

Abstract
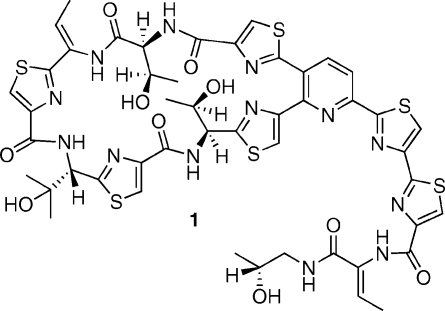
The total synthesis of the thiopeptide antibiotic, thiocillin I, is described. This work unequivocally defines the full structure (constitution and configuration) of the natural product as 1.
Introduction
Thiocillin I (1, Figure 1) and its congeners are thiopeptide antibiotics(i) isolated from Bacillus cereus.(ii) This substance has become of special significance in recent times, notably through the work of Walsh,(iii) in that biosynthetic investigations(iv) have enabled the production of analogues displaying useful biological properties through the genetic manipulation of producing organisms. Such efforts could lead to novel anti-infective agents with improved therapeutic properties, thereby alleviating the ongoing “antibiotic crisis.”(v)
Figure 1.

Structure of Thiocillin I.
A parallel approach based on chemical synthesis may become competitive with the recombinant strategy, if improved methods for the construction of key molecular subunits were to resolve at least some of the problems associated with the “formidable challenge”(iii) of a de novo assembly of thiopeptide architectures. A nettlesome aspect of such endeavors is the preparation of the pyridine−thiazole cluster found at the core of those antibiotics, noteworthy solutions for which have materialized over the past few years. For instance, the construction of the core of thiostrepton and related compounds incorporating a reduced pyridine inspired the development of remarkable biomimetic cycloadditions of azadienes;(vi) efforts aiming to produce fully aromatic cores by organometallic functionalization of a preexisting pyridine have engendered important advances in Pd-mediated coupling reactions,(vii) while approaches to a de novo construction of a pyridine nucleus fully decorated with the requisite complement of thiazolyl substituents, or at least with as many such thiazoles as possible, have produced significant refinements of the Hantzsch(viii) and the Bohlmann−Rahtz(ix) reactions. The foregoing notwithstanding, the construction of thiopeptide cores remains a lengthy proposition, generally requiring more than 10 linear steps. This difficulty continues to stimulate research aiming to simplify the task.
In that connection, the work of Bagley has shown that a Bohlmann-Rahtz avenue to the pyridine nucleus provides much opportunity for the development of more concise pathways.(x) Expanding upon these findings, we devised a method to prepare pyridines 4 through the one-step, three-component condensation of fragments 2 and 3 in the presence of NH4OAc (Scheme 1).(xi) Heterocyclic clusters such as 4 embody the core structures of a number of thiopeptides incorporating a fully aromatic pyridine. In this article, we illustrate an application of the new technique in what appears to be the first total synthesis(xii) of 1. In addition to resolving a number of technical difficulties inherent to past synthetic schemes, this exercise also provides full structural proof for the natural product. Indeed, the constitution of 1 was reasonably secure at the onset of the present work; however, its configuration remained to be ascertained. The similarity of thiocillin to micrococcin P1(xiii) suggested the configuration depicted in Figure 1: a surmise that was ultimately confirmed as detailed herein.
Scheme 1. One-Step Bohlmann−Rahtz Synthesis of Thiopeptide Cores.

