1909. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Kieffer BL, Shippenberg TS. Basal and morphine-evoked dopaminergic neurotransmission in the nucleus accumbens of MOR-and DOR-knockout mice. Eur J Neurosci. 2003;18:1915–1922. doi: 10.1046/j.1460-9568.2003.02912.x. [DOI] [PubMed] [Google Scholar]
- Cicero TJ, Nock B, Meyer ER. Gender-related differences in the antinociceptive properties of morphine. J Pharmacol Exp Ther. 1996;279:767–773. [PubMed] [Google Scholar]
- Cicero TJ, Ennis T, Ogden J, Meyer ER. Gender differences in the reinforcing properties of morphine. Pharmacol Biochem Behav. 2000;65:91–96. doi: 10.1016/s0091-3057(99)00174-4. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Okorn DM, Howard CE. Ethanol-induced conditioned place preference and activation in 15 inbred mouse strains. Alcohol Clin Exp Res. 1996;20:59A. [Google Scholar]
- Dilts RP, Kalivas PW. Autoradiographic localization of delta opioid receptors within the mesocorticolimbic dopamine system using radio-iodinated [2-D-penicillamine, 5-D-penicillamine]enkephalin (125I-DPDPE) Synapse. 1990;6:121–132. doi: 10.1002/syn.890060203. [DOI] [PubMed] [Google Scholar]
- Doyon WM, York JL, Diaz LM, Samson HH, Czachowski CL, Gonzales RA. Dopamine activity in the nucleus accumbens during consummatory phases of oral ethanol self-administration. Alcohol Clin Exp Res. 2003;27:1573–1582. doi: 10.1097/01.ALC.0000089959.66222.B8. [DOI] [PubMed] [Google Scholar]
- Dray A, Nunan L. Evidence that naloxonazine produces prolonged antagonism of central delta opioid receptor activity in vivo. Brain Res. 1984;323:123–127. doi: 10.1016/0006-8993(84)90273-7. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ, Kranzler HR. The contribution of genetics to addiction therapy approaches. Pharmacol Ther. 2005;108:86–93. doi: 10.1016/j.pharmthera.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4. Essex; England, Longman: 1996. [Google Scholar]
- Garbutt JC, Kranzler HR, O’Malley SS, Gastfriend DR, Pettinati HM, Silverman BL, et al. Efficacy and tolerability of long-acting injectable naltrexone for alcohol dependence: a randomized controlled trial. JAMA. 2005;293:1617–1625. doi: 10.1001/jama.293.13.1617. [DOI] [PubMed] [Google Scholar]
- Gianoulakis C. Characterization of the effects of acute ethanol administration on the release of beta-endorphin peptides by the rat hypothalamus. Eur J Pharmacol. 1990;180:21–29. doi: 10.1016/0014-2999(90)90588-w. [DOI] [PubMed] [Google Scholar]
- Gianoulakis C. Influence of the endogenous opioid system on high alcohol consumption and genetic predisposition to alcoholism. J Psychiatry Neurosci. 2001;26:304–318. [PMC free article] [PubMed] [Google Scholar]
- Gonzales RA, Weiss F. Suppression of ethanol-reinforced behavior by naltrexone is associated with attenuation of the ethanol-induced increase in dialysate dopamine levels in the nucleus accumbens. J Neurosci. 1998;18:10663–10671. doi: 10.1523/JNEUROSCI.18-24-10663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwell TN, Zangen A, Martin-Schild S, Wise RA, Zadina JE. Endomorphin-1 and −2 immunoreactive cells in the hypothalamus are labeled by fluoro-gold injections to the ventral tegmental area. J Comp Neurol. 2002;454:320–328. doi: 10.1002/cne.10464. [DOI] [PubMed] [Google Scholar]
- Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 2001;154:43–49. doi: 10.1007/s002130000622. [DOI] [PubMed] [Google Scholar]
- Honkanen A, Vilamo L, Wegelius K, Sarviharju M, Hyytia P, Korpi ER. Alcohol drinking is reduced by a mu 1-but not by a delta-opioid receptor antagonist in alcohol-preferring rats. Eur J Pharmacol. 1996;304:7–13. doi: 10.1016/0014-2999(96)00118-5. [DOI] [PubMed] [Google Scholar]
- Houghten RA, Johnson N, Pasternak GW. [3H]-beta-endorphin binding in rat brain. J Neurosci. 1984;4:2460–2465. doi: 10.1523/JNEUROSCI.04-10-02460.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyytia P. Involvement of mu-opioid receptors in alcohol drinking by alcohol-preferring AA rats. Pharmacol Biochem Behav. 1993;45:697–701. doi: 10.1016/0091-3057(93)90527-z. [DOI] [PubMed] [Google Scholar]
