

Abstract
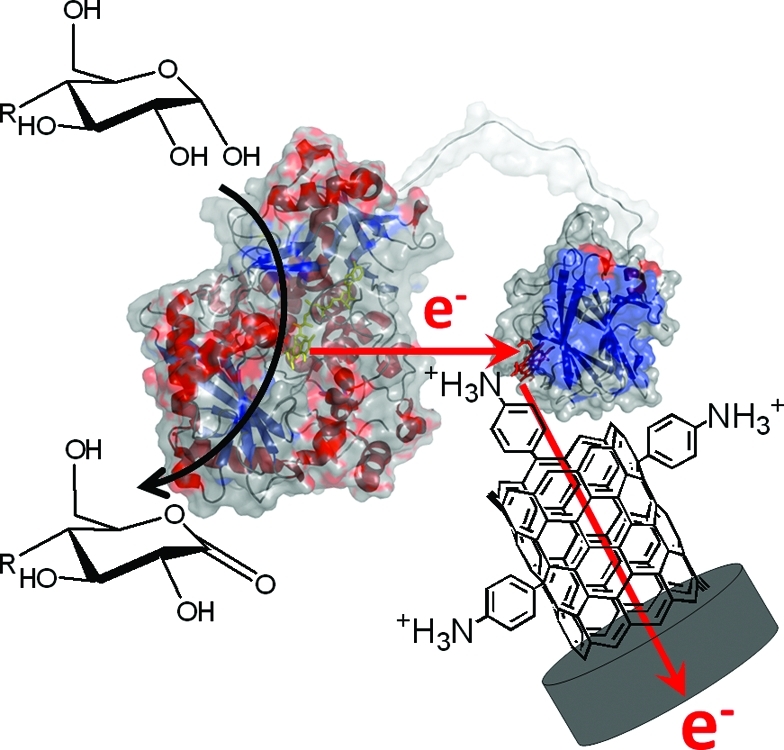
One of the challenges in the field of biosensors and biofuel cells is to establish a highly efficient electron transfer rate between the active site of redox enzymes and electrodes to fully access the catalytic potential of the biocatalyst and achieve high current densities. We report on very efficient direct electron transfer (DET) between cellobiose dehydrogenase (CDH) from Phanerochaete sordida (PsCDH) and surface modified single walled carbon nanotubes (SWCNT). Sonicated SWCNTs were adsorbed on the top of glassy carbon electrodes and modified with aryl diazonium salts generated in situ from p-aminobenzoic acid and p-phenylenediamine, thus featuring at acidic pH (3.5 and 4.5) negative or positive surface charges. After adsorption of PsCDH, both electrode types showed excellent long-term stability and very efficient DET. The modified electrode presenting p-aminophenyl groups produced a DET current density of 500 μA cm−2 at 200 mV vs normal hydrogen reference electrode (NHE) in a 5 mM lactose solution buffered at pH 3.5. This is the highest reported DET value so far using a CDH modified electrode and comes close to electrodes using mediated electron transfer. Moreover, the onset of the electrocatalytic current for lactose oxidation started at 70 mV vs NHE, a potential which is 50 mV lower compared to when unmodified SWCNTs were used. This effect potentially reduces the interference by oxidizable matrix components in biosensors and increases the open circuit potential in biofuel cells. The stability of the electrode was greatly increased compared with unmodified but cross-linked SWCNTs electrodes and lost only 15% of the initial current after 50 h of constant potential scanning.
Efficient electron transfer between redox enzymes and electrodes is a major issue in the development of biosensors and biofuel cells. Most commonly, large distances (above 20 Å) prevent direct electron transfer (DET) between the prosthetic group in the active site and the electrode.1,2 Mediated electron transfer (MET) employs redox mediators to shuttle redox equivalents or allow electron hopping between the enzyme and the electrode and has been widely applied to solve this problem. Good sensitivities and high current densities have been demonstrated for different biosensors and biofuel cells based on MET.3−8 On the other side, DET between the enzyme and the electrode surface would obviate problems associated with MET (e.g., leaking or limited stability of redox mediators, expensive heavy metal complexes, potential toxicity of mediator), allow a more simple design of the electrode, and reduce the number of additional components, which have to be added and optimized.(9) When implanted or deposited after use, DET based biosensors or biofuel cells cannot release environmentally hazardous, potentially toxic mediators or their degradation products.
It is well-known that DET either requires a close proximity of the active site of the redox enzyme to the protein surface (e.g., cytochrome c peroxidase)3,10 or needs a connection with the protein surface by a built in electron transfer pathway constructed by a chain of redox active cofactors (e.g., NiFe hydrogenaes)1,2,11 or by an electric wire.12−16 DET is therefore rarely observed; it was shown for only 5% of the currently known redox enzymes.3,17 Moreover, even if the prerequisites for DET are given, the correct orientation of the active site and vicinity to the electrode surface are required for high electron transfer rates.(2) DET is therefore difficult to achieve, and an optimized electrode surface is required. The most commonly employed electrode modifiers for DET are self-assembled monolayers (SAM) of functionalized thiols on gold, which facilitate the ordered immobilization of proteins.18−22 Similar to the modification with thiols, but more stable and with the possibility to be employed on both metal and carbon surfaces, is the modification with functionalized aryl diazonium salts.23−26 Corgier et al. introduced in 2005 the use of diazonium-modified antibodies for the direct electrically addressable immobilization of proteins.(27) In 2006, Polsky et al. showed a high direct heterogenenous electron transfer for horseradish peroxidase covalently bonded to carboxyl terminated aryl diazonium salt modified electrodes.(28) More recently, Blanford et al. used aryl diazonium coupling to attach anthracene-2-diazonium on a glassy carbon electrode surface to create the equivalent of an “electric plug” to achieve high DET currents with Pycnoporus cinnabarinus laccase.(29) In recent years, the currents obtained from both DET and MET based electrodes were increased by modifications based on nanomaterials such as single walled carbon nanotubes (SWCNTs).3,30,31 These can improve current densities by two mechanisms: (i) by functioning as nanoscaled electrical wires, which facilitate the contact of the active sites of redox enzymes32,33 and (ii) by generating a large surface area for protein binding.6,7,30−34 In 2001, Bahr and Tour modified SWCNTs with in situ generation of functionalized aryl diazonium species.(35)
