

Abstract
Cystic fibrosis (CF) transmembrane conductance regulator (CFTR)-dependent airway epithelial bicarbonate transport is hypothesized to participate in airway surface liquid pH regulation and contribute to lung defense. We measured pH and ionic composition in apical surface liquid (ASL) on polarized normal (NL) and CF primary bronchial epithelial cell cultures under basal conditions, after cAMP stimulation, and after challenge with luminal acid loads. Under basal conditions, CF epithelia acidified ASL more rapidly than NL epithelia. Two ASL pH regulatory paths that contributed to basal pH were identified in the apical membrane of airway epithelia, and their activities were measured. We detected a ouabain-sensitive (nongastric) H+,K+-ATPase that acidified ASL, but its activity was not different in NL and CF cultures. We also detected the following evidence for a CFTR-dependent 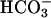 secretory pathway that was defective in CF: (i) ASL [
secretory pathway that was defective in CF: (i) ASL [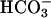 ] was higher in NL than CF ASL; (ii) activating CFTR with forskolin/3-isobutyl-1-methylxanthine alkalinized NL ASL but acidified CF ASL; and (iii) NL airway epithelia more rapidly and effectively alkalinized ASL in response to a luminal acid challenge than CF epithelia. We conclude that cultured human CF bronchial epithelial pHASL is abnormally regulated under basal conditions because of absent CFTR-dependent
] was higher in NL than CF ASL; (ii) activating CFTR with forskolin/3-isobutyl-1-methylxanthine alkalinized NL ASL but acidified CF ASL; and (iii) NL airway epithelia more rapidly and effectively alkalinized ASL in response to a luminal acid challenge than CF epithelia. We conclude that cultured human CF bronchial epithelial pHASL is abnormally regulated under basal conditions because of absent CFTR-dependent 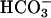 secretion and that this defect can lead to an impaired capacity to respond to airway conditions associated with acidification of ASL.
secretion and that this defect can lead to an impaired capacity to respond to airway conditions associated with acidification of ASL.
Cystic fibrosis (CF) is a fatal hereditary disease caused by lack of functional expression of the CF transmembrane conductance regulator (CFTR) in the apical membrane of airway epithelial cells (1). Although CFTR is a cAMP-regulated apical anion channel (2), a universally accepted paradigm linking abnormal ion transport to fatal lung disease in CF is lacking. Abnormal pH regulation of apical surface liquid (ASL) could contribute to CF pathogenesis, because biological processes on airway surfaces are pH-sensitive. Observations of more acidic pancreatic and seminal secretions in CF patients (3, 4) suggest that a defect in CFTR function could yield abnormally acidic luminal solutions, reflecting the fact that bicarbonate can permeate CFTR (5).
Difficulties sampling human ASL have limited ASL pH measurements in vivo. Studies of cultured human airway epithelial cells support CFTR-dependent apical bicarbonate conductance (6-8), but have not demonstrated pHASL dysregulation on CF airway epithelia. Although in vivo measurements of tracheal surface liquid in normal and CFTR knockout mice failed to reveal a significant pH difference (9), the lack of an abnormal bioelectric or pathologic pulmonary phenotype in the CF knockout mouse (10) and the paucity of CFTR expression in the murine trachea (11) argue that these data may have limited relevance to human normal and CF airway epithelium.
The inability to speculate on consequences of reduced CFTR-dependent 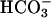 secretion into CF ASL reflects the paucity of data on other pH regulatory pathways in airway epithelia; no evidence for an apical membrane Na+/H+ exchanger (12, 13) or anion exchanger has been reported. A luminal H+,K+-ATPase (exchanging luminal K+ for cytosolic protons, and acidifying pHASL) was postulated in nasal airway epithelia (14), although its molecular identification and functional role were not tested, and recent data did not confirm its presence (15). A paracellular pathway could also contribute to pHASL regulation, but the permeability of this pathway to H+ or
secretion into CF ASL reflects the paucity of data on other pH regulatory pathways in airway epithelia; no evidence for an apical membrane Na+/H+ exchanger (12, 13) or anion exchanger has been reported. A luminal H+,K+-ATPase (exchanging luminal K+ for cytosolic protons, and acidifying pHASL) was postulated in nasal airway epithelia (14), although its molecular identification and functional role were not tested, and recent data did not confirm its presence (15). A paracellular pathway could also contribute to pHASL regulation, but the permeability of this pathway to H+ or 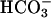 has not been studied.
has not been studied.
We hypothesized that airway pHASL regulation involves apical membrane ion transport processes mediated by CFTR and by a H+,K+-ATPase, in parallel with the paracellular movement of H+ and 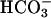 , and that absence of CFTR-dependent
, and that absence of CFTR-dependent 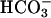 secretion in CF airway epithelia leads to an inability to balance proton secretion and alkalinize ASL. Therefore, we measured the pH and ionic composition of the ASL of primary cultures of CF and NL respiratory epithelia under basal conditions and after airway luminal acid challenge.
secretion in CF airway epithelia leads to an inability to balance proton secretion and alkalinize ASL. Therefore, we measured the pH and ionic composition of the ASL of primary cultures of CF and NL respiratory epithelia under basal conditions and after airway luminal acid challenge.
Methods
Cell Culture. Human bronchial epithelial cells were obtained at the time of lung transplantation from main stem/lobar bronchi of CF (n = 20), normal (NL) healthy control (n = 24), and disease control (n = 4, one primary ciliary dyskinesia, two non-CF bronchiectasis, one primary pulmonary hypertension) lungs, using protocols approved by the Institutional Committee on the Protection of the Rights of Human Subjects. Nasal epithelial cells were obtained from resected nasal polyps. Disaggregated airway epithelial cells were harvested, seeded, and cultured on 1.13-cm2 Transwell Col porous filters (pore diameter = 0.45 μm, Costar, Cambridge, MA) as described (16, 17), and studied 10-14 days after becoming confluent. Function was assessed by measuring transepithelial bioelectric potential difference (PD). The transepithelial resistance of CF and NL cultures was similar (CF 510 ± 24 vs. NL 534 ± 36 Ω·cm2). For studies of intracellular pH (pHi) regulation, airway cells were studied as described (12). Freshly excised bronchial tissue specimens were used for H+,K+-ATPase expression studies.
Collection of ASL and Measurement of Ionic Composition. The apical culture surface was washed (PBS). Test media was added to the apical compartment, aspirated, and reapplied. After specified time intervals, aliquots (1-5 μl) of ASL were removed with a constant bore microcapillary pipette as described (17). Na+ and K+ were measured by flame emission spectroscopy and Cl- was measured by amperometric titration (18). 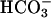 was measured by coupled enzymatic reactions and spectrophotometric analysis of NAD. Lactate was measured by a NAD-coupled assay (Sigma).
was measured by coupled enzymatic reactions and spectrophotometric analysis of NAD. Lactate was measured by a NAD-coupled assay (Sigma).