Results and Discussion
Current methods for the preparation thiopeptide cores via a Bohlmann−Rahtz pyridine synthesis could be improved in two respects. First, ynones 3 required for this chemistry derive from 2-formylthiazoles such as 5 and 6 (Scheme 2). Past routes to these materials have evolved from glycolonitrile (7)(xiv) or diethoxyacetonitrile (8),(xv) the elaboration of which to the desired subgoals, especially to 6, requires up to 8 steps. A more concise avenue to 5 and 6 is surely desirable. A good solution in that sense appeared to be the SeO2 oxidation of 2-methylthiazoles such as 9 and 10: a transformation that was undocumented at the onset of our endeavor. Indeed, substrates 9 and 10 resisted the action of SeO2 in refluxing ethanol or dioxane. However, oxidation to the corresponding 2-formylthiazoles occurred easily in refluxing acetic acid over 12 h.(xi) The reaction of 9 was best carried out at a concentration of 0.5 M, whereupon the desired 5 was obtained in 55% yield after purification. A significant byproduct of the reaction was 4-carbethoxythiazole (21% yield), which was readily removed by chromatography. This compound presumably arose through overoxidation of 5 to the corresponding acid, followed by decarboxylation. Higher concentrations (1 M or greater) caused the formation of unacceptably large quantities of 4-carbethoxythiazole, while more dilute conditions slowed down the rate of the reaction to a considerable degree. The oxidation of 10 under the same conditions also proceeded in 55% yield (after chromatography), but no discrete byproducts could be detected in this case.(xvi) The formylthiazoles thus obtained were smoothly advanced to ynones 13 and 15 by addition of ethynylmagnesium bromide and oxidation of the resultant carbinols with IBX(xvii) or with Dess−Martin reagent, respectively (Scheme 3).(xi)
Scheme 2. Oxidation of 2-Methylthiazoles to 2-Formylthiazoles.

Scheme 3. Preparation of Ynones Required for the Bohlmann−Rahtz Reaction.

Second, a conventional Bohlmann−Rahtz reaction requires the prior conversion of the enolizable component (2 in the present case) into an enamine, which subsequently undergoes cyclocondensation with the ynone in a second step of the sequence. A variant of this chemistry devised by Bagley(xa) circumvents the need for a two-step synthesis by causing an enolizable ketone, 16, an ynone, 17, and NH4OAc to combine in refluxing ethanol, leading to the expected pyridine 20 in excellent yield (Scheme 4). The transformation presumably proceeds via in situ formation of enamine 18, which then enters the actual Bohlman−Rahtz pathway. We have utilized this procedure for various synthetic objectives, and indeed we have found that the Bagley method generally affords better results than the traditional, stepwise protocol. However, the chemistry appears to perform well only if the enolizable component is part of a 1,3-dicarbonyl system, for example, if R2 = COOEt; otherwise, the enamine must still be prepared separately, and then combined with the ynone in refluxing EtOH. To illustrate, the Bagley synthesis of the amythiamicin core(xviii) required the conversion of a ketone structurally related to 2 into the corresponding enamine, which then reacted with a congener of 3 to afford a pyridine of the type 4. A method for the direct merger of 2, 3, and NH4OAc would clearly be advantageous.
Scheme 4. The Bagley One-Step Variant of the Bohlmann−Rahtz Reaction.

Initial attempts to combine 13 or 15 with the known 21(viii) under Bagley conditions met with failure. Analogy with the difficulties encountered in the Ugi reaction of α-amino acids with aromatic aldehydes(xix) suggested that the problem was attributable to an insufficiently rapid rate of formation of an enamine such as 18. While in the Ugi context the appropriate remedy proved to be Bronsted acid-assisted Lewis acid (“BLA”) catalysis,(xx) plain Bronsted acid catalysis afforded a perfectly adequate solution in the present case. Indeed, Bagley had established that particular Bohlmann−Rahtz reactions proceed more efficiently in mixtures of toluene and acetic acid (typically 5:1) as the solvent.(xxi) Experiments aiming to induce the combination of 13 and 15 with 21 and NH4OAc in media of progressively greater acidity revealed that the transformation proceeded best in refluxing acetic acid,(xi) that is, in a manner analogous to the Eiden−Herdeis pyridine synthesis.(xxii) Pyridines 22 and 23 were thus obtained in 63 and 52% yield, respectively, after chromatographic purification (Scheme 5). It should be noted that under the present conditions the TBS group originally present in 21 was replaced by an acetyl unit, presumably through acid-promoted desilylation and subsequent Fischer esterification of the liberated alcohol. Compound 23 is recognized as a protected form of the core cluster of micrococcin P1−P2,(xiii) thiocillin I,(ii) and YM266183.(xxiii) Its preparation by the present method (4 linear steps from 10; 20% overall yield) compares favorably with earlier routes. On the other hand, commercial 10 is costly; so, subsequent work relied on synthetic 10 prepared from 9 in 3 steps.(xxiv) This notwithstanding, the route to 23 from 9 (7 linear steps, 16% overall yield) was still preferable to other known approaches.
Scheme 5. Preparation of the Core of Thiocillin I.