- Imperato A, Di Chiara G. Preferential stimulation of dopamine release in the nucleus accumbens of freely moving rats by ethanol. J Pharmacol Exp Ther. 1986;239:219–228. [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kest B, Wilson SG, Mogil JS. Sex differences in supraspinal morphine analgesia are dependent on genotype. J Pharmacol Exp Ther. 1999;289:1370–1375. [PubMed] [Google Scholar]
- Khachaturian H, Lewis ME, Watson SJ. Enkephalin systems in diencephalon and brainstem of the rat. J Comp Neurol. 1983;220:310–320. doi: 10.1002/cne.902200305. [DOI] [PubMed] [Google Scholar]
- Kiefer F, Jahn H, Wiedemann K. A neuroendocrinological hypothesis on gender effects of naltrexone in relapse prevention treatment. Pharmacopsychiatry. 2005;38:184–186. doi: 10.1055/s-2005-871244. [DOI] [PubMed] [Google Scholar]
- Kornet M, Goosen C, Van Ree JM. Effect of naltrexone on alcohol consumption during chronic alcohol drinking and after a period of imposed abstinence in free-choice drinking rhesus monkeys. Psychopharmacology (Berl) 1991;104:367–376. doi: 10.1007/BF02246038. [DOI] [PubMed] [Google Scholar]
- Kranzler HR, Van Kirk J. Efficacy of naltrexone and acamprosate for alcoholism treatment: a meta-analysis. Alcohol Clin Exp Res. 2001;25:1335–1341. [PubMed] [Google Scholar]
- Krishnan-Sarin S, Jing SL, Kurtz DL, Zweifel M, Portoghese PS, Li TK, Froehlich JC. The delta opioid receptor antagonist naltrindole attenuates both alcohol and saccharin intake in rats selectively bred for alcohol preference. Psychopharmacology (Berl) 1995;120:177–185. doi: 10.1007/BF02246191. [DOI] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Wand GS, Li XW, Portoghese PS, Froehlich JC. Effect of mu opioid receptor blockade on alcohol intake in rats bred for high alcohol drinking. Pharmacol Biochem Behav. 1998;59:627–635. doi: 10.1016/s0091-3057(97)00474-7. [DOI] [PubMed] [Google Scholar]
- Le AD, Poulos CX, Quan B, Chow S. The effects of selective blockade of delta and mu opiate receptors on ethanol consumption by C57BL/6 mice in a restricted access paradigm. Brain Res. 1993;630:330–332. doi: 10.1016/0006-8993(93)90672-a. [DOI] [PubMed] [Google Scholar]
- Ling GS, Simantov R, Clark JA, Pasternak GW. Naloxonazine actions in vivo. Eur J Pharmacol. 1986;129:33–38. doi: 10.1016/0014-2999(86)90333-x. [DOI] [PubMed] [Google Scholar]
- Loh HH, Liu HC, Cavalli A, Yang W, Chen YF, Wei LN. mu Opioid receptor knockout in mice: effects on ligand-induced analgesia and morphine lethality. Brain Res Mol Brain Res. 1998;54:321–326. doi: 10.1016/s0169-328x(97)00353-7. [DOI] [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Autoradiographic differentiation of mu, delta, and kappa opioid receptors in the rat forebrain and midbrain. J Neurosci. 1987;7:2445–2464. [PMC free article] [PubMed] [Google Scholar]
- Marinelli PW, Lam M, Bai L, Quirion R, Gianoulakis C. A microdialysis profile of dynorphin A (1–8) release in the rat nucleus accumbens following alcohol administration. Alcohol Clin Exp Res. 2006;30:982–990. doi: 10.1111/j.1530-0277.2006.00112.x. [DOI] [PubMed] [Google Scholar]
- Marinelli PW, Quirion R, Gianoulakis C. A microdialysis profile of beta-endorphin and catecholamines in the rat nucleus accumbens following alcohol administration. Psychopharmacology (Berl) 2003;169:60–67. doi: 10.1007/s00213-003-1490-2. [DOI] [PubMed] [Google Scholar]
- Mason BJ, Salvato FR, Williams LD, Ritvo EC, Cutler RB. A double-blind, placebo-controlled study of oral nalmefene for alcohol dependence. Arch Gen Psychiatry. 1999;56:719–724. doi: 10.1001/archpsyc.56.8.719. [DOI] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Melendez RI, Rodd-Henricks ZA, Engleman EA, Li TK, McBride WJ, Murphy JM. Microdialysis of dopamine in the nucleus accumbens of alcohol-preferring (P) rats during anticipation and operant self-administration of ethanol. Alcohol Clin Exp Res. 2002;26:318–325. [PubMed] [Google Scholar]