Measurement of ASL pH. We developed a novel technique to measure pH with pH-sensitive microelectrodes (Microelectrodes, Bedford, NH) in small-volume samples that could be quickly temperature, water vapor, and gas equilibrated. Microaliquots (0.3-1.0 μl) were aspirated from the microcapillary tube into the tip of a section (0.5 cm) of CO2-permeable silicone tubing (Helix Medical, Malvern, PA; 0.025 in inner diameter, 0.047 in outer diameter). The pH microelectrode was inserted into the sample by stretching the end of the tubing containing the sample over the microelectrode tip, the tight fit trapping a thin layer of liquid between the tubing wall and the electrode, so that reference and pH electrodes made contact with the sample. The microelectrode and tubing were placed in a water bath that was continually gassed and equilibrated with 5% CO2. A column of air in the tubing, distal to the electrode, prevented water from reaching the sample. CO2 equilibration was complete within 2 min, as evidenced by a stable pH. Measurements were accurate and reproducible within ±0.01 pH units.
Measurement of H+,K+-ATPase Expression. Freshly excised tissue specimens were rapidly dissected and snap-frozen in OCT embedding compound. Thin sections (6-10 μm) were cut by a cryotome, mounted on glass slides, and stored at -80°C until analysis. Well differentiated human bronchial cultures and frozen tissue sections were fixed in 4% paraformaldehyde (room temperature, 4 min), permeabilized in 100% ethanol (-20°C, 4 min), and blocked with 20% goat serum in 50 mM sodium phosphate, pH 7.4/150 mM NaCl (3 h), before overnight (4°C) incubation with a monoclonal antibody against H+,K+-ATPase (Sigma) or control IgG (Jackson ImmunoResearch). Specimens were washed in PBS and incubated (1 h, 23°C) with Texas Red-conjugated goat anti-mouse IgG (Jackson ImmunoResearch) and fluorescein-labeled phalloidin (Molecular Probes). After washing, cultures were excised from the plastic supports and mounted on glass by using Vectashield containing 4,6-diamidino-2-phenylindole (DAPI) to label nuclei blue (Vector Laboratories). Confocal analyses used a Leica TCS 4D confocal microscope. All of the channels (red, green, and blue) were registered by independent and sequential scan of the specimens. Cultures were scanned in the xz axis and tissue sections were scanned in the xy axis.
Nongastric H+,K+-ATPase mRNA Expression. RNA was isolated by using the Qiagen RNAeasy RNA purification kit with DNase treatment according to the manufacturer's instructions (Qiagen, Valencia, CA). RT-PCR was performed with SuperScript II reverse transcriptase (Life Technologies, Rockville, MD) using 1 μgoftotal RNA and random primers according to manufacturer's instructions. Control RNA (colon and stomach) was purchased from Ambion (Austin, TX). PCR was performed by using the Roche LightCyler Thermocycler and the LightCyler-FastStart DNA Master SYBR Green I kit (Roche Diagnostics), using the forward primer 5′-TCTGAAGAACAACTGCCTG and reverse primer 5′-TACACGTTGTTCAGGGATG. The PCR product corresponds to positions 266-604 of the ATP1AL1 sequence (the nongastric K+,H+-ATPase, GenBank accession no. U02076). Primers were designed to span known introns to eliminate signal from contaminating DNA. Positive control primers for the cyclophilin cDNA were 5′-CCGTGTTCTTCGACATTGCC (forward) and 5′-ACACCACATGCTTGCCATCC (reverse). For PCR, 1 μl of cDNA template was used in a 20-μl reaction volume containing 3 mM MgCl2, 500 nM of each primer, and 1× master mix. Amplification conditions were as follows: 1 cycle at 95°C for 10 min and 45 cycles of 95°C for 15 sec, 55°C for 5 sec, and 72°C for 18 sec. The amplified products were separated on a 1.2% agarose gel.
cAMP-Mediated Regulation of pHASL in CF and NL Cultures. After washing of the apical surfaces of CF and NL bronchial tissues, 100 μl of 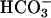 - and K+-free saline Ringer's solution (140 mM Na+/140 mM Cl-/1.2 mM Ca2+/1.2 mM Mg2+/2.5 mM
- and K+-free saline Ringer's solution (140 mM Na+/140 mM Cl-/1.2 mM Ca2+/1.2 mM Mg2+/2.5 mM 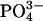 ,pH 5.6) was added apically. Forskolin (10-5 M) and 3-isobutyl-1-methylxanthine (IBMX) (2 × 10-4 M) or vehicle control were added to the apical and basolateral medium. ASL was sampled for measurements of pH and ionic composition.
,pH 5.6) was added apically. Forskolin (10-5 M) and 3-isobutyl-1-methylxanthine (IBMX) (2 × 10-4 M) or vehicle control were added to the apical and basolateral medium. ASL was sampled for measurements of pH and ionic composition.
Assessment of Paracellular 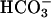 Permselectivity. Cultures of CF nasal epithelium (n = 8, triplicate preparations) were mounted in a modified Ussing chamber. The CF apical membrane ionic conductance is virtually zero in the presence of luminal amiloride (10-4 M), because CFTR is absent and Na+ channels are blocked (19), and basal calcium activated chloride conductance is minimal in the absence of a signal for increased intracellular Ca2+ (20). Consequently, voltage changes induced by changes in apical vs. basolateral anion bath concentrations are likely dominated by bi-ionic PDs across the paracellular shunt (21). The basolateral compartment was continually perfused with KBR solution (140 mM Na+/125 mM Cl-/25 mM
Permselectivity. Cultures of CF nasal epithelium (n = 8, triplicate preparations) were mounted in a modified Ussing chamber. The CF apical membrane ionic conductance is virtually zero in the presence of luminal amiloride (10-4 M), because CFTR is absent and Na+ channels are blocked (19), and basal calcium activated chloride conductance is minimal in the absence of a signal for increased intracellular Ca2+ (20). Consequently, voltage changes induced by changes in apical vs. basolateral anion bath concentrations are likely dominated by bi-ionic PDs across the paracellular shunt (21). The basolateral compartment was continually perfused with KBR solution (140 mM Na+/125 mM Cl-/25 mM 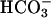 /5 mM K+/1.2 mM Ca2+/1.2 mM Mg2+/2.5 mM
/5 mM K+/1.2 mM Ca2+/1.2 mM Mg2+/2.5 mM 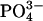 /2.5 mM glucose). To test the relative paracellular anion permselectivity of the shunt, the lumen was perfused with the following glucosefree solutions: (i) KBR, (ii) 25 mM Cl- and 125 mM
/2.5 mM glucose). To test the relative paracellular anion permselectivity of the shunt, the lumen was perfused with the following glucosefree solutions: (i) KBR, (ii) 25 mM Cl- and 125 mM 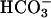 to test relative Cl- vs.
to test relative Cl- vs. 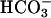 permeability, and (iii) 25 mM Cl-, 25 mM
permeability, and (iii) 25 mM Cl-, 25 mM 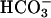 , and 100 mM gluconate to test Cl- vs. gluconate permeability. The solution's cationic composition was identical (140 mM Na+, 5 mM K+, 1.2 mM Ca2+, 1.2 mM Mg2+, 2.5 mM
, and 100 mM gluconate to test Cl- vs. gluconate permeability. The solution's cationic composition was identical (140 mM Na+, 5 mM K+, 1.2 mM Ca2+, 1.2 mM Mg2+, 2.5 mM 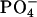 ). Transepithelial PDs were recorded from 3 M KCl agar bridges linked through calomel half-cells to an electrometer.