Cellobiose dehydrogenase (EC 1.1.99.18, CDH) is one of the redox enzymes capable of DET, which is a result of its two-domain structure.(36) The extracellular, glycosylated flavocytochrome is formed by basidiomycete and ascomycete fungi and is believed to be involved in lignocellulose degradation. Depending on the origin, CDHs show different substrate specificities, but in general, they all oxidize β-1,4-linked di- and oligosaccharides (e.g., cellobiose, cellodextrins, and lactose). Some ascomycete CDHs may also oxidize monosaccharides (e.g., glucose). In the catalytic reaction, 2H+/2e− are donated to the FAD cofactor in the larger flavodehydrogenase domain (DHCDH). The electrons are transferred either directly from the FAD to one- or two-electron acceptors or sequentially by intramolecular electron transfer (IET) to the haem b cofactor located in the smaller cytochrome domain (CYTCDH), which can act as an electron transfer mediator between DHCDH and an electrode (DET step). Because of their carbohydrate oxidizing activity, CDHs have been employed in DET based amperometric lactose biosensors37,38 and recently also in lactose or glucose powered biofuel cells.32−34,39,40 CDH from the basidiomycete Phanerochaete sordida (PsCDH) is an acidic enzyme with respect to its pH optima and its isoelectric point (pI).41,42 To improve the electron transfer efficiency between CYTPsCDH and electrodes, SWCNTs were adsorbed on the top of glassy carbon electrodes and further modified with in situ generated aryl diazonium salts starting from p-aminobenzoic acid and p-phenylenediamine. We have chosen phenyl-COOH (COOH-PD) and phenyl-NH2 (NH2-PD) moieties, because they are present in their deprotonated (carboxyphenyl group pKa = 2.8) or protonated state (aminophenyl group pKa = 4.6) at the investigated pH values of 4.5 and 3.5, respectively. PsCDH was specifically chosen for its very good DET properties10,22,32,33,37,38 and the low isoelectric point (a relatively high abundance of acidic amino acids). This study aimed at investigating the effect of negatively/positively charged SWCNTs on the DET characteristics obtained with a negatively charged protein moiety (CYTCDH) with respect to current density and stability of the produced electrodes. This study is also motivated by the fact that third generation biosensors and biofuel cell electrodes exhibit much lower current densities than their equivalents based on MET. If there is a way to drastically increase the current density through a higher percentage of immobilized redox enzymes orientated toward DET, much would be gained both for analytical as well as other types applications for third generation based bioelectrodes.
Experimental Part
Reagents and Equipment
All reagents were used as received, and aqueous solutions were prepared with Milli-Q water (>18 MΩcm−1, Millipore, Sydney, Australia). Ferricyanide (K4Fe(CN)6), hexaamineruthenium(III)chloride (Ru(NH3)6Cl3), p-aminobenzoic acid, polyethylene glycol diglycidylether (PEGDGE), sodium nitrate (NaNO2), dichloroindophenol (DCIP), and 1,4-benzoquinone (BQ) were obtained from Sigma Chemical Co. (Sydney, Australia). p-Phenylenediamine was from Hopkin and Williams LTD (Glenmilles, PA, USA). Single walled carbon nanotubes (SWCNT) were HiPco tubes from Carbon Nanotechnologies Inc. (Houston, TX, USA). All electrochemical measurements were performed with a BAS-100B electrochemical analyzer (Bioanalytical System Inc. West Lafayette, IL, USA) and a conventional three-electrode system using 3 mm diameter modified glassy carbon (GC) electrodes (BAS) as working electrodes, a homemade platinum plug as counter electrode, and an Ag|AgCl (3 M KCl) reference electrode. The potential is in all cases referred to the normal hydrogen reference electrode (NHE). The current densities were calculated with respect to the geometric electrode area. Unless otherwise stated, cyclic voltammetry (CVs) was carried out in 0.1 M sodium acetate buffers of various pH values. Buffer solutions were degassed by sparging with argon for at least 15 min prior to data acquisition and were blanketed with argon during the entire experiment.
Electrode Preparation and Electrochemical Measurements
GC electrodes were polished with aqueous alumina slurries (Al2O3) by stepwise decreasing the particle size (1 to 0.05 μm) on wet microcloth pads (Buehler, Lake Bluff, IL, USA) using Milli-Q water. The electrodes were thoroughly rinsed with and sonicated in Milli-Q water for 5 min between polishing steps. Ten μL of a 30 mg mL−1 overnight sonicated aqueous solution of SWCNTs was dropped on the top of a GC electrode dried in a desiccator. Carboxylate and amine groups on the GC electrodes were introduced by electrochemical reduction of the aryldiazonium cation generated in situ from either p-aminobenzoic acid (COOH-PD) or p-phenylenediamine (NH2-PD).(43) Briefly, 5 mM (final concentration) NaNO2 was added to a 5 mM acidic aqueous solution (0.5 M HCl) of the aryl amine (1 mM) to generate the aryldiazonium cation. The solution was kept in complete darkness in an ice bath and allowed to react for 5 min under a nitrogen stream and stirring. Surface derivatization was carried out using electrochemical reduction by scanning the SWCNTs-GC working electrode from 1.2 to −0.8 V vs NHE at a rate of 100 mV s−1 for 2 cycles in the aryldiazonium cation-generating solution. The resulting modified electrodes were removed and rinsed with large volumes of water and dried under a stream of nitrogen. Enzyme-modified electrodes were prepared by allowing 10 μL of a Phanerochaete sordida cellobiose dehydrogenase (PsCDH) solution to adsorb on the top (6.55 mg/mL; 206 U/mL, produced according the method from Ludwig et al.(44)) to adsorb on the top of the SWCNTs aryldiazonium modified GC electrodes (Figure 1). The electrodes were allowed to dry overnight at 4 °C under controlled humidity. The activity of PsCDH was 140 U mL−1, the protein concentration was 4.2 mg mL−1, and the specific activity was 33 U mg−1. The cultivation and purification of the enzyme was the same as described for Trametes villosa CDH.(44)
Figure 1.

Schematic representation of SWCNTs-GC electrodes modified with (A1) p-phenylenediamine or (B1) p-aminobenzoic acid. After modification, PsCDH is added and will adsorb onto the SWCNTs exhibiting either (A2) aniline moieties or (B2) benzoic acid moieties. The orientation of PsCDH is very likely influenced by the charged surfaces, with the highly negatively charged CYTPsCDH attracted by the positive charges of A2 or repulsed by the negative charges on B2.