- Nocjar C, Middaugh LD, Tavernetti M. Ethanol consumption and place-preference conditioning in the alcohol-preferring C57BL/6 mouse: relationship with motor activity patterns. Alcohol Clin Exp Res. 1999;23:683–692. [PubMed] [Google Scholar]
- Olive MF, Koenig HN, Nannini MA, Hodge CW. Stimulation of endorphin neurotransmission in the nucleus accumbens by ethanol, cocaine, and amphetamine. J Neurosci. 2001;21:RC184, 1–5. doi: 10.1523/JNEUROSCI.21-23-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive MF, Mehmert KK, Messing RO, Hodge CW. Reduced operant ethanol self-administration and in vivo mesolimbic dopamine responses to ethanol in PKC epsilon-deficient mice. Eur J Neurosci. 2000;12:4131–4140. doi: 10.1046/j.1460-9568.2000.00297.x. [DOI] [PubMed] [Google Scholar]
- Oslin DW, Berrettini W, Kranzler HR, Pettinati H, Gelernter J, Volpicelli JR, O’Brien CP. A functional polymorphism of the mu-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology. 2003;28:1546–1552. doi: 10.1038/sj.npp.1300219. [DOI] [PubMed] [Google Scholar]
- Paulson PE, Robinson TE. Relationship between circadian changes in spontaneous motor activity and dorsal versus ventral striatal dopamine neurotransmission assessed with on-line microdialysis. Behav Neurosci. 1994;108:624–635. doi: 10.1037//0735-7044.108.3.624. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego, CA: Academic Press; 2001. [Google Scholar]
- Ray LA, Hutchison KE. A polymorphism of the mu-opioid receptor gene (OPRM1) and sensitivity to the effects of alcohol in humans. Alcohol Clin Exp Res. 2004;28:1789–1795. doi: 10.1097/01.alc.0000148114.34000.b9. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, Gold LH. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther. 2000;293:1002–1008. [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ, et al. Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat Neurosci. 1999;2:151–156. doi: 10.1038/5706. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM, et al. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci U S A. 1997;94:1544–1549. doi: 10.1073/pnas.94.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci U S A. 1992;89:2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svingos AL, Clarke CL, Pickel VM. Localization of the delta-opioid receptor and dopamine transporter in the nucleus accumbens shell: implications for opiate and psychostimulant cross-sensitization. Synapse. 1999;34:1–10. doi: 10.1002/(SICI)1098-2396(199910)34:1<1::AID-SYN1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Tanda G, Di Chiara G. A dopamine-mu1 opioid link in the rat ventral tegmentum shared by palatable food (Fonzies) and nonpsychostimulant drugs of abuse. Eur J Neurosci. 1998;10:1179–1187. doi: 10.1046/j.1460-9568.1998.00135.x. [DOI] [PubMed] [Google Scholar]
- Tang A, George MA, Randall JA, Gonzales RA. Ethanol increases extracellular dopamine concentration in the ventral striatum in C57BL/6 mice. Alcohol Clin Exp Res. 2003;27:1083–1089. doi: 10.1097/01.ALC.0000075825.14331.65. [DOI] [PubMed] [Google Scholar]
- Tian M, Broxmeyer HE, Fan Y, Lai Z, Zhang S, Aronica S, et al. Altered hematopoiesis, behavior, and sexual function in mu opioid receptor-deficient mice. J Exp Med. 1997;185:1517–1522. doi: 10.1084/jem.185.8.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- Walther T, Voigt JP, Fink H, Bader M. Sex specific behavioural alterations in Mas-deficient mice. Behav Brain Res. 2000;107:105–109. doi: 10.1016/s0166-4328(99)00115-1. [DOI] [PubMed] [Google Scholar]
- Weiss F, Lorang MT, Bloom FE, Koob GF. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther. 1993;267:250–258. [PubMed] [Google Scholar]
- Yoshida Y, Koide S, Hirose N, Takada K, Tomiyama K, Koshikawa N, Cools AR. Fentanyl increases dopamine release in rat nucleus accumbens: involvement of mesolimbic mu- and delta-2-opioid receptors. Neuroscience. 1999;92:1357–1365. doi: 10.1016/s0306-4522(99)00046-9. [DOI] [PubMed] [Google Scholar]