). Transepithelial PDs were recorded from 3 M KCl agar bridges linked through calomel half-cells to an electrometer.
Response of NL and CF Epithelia to Acidification of Apical Liquid. To test the response to luminal acidification, we exposed the luminal surface of CF and NL cultures to a weakly buffered K+/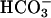 -free isotonic saline Ringer's solution (140 mM Na+/140 mM Cl- 1.2 mM Ca2+/1.2 mM Mg2+/2.5 mM
-free isotonic saline Ringer's solution (140 mM Na+/140 mM Cl- 1.2 mM Ca2+/1.2 mM Mg2+/2.5 mM 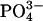 , pH adjusted to pH 3.0 with HCl). After washing with PBS and once with the acidified Ringer's solution, 100 μl of acidified Ringer's solution was added apically, and sampled for pH measurement. The basolateral bath was a KBR solution containing 25 mM
, pH adjusted to pH 3.0 with HCl). After washing with PBS and once with the acidified Ringer's solution, 100 μl of acidified Ringer's solution was added apically, and sampled for pH measurement. The basolateral bath was a KBR solution containing 25 mM 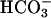 or 25 mM Hepes as buffer (pH of both was 7.4). pHi measurements during solution changes were made with a RatioMaster fluorimeter (Photon Technology, Brunswick, NJ) attached via fiber optics to a microscope (Zeiss), using 2′,7′-bis(carboxyethyl)-4 or 5-carboxyfluorescein (BCECF) fluorescence (22).
or 25 mM Hepes as buffer (pH of both was 7.4). pHi measurements during solution changes were made with a RatioMaster fluorimeter (Photon Technology, Brunswick, NJ) attached via fiber optics to a microscope (Zeiss), using 2′,7′-bis(carboxyethyl)-4 or 5-carboxyfluorescein (BCECF) fluorescence (22).
Data Presentation and Analysis. Results are presented as mean ± SE. When more than two groups were analyzed, a one-way analysis of variance analysis was performed with a Student-Newman-Keuls posttest to isolate differences between groups, whereas response of a variable in different groups over time was tested with two-way analysis of variance and Bonferroni correction using PRISM software (GraphPad, San Diego).
Results
ASL pH and 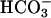 Concentrations on CF vs. NL Cultured Bronchial Epithelium. CF and NL cultures acidified ASL over time (Fig. 1A). However, acidification rates over the first 6 h were greater for CF than for NL cultures (ΔpH units/hr: CF-0.146 ± 0.011; NL-0.096 ± 0.029, P < 0.001). pH differences were maintained at 24 (P < 0.005) and 48 (P < 0.01) hours. In parallel, ASL
Concentrations on CF vs. NL Cultured Bronchial Epithelium. CF and NL cultures acidified ASL over time (Fig. 1A). However, acidification rates over the first 6 h were greater for CF than for NL cultures (ΔpH units/hr: CF-0.146 ± 0.011; NL-0.096 ± 0.029, P < 0.001). pH differences were maintained at 24 (P < 0.005) and 48 (P < 0.01) hours. In parallel, ASL 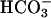 concentrations decreased at a greater rate in CF vs. NL cultures (Fig. 1B).
concentrations decreased at a greater rate in CF vs. NL cultures (Fig. 1B).
Fig. 1.

More acidic ASL on cultured primary human CF vs. normal bronchial epithelium. One hundred microliters of KBR was applied to the apical surface of cultures of CF and NL bronchial epithelium. Aliquots of liquid (1-5 μl, quadruplicate samples from cultures of 20 CF patients and 24 NL patients) were sampled at intervals and assayed for pH (A) and 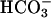 (B). *, P < 0.001 pHASL CF vs. NL.
(B). *, P < 0.001 pHASL CF vs. NL.
ASL acidification over a prolonged period could be affected by acidification of basolateral media. Basolateral pH had acidified at 24 h, likely reflecting substrate depletion and lactate accumulation, but was similar in CF and NL cultures (7.29 ± 0.02 vs. 7.31 ± 0.03, P = 0.8). Because blood flow would be expected to preserve substrate concentrations and basolateral pH at ≈7.4 in vivo, we tested the effect of replacing the basolateral solution with fresh media after 24 h. This led to an alkalinization of pHASL that was greater in NL vs. CF cultures (Δ pH units: NL, 0.33 ± 0.02 vs. CF, 0.23 ± 0.04, P < 0.05), and resulted in larger basal pHASL differences between CF and NL cultures.
To determine how cation/anion balance was maintained when 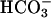 was depleted in ASL, we measured monovalent ion concentrations in ASL with time. From 0 to 24 h, ASL [Cl-] rose in CF (117 ± 4.3 mEq/liter vs. 134 ± 4.5 mEq/liter, P < 0.001) and NL (116.1 ± 4.4 vs. 131.6 ± 3.6 mEq/liter, P < 0.0001) cultures, balancing falls in ASL [
was depleted in ASL, we measured monovalent ion concentrations in ASL with time. From 0 to 24 h, ASL [Cl-] rose in CF (117 ± 4.3 mEq/liter vs. 134 ± 4.5 mEq/liter, P < 0.001) and NL (116.1 ± 4.4 vs. 131.6 ± 3.6 mEq/liter, P < 0.0001) cultures, balancing falls in ASL [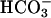 ] in CF (25.8 ± 1.5 vs. 1.28 ± 0.4 mEq/liter, P < 0.001) and NL (25.7 ± 1.3 vs. 2.6 ± 0.5, mEq/liter, P < 0.0001). [K+] concentrations fell in ASL from CF (5.1 ± 0.2 vs. 0.45 ± 35 mEq/liter, P < 0.0001) and NL (5.12 ± 0.4 vs. 0.42 ± 0.3 mEq/liter, P < 0.0001) whereas [Na+] remained stable in samples from CF (135.8 ± 7.8 vs. 132.9 ± 5.9 Eq/liter) and NL (135.8 ± 6.6 vs. 130 ± 6.4 mEq/liter). Comparison of measured anions (Cl- and
] in CF (25.8 ± 1.5 vs. 1.28 ± 0.4 mEq/liter, P < 0.001) and NL (25.7 ± 1.3 vs. 2.6 ± 0.5, mEq/liter, P < 0.0001). [K+] concentrations fell in ASL from CF (5.1 ± 0.2 vs. 0.45 ± 35 mEq/liter, P < 0.0001) and NL (5.12 ± 0.4 vs. 0.42 ± 0.3 mEq/liter, P < 0.0001) whereas [Na+] remained stable in samples from CF (135.8 ± 7.8 vs. 132.9 ± 5.9 Eq/liter) and NL (135.8 ± 6.6 vs. 130 ± 6.4 mEq/liter). Comparison of measured anions (Cl- and 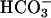 ) and cations (Na+ and K+) in ASL on CF and NL cultures at 24 h did not reveal significant anion gaps (see Fig. 7, which is published as supporting information on the PNAS web site).