Steady-State Kinetic Measurements
The pH-dependent activity of PsCDH in solution was measured using either the one-electron acceptor cytochrome c or the two-electron acceptors DCIP or BQ in 0.1 M sodium citrate buffer (measured pH range of 2.9−6.4) containing 30 mM lactose as electron donor according to published methods.(45) The apparent KM value of PsCDH for lactose was measured spectrophotometrically at 30 °C employing different lactose concentrations (0.1−50 mM) and cytochrome c (20 μM) as saturating electron acceptor(45) and calculated by linear least-squares regression using Sigma Plot 11 (Systat Software, San Jose, CA, USA). The kcat was calculated from the Vmax value by applying the molecular mass of PsCDH (81 000 Da), and the protein concentration was measured by the Bradford method using a manufactured protein assay reagent (Fermentas, St. Leon-Rot, Germany) and bovine serum albumin as protein standard.
Isoelectric Focusing
To determine the isoelectric point of PsCDH and its proteolytic cleavage products (papain cleavage), samples containing either the native enzyme or the flavodehydrogenase domain (DHPsCDH) and the cytochrome domain (CYTPsCDH) were applied on a Multiphor II system (GE Healthcare) using precast gels (Clean gel IEF) rehydrated with carrier ampholytes in the range between pH 2.0 and 9.0 (GE Healthcare and Serva, Heidelberg, Germany). The IEF protein standard 3−10 from Serva was used to determine the pI values. Enzymatically active bands were visualized by active staining. Therefore, the gel was first soaked in 3 mM DCIP solution before pouring 300 mM lactose solution over the surface. Shortly after, colorless bands appeared because of reduction of DCIP into its leucoform. Afterward, the gel was transferred into 20% acetic acid for overnight protein fixation. Proteins were visualized by silver staining.(46)
Results and Discussion
Aryl Diazonium Coupling
The process is based on aryl amines, which are converted into aryl diazonium salts by treatment with hydrochloric acid and nitrite at ice temperature. Subsequently, the aryl group is covalently attached to the electrode surface by electrochemical reduction, releasing N2.20,23,24 GC electrodes were used as robust supports for physical absorption of SWCNTs and further modification with the introduction of amine groups, which are protonated (positively charged) at pH 3.5 and carboxylate groups, which are deprotonated (negatively charged) at pH 4.5. Deposition of NH2-PD and COOH-PD, respectively, was achieved by two sequential potential CVs from 1.2 to −0.8 V vs NHE in aqueous solution containing 0.5 M HCl and 5 mM NaNO2. Two consecutive CVs are shown in Figure 2a for the NH2-PD modification of a SWCNTs-GC electrode and in Figure 2b for a corresponding COOH-PD one. The first cycle in Figure 2a presents two reduction peaks. During the second cycle, only a small reduction current at a lower potential is present, suggesting the presence of the grafted layer.23−25 The reduction of the p-aminobenzoic acid moiety is exhibited in Figure 2b and characterized by a well-defined, reproducible, and irreversible reduction peak at 0 mV vs NHE in the first cycle. The lower current obtained in the second cycle is evidence of surface saturation and suggests that a layer of covalently bound molecules was formed.
Figure 2.
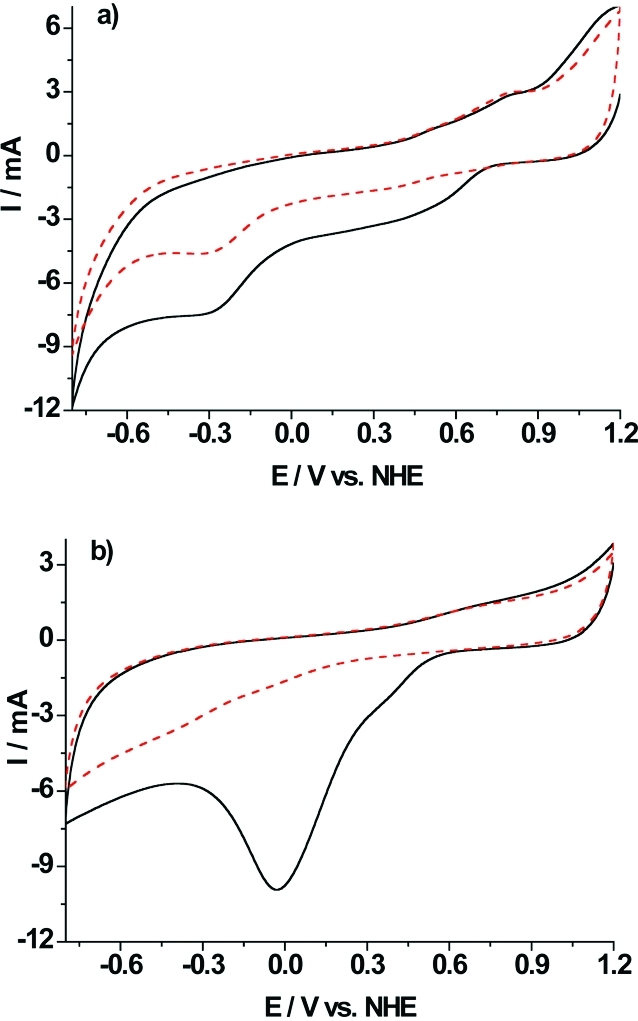
Cyclic voltammograms for in situ generation of aryl diazonium salts on SWCNTs-GC electrodes obtained from (a) 1 mM p-phenylenediamine and (b) 1 mM 4-aminobenzoic acid in 0.5 M HCl aqueous medium with 1 mM NaNO2 at a scan rate of 100 mVs−1. First scan (—), second scan (red − −).
Presence of the Grafted Layer
To demonstrate the presence of the grafted layer and investigate its properties, cyclic voltammetry with soluble electroactive species was performed and the influence of the electrode modification on the oxidation/reduction processes of Ru(NH3)63+ and Fe(CN)64- was investigated. Figure 3a shows the CVs recorded with the NH2-PD/SWCNTs-GC electrode in the presence of 5 mM Ru(NH3)63+ at pH 3.0 (—) and at pH 6.0 (red − −). The oxidation/reduction process of Ru(NH3)63+ presents a quasireversible behavior at pH 6.0 with an apparent redox potential of 0 mV vs NHE. At pH 3.0, the NH2-PD/SWCNTs-GC electrode shows a blocking behavior. The increase of positive charges on the NH2-PD modified electrode when decreasing the pH from 6.0 to 3.0 leads to electrostatic repulsion between Ru(NH3)63+ and the modified electrode surface. The surface pKa of NH2-PD/SWCNTs-GC should be close to that of surface bound p-aminothiophenol and to that of aniline adsorbed on gold pKa = 4.6.(47) A very large fraction of the amino groups will therefore be positively charged at pH 3.5 but neutral at pH 6.0. It could be argued that p-phenylenediamine can form a bifunctional diazonium salt, which should attack the graphite surface in an uncontrollable way. However, it was shown very recently(48) that upon exposure to sodium nitrite only one of the amine groups is converted. In Figure 3b, results from a COOH-PD/SWCNTs-GC electrode in the presence of Fe(CN)64- are shown. At pH 4.0, the carboxylate groups present at the electrode surface are negatively charged and block the Fe(CN)64- redox species from accessing the electrode. At pH 2.0, the carboxylate groups are mostly uncharged and the probe exhibits a well-defined redox wave with a midpoint potential of 425 mV vs NHE. This is in good agreement with previous studies by Saby et al.,(49) who reported a surface pKa of 2.8 for a glassy carbon electrode presenting p-carboxyphenyl groups.
Figure 3.
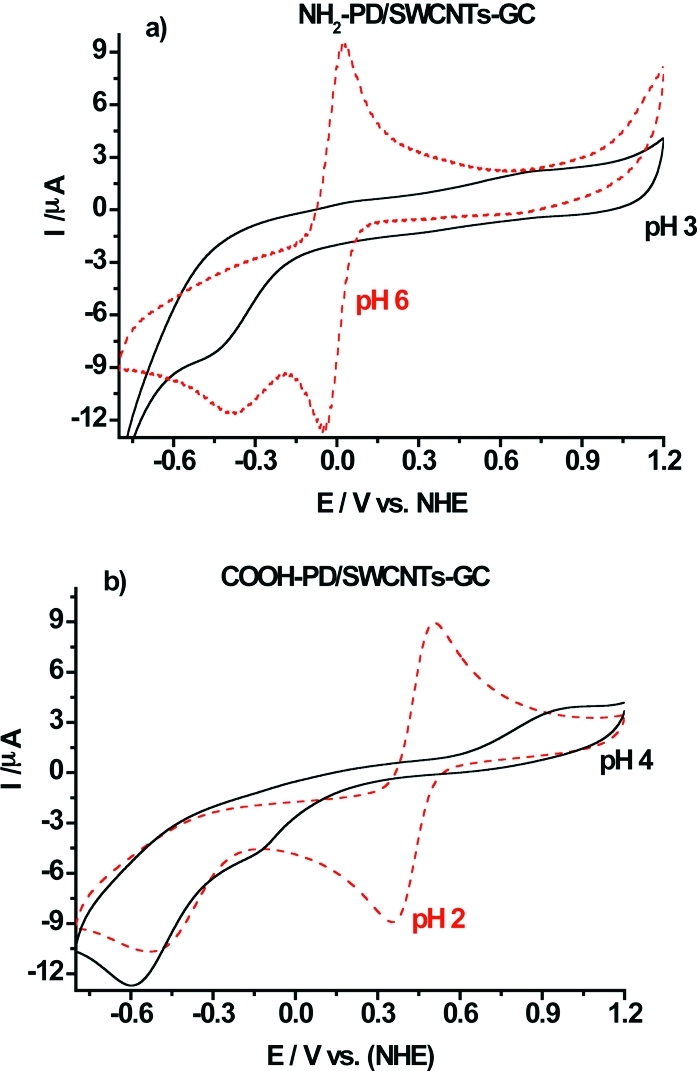
(a) Cyclic voltammograms recorded at NH2-PD/SWCNT-GC electrode in the presence of 5 mM Ru(NH3)63+ in acetate buffer at pH 3.0 (—) and at pH 6.0 (red − −). (b) Cyclic voltammograms recorded at COOH-PD/SWCNT-GC electrode in the presence of 5 mM Fe(CN)63-/4- at pH 4 (—) and at pH 2 (red − −).
Molecular and Catalytic Properties of P. sordida CDH
The isoelectric points (pI) of PsCDH and DHPsCDH were determined by isoelectric focusing using active staining and silver staining (Figure 4). The pI value of the purified PsCDH was estimated to be 4.1; for the proteolytic cleavage product DHPsCDH, a pI value of 5.7 was found. Additionally, minor bands were found in the silver stained gel, one representing a PsCDH isoform (pI = 4.0) and the other (pI = 4.8) could be the proteolytic cleavage product of either the minor isoform or DHPsCDH itself. Both bands are active and overemphasized by the sensitive active staining, which even shows a band for glucose oxidase in lane 1 (from the pI standard) although lactose was used as substrate. No CYTPsCDH band was found, it is likely to be further degraded by papain. By comparison of the values obtained for PsCDH to the values of the well characterized CDH from P. chrysosporium (CDH 4.18, DHPcCDH 5.45, CYTPcCDH 3.42),(50) it can be deduced that the pI of CYTPsCDH must be even lower than the pI of CYTPcCDH to compensate for the less negatively charged DHPsCDH.
Figure 4.
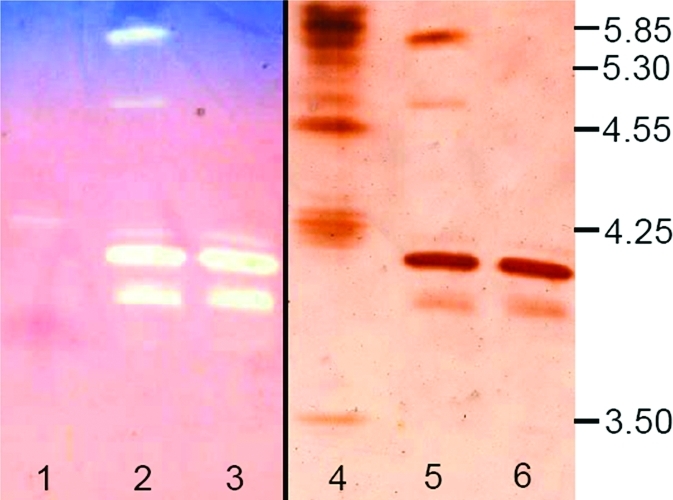
Isoelectric focusing of P. sordida CDH. Active staining with 2,6-dichloroindophenol and lactose is shown on the left side; silver staining is on the right side. Lanes 1 and 4, pI marker; lanes 2 and 5, proteolytic digest of PsCDH; lanes 3 and 6, purified PsCDH.