) and cations (Na+ and K+) in ASL on CF and NL cultures at 24 h did not reveal significant anion gaps (see Fig. 7, which is published as supporting information on the PNAS web site).
Role of H+,K+-ATPase in Regulation of pHASL in CF and NL Bronchial Epithelial Cultures. We detected no lactate in ASL (not shown). Hypothesizing that acidification of ASL on CF and NL cultures reflected the activity of a H+,K+-ATPase, we varied apical K+ concentrations (0, 5, or 20 mM K+) and demonstrated K+-dependent acidification in NL and CF cultures (Fig. 2 A and B). CF epithelial acidification rates were greater over 6 h at each K+ concentration than for NL epithelia (Fig. 2C). Two observations suggested similar H+,K+-ATPase activity in CF and NL cultures. First, differences in pHASL in CF and NL were independent of ASL [K+] (Fig. 2C). Second, CF and NL cultures exhibited identical rates of K+ removal from ASL (Fig. 2D).
Fig. 2.

CF and NL cultures exhibit K+-dependent acidification and ASL K+ depletion. One hundred microliters of KBR (0, 5, or 20 mM/liter [K+]) was applied to the apical surface of CF and NL bronchial epithelial cultures. Microaliquots of apical liquid were assayed for pH. NL (A) and CF (B) cultures exhibit 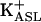 -dependent acidification over 6 h. †, P < 0.0001, 0 mM vs. 20 mM K+; *, P < 0.01, 0 mM vs. 5 mM K+. (C) Acidification rates measured in CF and 24 NL cultures in response to solutions containing 0, 5, and 20 mM K+ measured over 6 h. *, P < 0.01 rates of ASL acidification on CF vs. NL cultures at each [K+]. (D)[K+]ASL depletion by CF and NL cultures. One hundred microliters of KBR ([K+] 20 mM/liter) was applied to the apical surface of cultures of CF and NL bronchial epithelia. Microaliquots of apical liquid were assayed for [K+]. P = 0.56.
-dependent acidification over 6 h. †, P < 0.0001, 0 mM vs. 20 mM K+; *, P < 0.01, 0 mM vs. 5 mM K+. (C) Acidification rates measured in CF and 24 NL cultures in response to solutions containing 0, 5, and 20 mM K+ measured over 6 h. *, P < 0.01 rates of ASL acidification on CF vs. NL cultures at each [K+]. (D)[K+]ASL depletion by CF and NL cultures. One hundred microliters of KBR ([K+] 20 mM/liter) was applied to the apical surface of cultures of CF and NL bronchial epithelia. Microaliquots of apical liquid were assayed for [K+]. P = 0.56.
Molecular Identification of H+,K+-ATPase in Human Bronchial Epithelium. We localized H+,K+-ATPase protein to apical membranes of cultured NL bronchial epithelium and freshly isolated NL bronchial tissue with confocal immunofluorescence microscopy using a monoclonal antibody detecting both gastric and nongastric forms (Fig. 3A). H+,K+-ATPase was also detected in CF tissues (not shown). Because two H+,K+-ATPase isoforms, a gastric and a nongastric (colonic) form, are described (23, 24), pharmacological inhibitor studies were initiated with Sch28080 (which inhibits the gastric isoform) and ouabain (which inhibits the nongastric isoform) to determine which isoform functionally dominated in airway epithelium. Sch28080 was a gift from James Kaminski (Schering-Plough). K+-dependent acidification was inhibited by luminal ouabain, but not Sch28080 (Fig. 3B), as was the rate of K+ removal from surface liquid (control 7.4 ± 0.34 mEq/hr, ouabain 2.1 ± 0.4 mEq/hr, P < 0.05; Sch28080 7.1 ± 0.4 mEq/hr, P = 0.85). PD and transepithelial resistance were unchanged during the time course of these experiments, suggesting that ouabain had not traversed the epithelium and inhibited the basolateral Na+,K+-ATPase. These data support a predominant role for the nongastric form of the H+,K+-ATPase in airway, a conclusion supported by our detection of mRNA for nongastric H+,K+-ATPase (ATP1AL1) in cultured NL airway and colonic, but not gastric, epithelial tissues by using RT-PCR (Fig. 3C).
Fig. 3.

H+,K+-ATPase in cultured and freshly excised human bronchial epithelium. (A) Immunostaining of well differentiated primary bronchial cultures (Upper) and freshly excised bronchus (Lower) were analyzed with confocal microscopy. Specimens were stained with a monoclonal antibody recognizing the β-subunit of the ATPase, followed by a Texas Red-labeled secondary antibody and fluorescein-labeled phalloidin. Red and green channels were registered by sequential scanning in the xz axis (cultures; magnification, ×200) and xy axis (bronchial sections; magnification, ×80). H+,K+-ATPase is heavily expressed in the apical membrane of ciliated cells of the bronchial cultures, as well as in superficial epithelium and ciliated ducts in the human bronchus. Actin cytoskeleton (phalloidin) indicates the position of the plasma membrane. L, lumen; S, support; D, ciliated duct. (B) Ouabain, not Sch28080, inhibited K+-dependent ASL acidification. Fifty microliters of KBR containing 0 mM or 20 mM K+ was applied apically to NL bronchial epithelial cultures in the presence or absence of inhibitors. Ouabain (500 μM), an inhibitor of the nongastric isoform of the H+,K+-ATPase, and Sch28008 (10 μM), an inhibitor of the gastric isoform of the H+,K+-ATPase, were added. pHASL and K+ASL were monitored over 90 min. *, P < 0.001 pHASL control vs. timezero; †,pHASL sch28080 vs. Timezero.(C) mRNA expression of the nongastric isoform of the H+,K+-ATPase was detected by RT-PCR in cultured bronchial epithelia from three NL subjects as well as in normal colonic, but not gastric, tissue.
Although we have previously detected no resting apical membrane H+ or K+ conductance in human airway epithelium (12, 13, 21), we tested for a possible role of these conductances in pHASL regulation under conditions when H+,K+-ATPase activity was minimized, i.e., under 0 mM K+ conditions. The K+ channel blocker, barium, did not alter ASL acidification rates over 6 h (control 0.066 ± 0.003 vs. barium 0.063 ± 0.004 pH units/hr, n = 4), arguing against a role for an apical K+ conductance and membrane potential. We probed a role for a proton conductive pathway in the apical membrane for ASL pH regulation by adding amiloride (100 μM) to the lumen to hyperpolarize the apical membrane potential. Again, we detected no change in ASL acidification rates over 6 h with amiloride (control 0.058 ± 0.006 vs. amiloride 0.061 ± 0.004 pH units/hr, n = 4).