The pH-dependent activity for different electron acceptors was measured within the pH range of 2.9−6.4 (Figure 5). With the two-electron acceptors, BQ and DCIP, PsCDH exhibits a plateau-shaped pH profile between pH 3.5 and 5.0, which ensures that lactose turnover at DHPsCDH is relatively constant at pH 3.5 and 4.5. In contrast, the one-electron acceptor cytochrome c, which interacts solely with CYTPsCDH, shows a bell-shaped profile with a pH optimum at pH 4.0. The IET is strongly pH dependent and decreases dramatically above pH 4.5 (34% residual activity at pH 5.0, 8% at pH 5.5). However, at the investigated pH values, the IET is fairly constant with 95% of the maximum value at pH 3.5 and 80% at pH 4.5, which ensures that DET currents measured on the electrodes are not influenced by rate limiting or vastly differing IET rates.
Figure 5.

Activity versus pH profiles of PsCDH using (−▽−) cytochrome c, (−◻−) DCIP, and (−○−) BQ. The turnover numbers are given for the different electron acceptors. For lactose, the turnover number would be the same in the case of the two-electron acceptors but only half the value in the case of cytochrome c (stoichiometry = 2).
The KM value of PsCDH for lactose was determined to be 0.7 mM, and the kcat was 45 s−1, values which compare well with data reported for P. chrysosporium CDH measured for lactose with cytochrome c as electron acceptor at room temperature (KM = 0.63 mM, kcat = 28.8 s−1).(51) The lactose concentration used for all further experiments (5 mM) is therefore 7.1-fold the KM value of the enzyme and ensures a high catalytic substrate turnover (∼88% of Vmax). It is, on the other side, sufficiently low to avoid significant background current by nonspecific oxidation as reported for a SWCNT modified electrode and a lactose concentration of 100 mM.(32)
Effect of NH2-PD and COOH−PD Modifications on DET of PsCDH
PsCDH has an overall isoelectric point of 4.1, but its two domains differ strongly in their individual pI values. Whereas DHPsCDH is less negatively charged (pI = 5.7), CYTPsCDH has an isoelectric point below 3.5 and presents many negatively charged amino acid residues on its surface (when assuming a high structural similarity with P. chrysosporium CYTCDH, PDB ID: 1D7B).(52) We sought to take advantage of this uneven distribution of charges and investigate the influence of charged electrode surfaces on the possibility to orientate the enzyme to achieve efficient DET. Moreover, the pH optimum for activity and IET between pH 3.5 and 4.5 allowed us to neglect their influence on DET within this pH range. To obtain high currents, SWCNTs were used as nanoscaled electrical wires to facilitate DET between the CYTCDH of the native enzyme and the underlying electrode and to provide a large surface area for enzyme immobilization.32,33 Moreover, aryl diazonium coupling was used to create specific functional charged groups on the electrode nanowire surface to possibly increase the interaction forces and orientate the enzyme on the surface. The scheme in Figure 1 shows the modification route of SWCNTs-GC electrodes with p-aminobenzoic acid or p-phenylenediamine and subsequently with PsCDH. In Figure 6a, the CV of a PsCDH modified COOH-PD/SWCNTs-GC electrode shows the DET current in the presence and absence of substrate in acetate buffer, pH 4.5. In the CV in the absence of substrate, two redox waves can be observed. The first redox wave at −50 mV can be referred to FAD from DHPsCDH, while the second one at a more positive potential (100 mV) can be assigned to the haem b in CYTPsCDH. When 5 mM substrate is added, the onset of the electrocatalytic current for lactose oxidation can be observed around 70 mV vs NHE with a maximum increase in the slope of the wave at 170 mV and the current steadily increases up until around 200−210 mV and after that starts to level off. No electrocatalytic current was observed from the DHFAD domain. Therefore, the wave seen at −50 mV in the CV without substrate is most likely caused by FAD released from the protein or a denaturated domain, as we already reported in refs (32 and 33), rather than DET between the catalytically active site of the DHFAD domain and the electrode.
Figure 6.
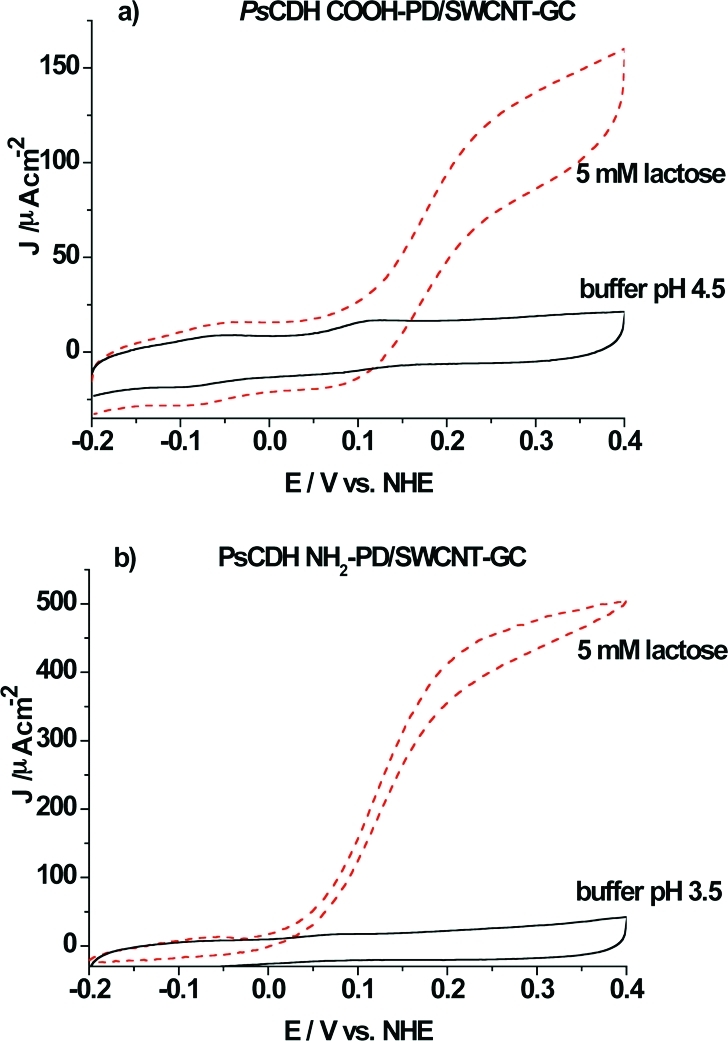
(a) Cyclic voltammogram of a PsCDH COOH-PD/SWCNT-GC electrode in the presence of 5 mM lactose (red − −) and in the absence of substrate (−). 0.1 M acetate buffer, pH 4.5. Scan rate 1 mV s−1. (b) Cyclic voltammogram of a PsCDH NH2-PD/SWCNTs-GC electrode in the presence of 5 mM lactose (red − −) and in the absence of substrate. 0.1 M acetate buffer, pH 3.5. Scan rate 1 mV s−1.