CFTR 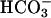 Secretion: Basal Role and Response to Raised Cellular cAMP. If more rapid acidification and lower [
Secretion: Basal Role and Response to Raised Cellular cAMP. If more rapid acidification and lower [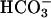 ] of CF ASL were not caused by abnormal H+ secretion rates by the H+,K+-ATPase, it may reflect absence of CFTR-mediated
] of CF ASL were not caused by abnormal H+ secretion rates by the H+,K+-ATPase, it may reflect absence of CFTR-mediated 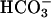 secretion. We probed the role of CFTR-mediated
secretion. We probed the role of CFTR-mediated 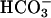 secretion by testing the effect of raising intracellular [cAMP] in CF, NL, and disease control epithelia on the pH of an apical unbuffered K+/
secretion by testing the effect of raising intracellular [cAMP] in CF, NL, and disease control epithelia on the pH of an apical unbuffered K+/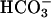 -free (pH ≈ 5.8) ASL solution (Fig. 4). NL and disease control epithelia rapidly alkalinized this unbuffered solution, consistent with
-free (pH ≈ 5.8) ASL solution (Fig. 4). NL and disease control epithelia rapidly alkalinized this unbuffered solution, consistent with 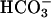 secretion. A greater and more sustained alkalinization accompanied treatment with forskolin/IBMX to raise intracellular cAMP and activate CFTR (Fig. 4 A and B). In CF cultures, although an identical rapid alkalinization was observed, presumably reflecting H+/
secretion. A greater and more sustained alkalinization accompanied treatment with forskolin/IBMX to raise intracellular cAMP and activate CFTR (Fig. 4 A and B). In CF cultures, although an identical rapid alkalinization was observed, presumably reflecting H+/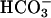 movement through the paracellular path (see below), a “paradoxical” acidification, rather than alkalinization of ASL, occurred (see Fig. 4C), reflecting in part the absence of CFTR-dependent
movement through the paracellular path (see below), a “paradoxical” acidification, rather than alkalinization of ASL, occurred (see Fig. 4C), reflecting in part the absence of CFTR-dependent 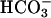 secretion.
secretion.
Fig. 4.

Increased intracellular cAMP alkalinizes ASL on NL bronchial cultures and acidifies ASL on CF bronchial cultures. We tested the effect of cAMP activation (10-5 M forskolin/200 μM IBMX bilaterally) on pHASL regulation in NL (Fig. 5A), disease control (Fig. 5B), and CF (Fig. 5C) bronchial epithelial cultures. One hundred microliters of bicarbonate/K+-free saline Ringer's solution (pH 5.6) was applied apically. Microaliquots of apical liquid were sampled and assayed for pHASL.(A) *, P < 0.001 NL control vs. NL forskolin/IBMX. (B) *, P < 0.001 disease control vs. disease control + forskolin/IBMX. (C) *, P < 0.005 CF control vs. CF forskolin/IBMX.
At 6 h, ASL [Cl-] from forskolin/IBMX-treated CF cultures was significantly higher than in ASL from forskolin/IBMX-treated NL cultures (CF-124.6 ± 1.70 mM vs. NL-118.7 ± 1.60 mM, P < 0.05). The reciprocal relationship between ASL Cl- and 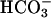 suggests that NL but not CF cultures secreted
suggests that NL but not CF cultures secreted 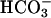 .
.
Paracellular Pathway Conductivity for 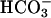 and Other Anions. Airway epithelia exhibit a relatively “leaky” paracellular pathway that could participate in pHASL regulation. As best known, the total ionic permeability of this path is similar in NL and CF (21). Amiloride-treated CF cultures, lack significant apical ionic conductances, and apical membrane resistances are >10 KΩ (21). Consequently, transepithelial PD changes in response to asymmetric bath ion compositions are dominated by paracellular ionic gradients. When the apical solution was switched from KBR to the high
and Other Anions. Airway epithelia exhibit a relatively “leaky” paracellular pathway that could participate in pHASL regulation. As best known, the total ionic permeability of this path is similar in NL and CF (21). Amiloride-treated CF cultures, lack significant apical ionic conductances, and apical membrane resistances are >10 KΩ (21). Consequently, transepithelial PD changes in response to asymmetric bath ion compositions are dominated by paracellular ionic gradients. When the apical solution was switched from KBR to the high 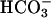 /low Cl- solution, creating equal but oppositely directed transepithelial concentration gradients for Cl- and
/low Cl- solution, creating equal but oppositely directed transepithelial concentration gradients for Cl- and 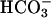 , the PD hyperpolarized (ΔPD = -1.4 ± 0.08 mV, P < 0.05 vs. bilateral KBR, Fig. 5), consistent with Cl- exceeding
, the PD hyperpolarized (ΔPD = -1.4 ± 0.08 mV, P < 0.05 vs. bilateral KBR, Fig. 5), consistent with Cl- exceeding 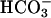 permeation via the paracellular path. We continued the sequence of anion permselectivity by switching the apical solution to one that substituted Cl- with the relatively impermeant anion gluconate, creating equal but oppositely directed transepithelial concentration gradients across the cultures for Cl- and gluconate but no gradient for
permeation via the paracellular path. We continued the sequence of anion permselectivity by switching the apical solution to one that substituted Cl- with the relatively impermeant anion gluconate, creating equal but oppositely directed transepithelial concentration gradients across the cultures for Cl- and gluconate but no gradient for 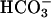 permeation. A larger hyperpolarization (ΔPD = -4.02 ± 0.12 mV, P < 0.05, vs. low Cl- solution) was observed, suggesting that the anion permeation sequence of the shunt is Cl- >
permeation. A larger hyperpolarization (ΔPD = -4.02 ± 0.12 mV, P < 0.05, vs. low Cl- solution) was observed, suggesting that the anion permeation sequence of the shunt is Cl- > 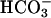 > gluconate.
> gluconate.
Fig. 5.

The paracellular path conducts 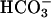 and exhibits selectivity for individual anions. Primary CF epithelial cultures were mounted in Ussing chambers. Inhibition of ENaC (Amiloride, 10-4 M) eliminated significant apical membrane conductance because these cultures also lack CFTR. Changes in potential difference, under these circumstances, thus reflect paracellular ion movement. The basolateral side was perfused with KBR. The changes in transepithelial potential difference (ΔPD) result from switching the apical solution from KBR to one where the chloride and bicarbonate concentrations were inverted (imposing equal “basolateral-to-apical” chloride and “apical-to-basolateral” bicarbonate gradients), and subsequently to one where 95 mM gluconate replaced 95 mM bicarbonate in the apical solution (i.e., imposing equal basolateral-to-apical chloride and apical-to-basolateral gluconate gradients). Results are shown as the PD. Results from duplicate cultures of nine CF epithelia were analyzed. *, P < 0.05 PD apical “high apical bicarbonate” solution vs. KBR. **, P < 0.05 apical “high gluconate” vs. “high bicarbonate” and KBR.
and exhibits selectivity for individual anions. Primary CF epithelial cultures were mounted in Ussing chambers. Inhibition of ENaC (Amiloride, 10-4 M) eliminated significant apical membrane conductance because these cultures also lack CFTR. Changes in potential difference, under these circumstances, thus reflect paracellular ion movement. The basolateral side was perfused with KBR. The changes in transepithelial potential difference (ΔPD) result from switching the apical solution from KBR to one where the chloride and bicarbonate concentrations were inverted (imposing equal “basolateral-to-apical” chloride and “apical-to-basolateral” bicarbonate gradients), and subsequently to one where 95 mM gluconate replaced 95 mM bicarbonate in the apical solution (i.e., imposing equal basolateral-to-apical chloride and apical-to-basolateral gluconate gradients). Results are shown as the PD. Results from duplicate cultures of nine CF epithelia were analyzed. *, P < 0.05 PD apical “high apical bicarbonate” solution vs. KBR. **, P < 0.05 apical “high gluconate” vs. “high bicarbonate” and KBR.