The electrocatalytic current density curve for substrate oxidation reaches a maximum at 150 μA cm−2. This value is comparable to the electrocatalytic current obtained for a PsCDH SWCNT-GC electrode (results not shown) and for a PsCDH SWCNT modified pyrolytic graphite electrode(32) (results not shown). These similar results are not surprising, since it is known that SWCNTs present already some carboxyl groups on their surface(53) and the COOH-PD modification is only expected to saturate the surface with such functionalities. The CVs reported here were obtained at a scan rate of 1 mVs−1. At this scan rate, there is still a noticeable capacitive current due to the high surface area of the electrode. When the amount of enzyme solution used for electrode preparation was varied (5 or 10 μL), the maximum electrocatalytic current was constant, indicating an excess of enzyme.
The modification of the SWCNTs-GC electrode with NH2-PD and PsCDH led to different results compared with earlier reported data for SWCNTs32,33 or the COOH-PD/SWCNTs-GC from this study, especially with respect to the very high catalytic current density obtained (Figure 6b). In fact, a current density of more than 500 μA cm−2 was obtained when the electrode was immersed in a solution containing 5 mM lactose in acetate buffer at pH 3.5 as seen in Figure 6b. This corresponds to an approximately 3.5-fold increase in current with respect to the modification with COOH-PD/SWCNTs. The electrocatalytic current for lactose oxidation can be observed to start at around 0 mV with a maximum for the wave at around 100 mV vs NHE. In the CV without substrate, only a well-defined wave with a midpoint potential at 70 mV can be observed. The onset of the catalytic anodic wave for lactose oxidation is around 50 mV lower than what we obtained for the COOH-PD/SWCNTs-GC (70 mV) and previously for unmodified SWCNTs (75 mV at pH 3.5).32,33 The reason for the lower onset of the anodic wave for lactose oxidation and for the haem b midpoint potential might be the obvious extremely good DET communication between CYTPsCDH and the NH2-PD/SWCNTs layer. The low isoelectric point of CYTPsCDH (pI <3.5) and the high surface concentration of negatively charged amino acid residues already at pH 3.5 can explain why a NH2-PD modification (together with the already existing COOH groups) can create a less electrostatic repulsive environment for CYTPsCDH and enhance DET. Similar current densities for PsCDH (500 μA cm−2 at pH 4.0) were obtained before only when a mediated system based on an osmium redox polymer in combination with PsCDH/SWCNTs modified pyrolytic graphite electrodes was used,(33) where the electrons are shuttled in a mediated mode from the enzyme to the electrode by the osmium redox polymer that was cross-linked to the enzyme. Previous studies of both basidiomycete (class I) and ascomycete (class II) CDH on thiol modified gold electrodes21,22,54,55 have revealed that a positively charged headgroup on the SAM yields much better electrochemical communication with the enzyme than a negatively charged SAM. As expected, the good electrochemical communication between PsCDH and the NH2-PD modified surface is in agreement with previous results, shown for PsCDH at positively charged 4,4′-aldrithiol SAM modified gold electrodes.(22) Two mechanisms can explain the enhanced current observed at NH2-PD-SWCNT modified electrodes. First, PsCDH can be orientated in a better way for DET by electrostatic forces from the positively charged SWCNT surface. This may also lead to that a higher percentage of the immobilized CDH molecules are orientated for DET with the electrode material. Second, the electrostatic repulsion between the negative surface charges of unmodified or COOH-PD SWCNTs and the also negatively charged CYTPsCDH at low pH might reduce the DET rate.
Stability Measurements
When investigating the stability of PsCDH NH2-PD/SWCNTs-GC electrodes in 5 mM lactose at pH 3.5, it performed extremely well. The catalytic current decreased only by 15% in 50 h during continuous recording of multicycle voltammograms at a scan rate of 0.1 mV s−1 between −0.2 and 0.4 V. In Figure 7, 15 scans are shown, which cover a time span of 50 h. Every 10 h, the solution was exchanged for a freshly prepared lactose solution. These results were obtained without a cross-linker. The long-term stability of the electrode was neither positively nor negatively affected when a cross-linker (polyethylene glycol diglycidylether, PEGDGE) was added in a control experiment. This is different to previous reports for CDH/SWCNTs modified carbon electrodes, where always a poor long-term stability was observed in the absence of a cross-linker. The NH2-PD modification seems to prevent the release of PsCDH from the SWCNTs layer, which is most probably due to attractive electrostatic interactions. Also, in this case, for the COOH-PD/SWCNTs modification, no cross-linker had to be added, suggesting an environment which strongly binds PsCDH and supports its stability. The stability was much higher than what has been reported before for PsCDH cross-linked to SWCNTs modified pyrolytic graphite electrodes working under DET conditions (20% decrease after 12 h when scanning at a scan rate of 0.1 mV s−1 between 294 and 394 mV).(32) A comparable stability to the one reported here was only exhibited previously for PsCDH cross-linked to an Os redox polymer and SWCNTs on graphite working under MET conditions (33% decrease after 100 h of running multicyclic voltammograms at different pH values).(33) We have previously seen that, when increasing the ionic strength of the supporting electrolyte, then the DET response current rather increases than decreases,(56) as could have been expected if the interaction between the adsorbed enzyme and the electrode surface is solely based on electrostatic interactions. Obviously, the influence of the ionic strength is much more complex, as was also recently shown for another redox enzyme, sulfite oxidase, when immobilized onto SAM-coated silver electrodes(57) and is currently under intensive studies.
Figure 7.
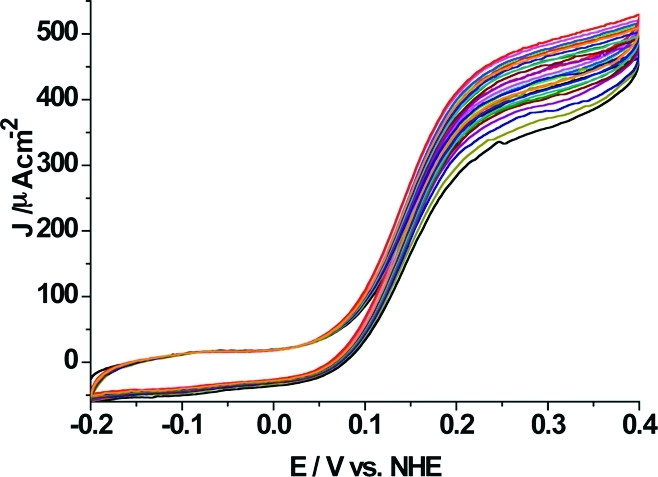
Multiple CVs (15 cycles are shown registered at even intervals during 50 h) of a PsCDH NH2-PD/SWCNTs-GC electrode in the presence of 5 mM lactose in 0.1 M acetate buffer, pH 3.5. Scan rate 0.1 mVs−1.