Comparison of Response of CF and NL Airway Epithelium to Acid Challenge. We speculated that the response of CF cultures to luminal acid challenge was abnormal. We investigated the NL and CF culture capacity to alkalinize a luminal acidic 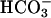 - and K+-free isosmotic solution (pH 3.0) in the presence and absence of basolateral bath
- and K+-free isosmotic solution (pH 3.0) in the presence and absence of basolateral bath 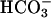 . After apical acid loading, pHASL recovered more slowly and incompletely in CF than NL cultures (Fig. 6). Comparison experiments performed without basolateral
. After apical acid loading, pHASL recovered more slowly and incompletely in CF than NL cultures (Fig. 6). Comparison experiments performed without basolateral 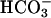 demonstrated that basolateral
demonstrated that basolateral 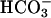 contributed relatively more to ASL alkalinization in response to acid challenge on NL than CF cultures (compare Fig. 6 A and B).
contributed relatively more to ASL alkalinization in response to acid challenge on NL than CF cultures (compare Fig. 6 A and B).
Fig. 6.

Transepithelial 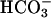 movement after ASL acid-challenge is reduced in CF vs. NL cultures. One hundred microliters of saline Ringer's solution (pH 3.3) was applied apically to NL and CF bronchial epithelial cultures. The basolateral medium was KBR or similar solution where 25 mM Na+ Hepes replaced 25 mM Na+
movement after ASL acid-challenge is reduced in CF vs. NL cultures. One hundred microliters of saline Ringer's solution (pH 3.3) was applied apically to NL and CF bronchial epithelial cultures. The basolateral medium was KBR or similar solution where 25 mM Na+ Hepes replaced 25 mM Na+  , pH both = 7.45. Recovery from luminal acid challenge in the presence and absence of basolateral
, pH both = 7.45. Recovery from luminal acid challenge in the presence and absence of basolateral 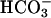 is shown for NL (A) and CF (B). A significantly greater basolateral
is shown for NL (A) and CF (B). A significantly greater basolateral 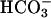 dependence is observed in NL vs. CF cultures.
dependence is observed in NL vs. CF cultures.
The acidic luminal solutions induced small and transient increases in equivalent short circuit current and transient falls in resistance at 5 min (NL 551 ± 28 before vs. 490 ± 22 after Ω·cm2; CF 529 ± 36 before vs. 480 ± 28 after Ω·cm2, n = 12, P < 0.05). However, no bioelectric evidence of epithelial damage was apparent at this or later time points, consistent with previously published data (25). Cell viability (by vital dye exclusion) was unaltered.
We tested effects of apical acid challenge on cytosolic pH (pHi) to ascertain whether pHASL differences in CF and NL preparations after acid challenge reflected differential effects on pHi and, thus, potentially other pH regulatory paths. No difference in basal pHi in CF (n = 3) and NL (n = 4) culture was seen, nor was baseline pHi affected by substitution of Hepes for 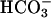 in the basolateral bath (basal pHi: CF-
in the basolateral bath (basal pHi: CF-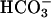 6.960 ± 0.2 vs. NL-
6.960 ± 0.2 vs. NL-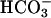 6.940 ± 0.2, P = 0.85; basal pHi: CF Hepes 6.960 ± 0.2 vs. NL Hepes 6.970 ± 0.1, P = 0.9). CF and NL cultures exhibited similar, moderate, transient acidification of pHi in response to apical acidic solutions in the presence or absence of
6.940 ± 0.2, P = 0.85; basal pHi: CF Hepes 6.960 ± 0.2 vs. NL Hepes 6.970 ± 0.1, P = 0.9). CF and NL cultures exhibited similar, moderate, transient acidification of pHi in response to apical acidic solutions in the presence or absence of 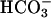 (nadir pHi: CF
(nadir pHi: CF 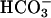 6.750 ± 0.3 vs. NL
6.750 ± 0.3 vs. NL 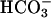 6.750 ± 0.2, P = 0.85, nadir pHi: CF Hepes 6.760 ± 0.2 vs. NL Hepes 6.780 ± 0.1, P = 0.9). pHi recovery after luminal acid challenge was similar in both groups under each condition. Thus, more effective regulation of pHASL in response to luminal acid load in NL preparations likely reflected CFTR-mediated
6.750 ± 0.2, P = 0.85, nadir pHi: CF Hepes 6.760 ± 0.2 vs. NL Hepes 6.780 ± 0.1, P = 0.9). pHi recovery after luminal acid challenge was similar in both groups under each condition. Thus, more effective regulation of pHASL in response to luminal acid load in NL preparations likely reflected CFTR-mediated 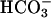 secretion, not effects on pHi.
secretion, not effects on pHi.
Discussion
Airway surface liquid is a critical component of lung host defense. Its pH appears to reflect a balance between active transcellular ion transport and passive paracellular ion movement. Our experiments defined pathways participating in pHASL regulation by NL- and CF-cultured bronchial epithelium under basal and acid-stressed conditions.
Airway epithelial cultures acidified lumenally applied KBR under basal conditions (Fig. 1). The basal acidification (and  depletion) rate was more rapid in CF cultures (Fig. 1), and pHASL at 24 and 48 h was lower in CF than NL. A small component of ASL acidification likely reflected selective lactate accumulation in the basolateral compartment. When basal media was replaced with fresh media at 24 h, the pHASL difference between CF and NL ASL was accentuated. Taken together, these data show that pHASL regulation under basal conditions is abnormal in CF airway epithelia.
depletion) rate was more rapid in CF cultures (Fig. 1), and pHASL at 24 and 48 h was lower in CF than NL. A small component of ASL acidification likely reflected selective lactate accumulation in the basolateral compartment. When basal media was replaced with fresh media at 24 h, the pHASL difference between CF and NL ASL was accentuated. Taken together, these data show that pHASL regulation under basal conditions is abnormal in CF airway epithelia.
ASL acidification reflects, at least in part, apical membrane H+,K+-ATPase activity. CF and NL cultures exhibited K+-dependent ASL acidification (Fig. 2). Our studies demonstrated that (i) K+ depletion rates in CF and NL cultures were identical (Fig. 2D); and (ii) differences in acidification rates between CF and NL cultures were similar in magnitude, regardless of K+ASL (Fig. 2C), suggesting that H+,K+-ATPase activity was similar in CF and NL cultures and, thus, not the basis for abnormal pHASL on CF airway epithelia.
We characterized the molecular and functional activity of the H+,K+-ATPase in airway epithelia with complementary approaches. Immunocytochemical studies verified the apical membrane location of H+,K+-ATPase in cultured proximal human bronchial epithelium and freshly excised superficial bronchial epithelium. Pharmacological inhibitor studies showed that that the nongastric (colonic) isoform was dominant functionally in airway epithelia, consistent with the demonstration that cultured proximal human bronchial epithelium expressed mRNA for the nongastric isoform of the H+,K+-ATPase (Fig. 3).
The simplest explanation for basal pHASL differences between CF and NL cultures is that it reflected the lack of 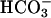 secretion by CF airway epithelia, because it was not caused by increased H+,K+-ATPase-mediated H+ secretion. The pHASL alkalinization in response to raising cell cAMP observed in NL but not CF cultures is consistent with CFTR-dependent
secretion by CF airway epithelia, because it was not caused by increased H+,K+-ATPase-mediated H+ secretion. The pHASL alkalinization in response to raising cell cAMP observed in NL but not CF cultures is consistent with CFTR-dependent 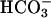 secretion (Fig. 4). With respect to possible alternative mechanisms of airway epithelial
secretion (Fig. 4). With respect to possible alternative mechanisms of airway epithelial  secretion, we failed to detect evidence of an anion exchanger that might be regulated by CFTR on the apical membrane of airway epithelium (22), supporting the hypothesis that
secretion, we failed to detect evidence of an anion exchanger that might be regulated by CFTR on the apical membrane of airway epithelium (22), supporting the hypothesis that 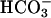 secretion is mediated by CFTR itself and, thus, is electrogenic.
secretion is mediated by CFTR itself and, thus, is electrogenic.
An electrogenic CFTR-dependent mechanism would provide an effective means to constantly adjust pHASL. For example, with 25 mM 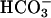 in ASL, there is little or no electrochemical driving force for
in ASL, there is little or no electrochemical driving force for 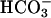 secretion (13). However, if ASL [
secretion (13). However, if ASL [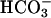 ] falls, the electrochemical gradient will favor CFTR-mediated apical
] falls, the electrochemical gradient will favor CFTR-mediated apical 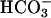 secretion, limiting acidification. In CF, the
secretion, limiting acidification. In CF, the 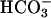 exit pathway is absent, leading to a failure to buffer H+,K+-ATPase-mediated proton secretion and acidification of ASL. Note that the paradoxical acidification of ASL on CF cultures after forskolin/IBMX (Fig. 4C) may reflect the cAMP activation of H+,K+-ATPase activity, as reported in the gastric parietal cell (26). Taken together, these data indicate that CF cultures lack the capacity to secrete
exit pathway is absent, leading to a failure to buffer H+,K+-ATPase-mediated proton secretion and acidification of ASL. Note that the paradoxical acidification of ASL on CF cultures after forskolin/IBMX (Fig. 4C) may reflect the cAMP activation of H+,K+-ATPase activity, as reported in the gastric parietal cell (26). Taken together, these data indicate that CF cultures lack the capacity to secrete 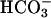 to match H+,K+-ATPase-mediated proton secretion into ASL under basal and cAMP-stimulated conditions, resulting in “hyperacidification” of ASL.
to match H+,K+-ATPase-mediated proton secretion into ASL under basal and cAMP-stimulated conditions, resulting in “hyperacidification” of ASL.
pHASL regulation in response to luminal acid challenges is likely important in lung defense. It has been speculated that the first CF bacterial infections in children follow gastric aspiration (27, 28). The observation that CF bronchial epithelium less effectively alkalinized an acid challenge to ASL is consistent with this (Fig. 6).
The response to acid challenge was complex. Our data suggested participation of CFTR-dependent cellular as well as paracellular pathways. Realkalinization was basolateral bath 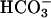 -dependent, likely reflecting transcellular and paracellular
-dependent, likely reflecting transcellular and paracellular 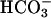 transport (Fig. 6). Impairment of realkalinization in CF, compared to NL, cultures was greatest in the presence of basolateral
transport (Fig. 6). Impairment of realkalinization in CF, compared to NL, cultures was greatest in the presence of basolateral 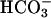 , consistent with impaired cellular (transapical)
, consistent with impaired cellular (transapical) 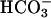 movement across CF epithelia. In the absence of any known mechanism of apical membrane
movement across CF epithelia. In the absence of any known mechanism of apical membrane 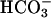 transport in CF epithelium, the residual ability to alkalinize pHASL in CF may reflect net H+ absorption and/or
transport in CF epithelium, the residual ability to alkalinize pHASL in CF may reflect net H+ absorption and/or 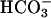 secretion via the paracellular pathway (Fig. 6B), a notion consistent with our paracellular permselectivity experiments, which suggested that the paracellular pathway is permeable to
secretion via the paracellular pathway (Fig. 6B), a notion consistent with our paracellular permselectivity experiments, which suggested that the paracellular pathway is permeable to 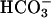 . Indeed, if CFTR is the sole path of
. Indeed, if CFTR is the sole path of 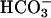 translocation across the apical membrane of NL airway epithelial cells, the paracellular pathway may be the only mode of alkalinizing acidified CF ASL.
translocation across the apical membrane of NL airway epithelial cells, the paracellular pathway may be the only mode of alkalinizing acidified CF ASL.
Our paracellular permselectivity experiments support a small 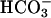 current and may be best explained by a transient increase in paracellular permeability, perhaps dependent on the extreme deviation of mucosal pH from “basal” values. The surface glycocalyx/mucus layer may have intrinsic buffering properties that contribute to the initial increase in pH. However, when we rechallenged the preparations with a second acid load, we observed a similar pattern of recovery (unpublished data), suggesting that pH recovery truly reflected transepithelial ion movement.
current and may be best explained by a transient increase in paracellular permeability, perhaps dependent on the extreme deviation of mucosal pH from “basal” values. The surface glycocalyx/mucus layer may have intrinsic buffering properties that contribute to the initial increase in pH. However, when we rechallenged the preparations with a second acid load, we observed a similar pattern of recovery (unpublished data), suggesting that pH recovery truly reflected transepithelial ion movement.
Alterations in pHASL might contribute to CF pathogenesis. An abnormally low surface pH may adversely affect mucus viscosity by altering exposure of hydrophobic regions of mucin molecules as well as changing the electrostatic charge of their carbohydrate side chains (29). Thus, in the characteristically depleted CF periciliary liquid layer, low pHASL would “tighten” adhesive interactions between mucins in the mucus layer and membrane surface tethered mucins (17), rendering cough clearance of adherent mucus less effective (30). Failure of CF epithelia to compensate for an intraluminal acid load may heighten the inflammatory response in the airway by interfering with bactericidal activity (31, 32), promoting secretion by immune cells of substances harmful to the lung (33) and reducing the efficacy of commonly used antibiotics against Pseudomonas aeruginosa (34).
In conclusion, we have shown that in NL airway epithelia the activity of a H+,K+-ATPase that acidifies ASL is balanced by the 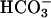 secretory function of CFTR. In addition, the paracellular path can serve to buffer pHASL with respect to plasma, in part via H+/
secretory function of CFTR. In addition, the paracellular path can serve to buffer pHASL with respect to plasma, in part via H+/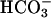 permeation. In CF, the airway epithelial cellular alkalinization function is lost by virtue of absent CFTR-mediated
permeation. In CF, the airway epithelial cellular alkalinization function is lost by virtue of absent CFTR-mediated 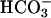 secretory transport, rendering pHASL more acidic under basal conditions and limiting the response to acidic challenges to airway surfaces.
secretory transport, rendering pHASL more acidic under basal conditions and limiting the response to acidic challenges to airway surfaces.