Conclusions
In PsCDH modified GC electrodes, both the COOH-PD/SWCNT and the NH2-PD/SWCNT modifications were shown to provide a favorable environment for enzyme deposition. Also, the long-term stability of the modified electrode was positively affected by the aryl diazonium salt modification. We can report a loss of 15% of the original activity after 50 h of cycling the electrode between −200 and +400 mV vs NHE. The PsCDH COOH-PD/SWCNTs-GC electrode resulted in a lower current density (150 μAcm−2, 5 mM lactose, pH 4.5) than the PsCDH NH2-PD/SWCNTs-GC electrode exhibiting a current density of 500 μAcm−2 (5 mM lactose, pH 3.5). We conclude that the strongly negatively charged CYTPsCDH interacts better with the positively charged surface of NH2-PD modified electrodes. To the best of our knowledge, this is the first time that such a high a current density is reported for an oxidizing enzyme modified electrode working under DET conditions. Similar current densities were previously reported only for enzymes working in MET conditions.8,33,34 Moreover, the onset of the electrocatalytic current for lactose oxidation started at a potential more than 50 mV lower than when the SWCNTs-GC electrodes were modified with COOH-PD.
Acknowledgments
F.T. would like to acknowledge the Solander Program and the Swedish Chemical Society for supporting travelling and living expenses during his stay at the School of Chemistry at UNSW, Sydney, Australia. R.L. gratefully acknowledges financial support from the Austrian Academy of Sciences (APART-fellowship 11322). L.G. gratefully acknowledges financial support from the The Swedish Research Council (VR) and the Foundation for Research, Science, and Technology, New Zealand. R.L. and L.G. gratefully acknowledge financial support from the European Union (“3D-Bionanodevice” NMP4-SL-2009-229255). J.J.G. gratefully acknowledges financial support from the Australian Research Council Discovery Projects funding scheme (project number DP1094564).
References
- Cracknell J. A.; Vincent K. A.; Armstrong F. A. Chem. Rev. 2008, 108, 2439–2461. [DOI] [PubMed] [Google Scholar]
- Léger C.; Bertrand P. Chem. Rev. 2008, 108, 2379–2438. [DOI] [PubMed] [Google Scholar]
- Tasca F.; Timur S.; Ludwig R.; Haltrich D.; Volc J.; Antiochia R.; Gorton L. Electroanalysis 2007, 19, 294–302. [Google Scholar]
- Heller A. Curr. Opin. Chem. Biol. 2006, 10, 664–672. [DOI] [PubMed] [Google Scholar]
- Mano N.; Heller A. Anal. Chem. 2005, 77, 729–732. [DOI] [PubMed] [Google Scholar]
- Zafar M. N.; Tasca F.; Gorton L.; Patridge E. V.; Ferry J. G.; Nöll G. Anal. Chem. 2009, 81, 4082–4088. [DOI] [PubMed] [Google Scholar]
- Tasca F.; Gorton L.; Wagner J. B.; Nöll G. Biosens. Bioelectron. 2008, 24, 272–278. [DOI] [PubMed] [Google Scholar]
- Tasca F.; Gorton L.; Kujawa M.; Patel I.; Harreither W.; Peterbauer C. K.; Ludwig R.; Nöll G. Biosens. Bioelectron. 2010, 25, 1710–1716. [DOI] [PubMed] [Google Scholar]
- Barton S. C.; Gallaway J.; Atanassov P. Chem. Rev. 2004, 104, 4867–4886. [DOI] [PubMed] [Google Scholar]
- Christenson A.; Dimcheva N.; Ferapontova E. E.; Gorton L.; Ruzgas T.; Stoica L.; Shleev S.; Yaropolov A. L.; Haltrich D.; Thorneley R. N. F.; Aust S. D. Electroanalysis 2004, 16, 1074–1092. [Google Scholar]
- Vincent K. A.; Parkin A.; Armstrong F. A. Chem. Rev. 2007, 107, 4366–4413. [DOI] [PubMed] [Google Scholar]
- Wei J. J.; Liu H. Y.; Dick A. R.; Yamamoto H.; He Y. F.; Waldeck D. H. J. Am. Chem. Soc. 2002, 124, 9591–9599. [DOI] [PubMed] [Google Scholar]
- Gooding J. J.; Wibowo R.; Liu J. Q.; Yang W. R.; Losic D.; Orbons S.; Mearns F. J.; Shapter J. G.; Hibbert D. B. J. Am. Chem. Soc. 2003, 125, 9006–9007. [DOI] [PubMed] [Google Scholar]
- Hess C. R.; Juda G. A.; Dooley D. M.; Amii R. N.; Hill M. G.; Winkler J. R.; Gray H. B. J. Am. Chem. Soc. 2003, 125, 7156–7157. [DOI] [PubMed] [Google Scholar]
- Liu G. Z.; Gooding J. J. Langmuir 2006, 22, 7421–7430. [DOI] [PubMed] [Google Scholar]
- Willner I.; Yan Y. M.; Willner B.; Tel-Vered R. Fuel Cells 2009, 9, 7–24. [Google Scholar]
- Wollenberger U.; Spricigo R.; Leimkuhler S.; Schronder K.. Protein electrodes with direct electrochemical communication In Biosensing for the 21st Century; Springer-Verlag: Berlin, 2008; Vol. 109; pp 19−64. [DOI] [PubMed] [Google Scholar]
- Finklea H. O.Electrochemistry of organized monolayers of thiols and related molecules on electrodes. In Electroanalytical Chemistry; Bard A. J, Ed.; Marcel Dekker: New York, 1996; Vol. 19; pp 109−335. [Google Scholar]
- Gooding J. J.; Hibbert D. B. TrAC, Trends Anal. Chem. 1999, 18, 525–533. [Google Scholar]
- Gooding J. J.; Mearns F.; Yang W. R.; Liu J. Q. Electroanalysis 2003, 15, 81–96. [Google Scholar]
- Lindgren A.; Larsson T.; Ruzgas T.; Gorton L. J. Electroanal. Chem. 2000, 494, 105–113. [Google Scholar]
- Stoica L.; Dimcheva N.; Haltrich D.; Ruzgas T.; Gorton L. Biosens. Bioelectron. 2005, 2010–2018. [DOI] [PubMed] [Google Scholar]
- Delamar M.; Hitmi R.; Pinson J.; Savéant J. M. J. Am. Chem. Soc. 1992, 114, 5883–5884. [Google Scholar]
- Downard A. J. Electroanalysis 2000, 12, 1085–1096. [Google Scholar]
- Gooding J. J. Electroanalysis 2008, 20, 573–582. [Google Scholar]
- Liu Y. C.; McCreery R. L. J. Am. Chem. Soc. 1995, 117, 11254–11259. [Google Scholar]
- Corgier B. P.; Marquette C. A.; Blum L. J. J. Am. Chem. Soc. 2005, 127, 18328–18332. [DOI] [PubMed] [Google Scholar]
- Polsky R.; Harper J. C.; Dirk S. M.; Arango D. C.; Wheeler D. R.; Brozik S. M. Langmuir 2007, 23, 364–366. [DOI] [PubMed] [Google Scholar]
- Blanford C. F.; Heath R. S.; Armstrong F. A. Chem. Commun. 2007, 1710–1712. [DOI] [PubMed] [Google Scholar]
- Yan Y. M.; Yehezkeli O.; Willner I. Chem.—Eur. J. 2007, 13, 10168–10175. [DOI] [PubMed] [Google Scholar]
- Guiseppi-Elie A.; Lei C.; Baughman Ray H. Nanotechnology 2002, 13, 559–564. [Google Scholar]
- Tasca F.; Gorton L.; Harreither W.; Haltrich D.; Ludwig R.; Nöll G. J. Phys. Chem. C 2008, 112, 9956–9961. [Google Scholar]
- Tasca F.; Gorton L.; Harreither W.; Haltrich D.; Ludwig R.; Nöll G. Anal. Chem. 2009, 81, 2791–2798. [DOI] [PubMed] [Google Scholar]
- Tasca F.; Gorton L.; Harreither W.; Haltrich D.; Ludwig R.; Nöll G. J. Phys. Chem. C 2008, 112, 13668–13673. [Google Scholar]
- Bahr J. L.; Tour J. M. Chem. Mater. 2001, 13, 3823–3824. [Google Scholar]
- Ludwig R.; Harreither W.; Tasca F.; Gorton L. ChemPhysChem 2010, 11, 2674–2697. [DOI] [PubMed] [Google Scholar]
- Stoica L.; Ludwig R.; Haltrich D.; Gorton L. Anal. Chem. 2006, 78, 393–398. [DOI] [PubMed] [Google Scholar]
- Safina G.; Ludwig R.; Gorton L. Electrochim. Acta 2010, 55, 7690–7695. [Google Scholar]
- Coman V.; Vaz-Domínguez C.; Ludwig R.; Harreither W.; Haltrich D.; De Lacey A. L.; Ruzgas T.; Gorton L.; Shleev S. Phys. Chem. Chem. Phys. 2008, 10, 6093–6096. [DOI] [PubMed] [Google Scholar]
- Coman V.; Ludwig R.; Harreither W.; Haltrich D.; Gorton L.; Ruzgas T.; Shleev S. Fuel Cells 2010, 10, 9–16. [DOI] [PubMed] [Google Scholar]
- Zamocky M.; Ludwig R.; Peterbauer C.; Hallberg B. M.; Divne C.; Nicholls P.; Haltrich D. Curr. Protein Pept. Sci. 2006, 7, 255–280. [DOI] [PubMed] [Google Scholar]
- Harreither W.; Sygmund C.; Dunhofen E.; Vicuna R.; Haltrich D.; Ludwig R. Appl. Environ. Microbiol. 2009, 75, 2750–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranton S.; Belanger D. J. Phys. Chem. B 2005, 109, 24401–24410. [DOI] [PubMed] [Google Scholar]
- Ludwig R.; Salamon A.; Varga J.; Zamocky M.; Peterbauer; K. C.; Kulbe K. D.; Haltrich D. Appl. Microbiol. Biotechnol. 2004, 64, 213–222. [DOI] [PubMed] [Google Scholar]
- Baminger U.; Subramaniam S. S.; Renganathan V.; Haltrich D. Appl. Environ. Microbiol. 2001, 67, 1766–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge W. J. Biochem. Biophys. Methods 1985, 11, 13–20. [DOI] [PubMed] [Google Scholar]
- Sun L.; Johnson B.; Wade T.; Crooks R. M. J. Phys. Chem. 1990, 94, 8869–8871. [Google Scholar]
- Liu G.; Luais E.; Gooding J. J.. Langmuir 2011, DOI:org/10.1021/la104373v, in press. [Google Scholar]
- Saby C.; Ortiz B.; Champagne G. Y.; Belanger D. Langmuir 1997, 13, 6805–6813. [Google Scholar]
- Henriksson G.; Pettersson G.; Johansson G.; Ruiz A.; Uzcategui E. Eur. J. Biochem. 1991, 196, 101–106. [DOI] [PubMed] [Google Scholar]
- Bao W.; Usha S. N.; Renganathan V. Arch. Biochem. Biophys. 1993, 300, 705–713. [DOI] [PubMed] [Google Scholar]
- Hallberg B. M.; Bergfors T.; Bäckbro K.; Pettersson G.; Henriksson G.; Divna C. Structure 2001, 8, 79–88. [DOI] [PubMed] [Google Scholar]
- Yu X. F.; Mu T.; Huang H. Z.; Liu Z. F.; Wu N. Z. Surf. Sci. 2000, 461, 199–207. [Google Scholar]
- Lindgren A.; Gorton L.; Ruzgas T.; Baminger U.; Haltrich D.; Schülein M. J. Electroanal. Chem. 2001, 496, 76–81. [Google Scholar]
- Coman V.; Harreither W.; Ludwig R.; Haltrich D.; Gorton L. Chem. Anal. (Warsaw) 2007, 52, 945–960. [Google Scholar]
- Larsson T.; Lindgren A.; Ruzgas T.; Lindquist S. E.; Gorton L. J. Electroanal. Chem. 2000, 482, 1–10. [Google Scholar]
- Sezer M.; Spricigo R.; Utesch T.; Millo D.; Leimkuehler S.; Mroginski M. A.; Wollenberger U.; Hildebrandt P.; Weidiger I. M. Phys. Chem. Chem. Phys. 2010, 12, 7894–7903. [DOI] [PubMed] [Google Scholar]