Supplementary Material
Acknowledgments
We acknowledge the technical assistance of J. Chadburn, M. Kelly, and L. Brown. This work was supported by National Institutes of Health Grant P01 HL 42384 and Cystic Fibrosis Foundation Grant R026.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ASL, apical surface liquid; CF, cystic fibrosis; CFTR, CF transmembrane conductance regulator; NL, normal; PD, potential difference; pHi, intracellular pH; IBMX, 3-isobutyl-1-methylxanthine.
References
- 1.Cheng, S. H., Gregory, R. J., Marshall, J., Paul, S., Souza, D. W., White, G. A., O'Riordan, C. R. & Smith, A. E. (1990) Cell 63, 827-834. [DOI] [PubMed] [Google Scholar]
- 2.Kartner, N., Hanrahan, J. W., Jensen, T. J., Naismith, A. L., Sun, S. Z., Ackerley, C. A., Reyes, E. F., Tsui, L. C., Rommens, J. M., Bear, C. E., et al. (1991) Cell 64, 681-691. [DOI] [PubMed] [Google Scholar]
- 3.Johansen, P. G., Anderson, C. M. & Hadorn, B. (1968) Lancet 1, 455-460. [PubMed] [Google Scholar]
- 4.Kopelman, H., Durie, P., Gaskin, K., Weizman, Z. & Forstner, G. (1985) N. Engl. J. Med. 312, 329-334. [DOI] [PubMed] [Google Scholar]
- 5.Linsdell, P., Tabcharani, J. A., Rommens, J. M., Hou, Y. X., Chang, X. B., Tsui, L. C., Riordan, J. R. & Hanrahan, J. W. (1997) J. Gen. Physiol. 110, 355-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devor, D. C., Singh, A. K., Lambert, L. C., DeLuca, A., Frizzell, R. A. & Bridges, R. J. (1999) J. Gen. Physiol. 113, 743-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devor, D. C., Bridges, R. J. & Pilewski, J. M. (2000) Am. J. Physiol. 279, C461-C479. [DOI] [PubMed] [Google Scholar]
- 8.Smith, J. J. & Welsh, M. J. (1992) J. Clin. Invest. 89, 1148-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jayaraman, S., Song, Y., Vetrivel, L., Shankar, L. & Verkman, A. S. (2001) J. Clin. Invest. 107, 317-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grubb, B. R. & Boucher, R. C. (1999) Physiol Rev. 79, S193-S214. [DOI] [PubMed] [Google Scholar]
- 11.Rochelle, L. G., Li, D. C., Ye, H., Lee, E., Talbot, C. R. & Boucher, R. C. (2000) Am. J. Physiol. 279, L14-L24. [DOI] [PubMed] [Google Scholar]
- 12.Paradiso, A. M. (1997) Am. J. Physiol. 273, L148-L158. [DOI] [PubMed] [Google Scholar]
- 13.Willumsen, N. J. & Boucher, R. C. (1992) J. Physiol. 455, 247-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith, J. J. & Welsh, M. J. (1993) J. Clin. Invest. 91, 1590-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer, H., Widdicombe, J. H. & Illek, B. (2002) Am. J. Physiol. 282, C736-C743. [DOI] [PubMed] [Google Scholar]
- 16.Matsui, H., Randell, S. H., Peretti, S. W., Davis, C. W. & Boucher, R. C. (1998) J. Clin. Invest. 102, 1125-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsui, H., Grubb, B. R., Tarran, R., Randell, S. H., Gatzy, J. T., Davis, C. W. & Boucher, R. C. (1998) Cell 95, 1005-1015. [DOI] [PubMed] [Google Scholar]
- 18.Knowles, M. R., Robinson, J. M., Wood, R. E., Pue, C. A., Mentz, W. M., Wager, G. C., Gatzy, J. T. & Boucher, R. C. (1997) J. Clin. Invest. 100, 2588-2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willumsen, N. J., Davis, C. W. & Boucher, R. C. (1989) Am. J. Physiol 256, C1045-C1053. [DOI] [PubMed] [Google Scholar]
- 20.Willumsen, N. J. & Boucher, R. C. (1989) Am. J. Physiol 256, C226-C233. [DOI] [PubMed] [Google Scholar]
- 21.Willumsen, N. J. & Boucher, R. C. (1989) Am. J. Physiol. 256, C1054-C1063. [DOI] [PubMed] [Google Scholar]
- 22.Paradiso, A. M., Coakley, R. D. & Boucher, R. C. (2003) J. Physiol. 548, 203-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Binder, H. J., Sangan, P. & Rajendran, V. M. (1999) Semin. Nephrol. 19, 405-414. [PubMed] [Google Scholar]
- 24.Silver, R. B. & Soleimani, M. (1999) Am. J. Physiol. 276, F799-F811. [DOI] [PubMed] [Google Scholar]
- 25.Boucher, R. C. (1981) Clin. Chest Med. 2, 377-392. [PubMed] [Google Scholar]
- 26.Levine, R. A., Nandi, J. & King, R. L. (1990) J. Clin. Invest. 86, 400-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott, R. B., O'Loughlin, E. V. & Gall, D. G. (1985) J. Pediatr. 106, 223-227. [DOI] [PubMed] [Google Scholar]
- 28.Stringer, D. A., Sprigg, A., Juodis, E., Corey, M., Daneman, A., Levison, H. J. & Durie, P. R. (1988) Can. Assoc. Radiol. J. 39, 100-102. [PubMed] [Google Scholar]
- 29.Bhaskar, K. R., Gong, D. H., Bansil, R., Pajevic, S., Hamilton, J. A., Turner, B. S. & LaMont, J. T. (1991) Am. J. Physiol 261, G827-G832. [DOI] [PubMed] [Google Scholar]
- 30.King, M. (1987) Biorheology 24, 589-597. [DOI] [PubMed] [Google Scholar]
- 31.Nakayama, K., Jia, Y. X., Hirai, H., Shinkawa, M., Yamaya, M., Sekizawa, K. & Sasaki, H. (2002) Am. J. Respir. Cell Mol. Biol. 26, 105-113. [DOI] [PubMed] [Google Scholar]
- 32.Allen, D. B., Maguire, J. J., Mahdavian, M., Wicke, C., Marcocci, L., Scheuenstuhl, H., Chang, M., Le, A. X., Hopf, H. W. & Hunt, T. K. (1997) Arch. Surg. 132, 991-996. [DOI] [PubMed] [Google Scholar]
- 33.Trevani, A. S., Andonegui, G., Giordano, M., Lopez, D. H., Gamberale, R., Minucci, F. & Geffner, J. R. (1999) J. Immunol. 162, 4849-4857. [PubMed] [Google Scholar]
- 34.Corkill, J. E., Deveney, J., Pratt, J., Shears, P., Smyth, A., Heaf, D. & Hart, C. A. (1994) Pediatr. Res. 35, 299-302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.