The total synthesis of thiocillin I continued with Parekh−Doering oxidation of the known (R)-alcohol 24(xxv) (Scheme 6). Immediate condensation of the sensitive aldehyde 25 with methyl cysteinate hydrochloride, under the conditions of the Shioiri thiazole synthesis,(xxvi) gave a mixture of diastereomers of the expected thiazoline 26.(xxvii) In crude form, the latter was immediately aromatized to thiazole 27. As thoroughly detailed in the literature, only so-called “chemical” MnO2(xxviii) performed adequately in this oxidative step. The BOC group in 27 was then released without incident by the customary TFA treatment. No evidence of acid-promoted elimination of the tertiary alcohol, either in 27 or in later synthetic intermediates incorporating such a fragment, was ever garnered as a consequence of TFA treatment. Amine 28 was then coupled with the previously described acid 29.(xiv) The resultant 30 was again N-deblocked and amine 31 was coupled with the known(xxix) acid 32, resulting in formation of 33. This material was advanced to amine trifluoroacetate salt 35 by mesylation/dehydration (introduction of the dehydroaminoacid motif) followed by deblocking of the serinyl segment.
Scheme 6. Synthesis of the “Western” Segment of Thiocillin I.

(a) SO3·pyr, DMSO, Et3N, CH2Cl2 0 °C to rt; (b) methyl cysteinate ·HCl, NaHCO3, 1:1 MeOH, H2O, rt, 6 h; (c) MnO2, MeCN, reflux, 6 h, 30% (a)−(c); (d) 1:4 TFA/CH2Cl2, rt, 2 h; (e) EDCI, HOBt, CH2Cl2, rt, 12 h, 65% of 30 from 27 (d)−(e); (f) MsCl, Et3N, then DBU; 32% of 34 from 30 (d)−(f); (g) TFA, MeOH.
At this juncture, we faced a minor dilemma. In principle, the synthesis of 1 could be completed through the union of fragments 23 and 35 by methodology analogous to that devised earlier for the synthesis of micrococcin.(xiii) However, the modified Bohlmann−Rahtz reaction outlined in Scheme 5 had afforded compound 23 as an acetate ester. Using this compound as such in subsequent steps would have forced either a revision of late-stage protecting group tactics (Scheme 7: the appropriate hydroxyl groups in fragments 38 and 39 must be orthogonally protected), or a reoptimization of the final sequence with an acetyl-protected pyridine segment. In the interest of time, we opted to convert 23 into 36, so that thiocillin I could be reached by retracing the chemistry devised earlier (Scheme 7). Thus, ester saponification and N-BOC activation of the oxazolidinone prepared the molecule for coupling with the known 39.xiii,xiv The emerging 40 underwent smooth dehydration to 41 upon reaction with methanesulfonyl chloride in the presence of DBU, as a prelude to deblocking of the primary alcohol and stepwise oxidation thereof to carboxylic acid 44. This material was very polar and difficult to purify; so it was best advanced directly to the final steps of the synthesis. Crude 44 was thus coupled with fragment 35 to yield 45 (Scheme 8). Global deblocking followed by macrocyclization of monoseco intermediate 46 with DPPA(xxx) afforded 1, which proved to be identical(xxxi) to natural thiocillin I, kindly provided by the Shionogi Co. Japan.
Scheme 7. Elaboration of the Complete “Eastern” Portion of Thiocillin I.

(a) K2CO3, 1:1 CH2Cl2-EtOH, 48 h; (b) TBS-Cl, imidazole DMF, rt, 12 h; (c) LiOH, 1:1 THF-H2O, rt, 4 h; (d) BOC2O, Et3N, CH2Cl2; (e) BOP-Cl, Et3N, MeCN, rt, 6 h, 20% (a)−(e); (f) MsCl, Et3N, CH2Cl2, 2 h, then DBU, 3 h; (g) TBAF, THF, 3 h; (h) Dess−Martin periodinane, CH2Cl2, rt, 5 h, 84% (f)−(h); (i) NaClO2, 2-methyl-2-butene, NaH2PO4, aq. THF, rt, 4 h.
Scheme 8. Total Synthesis of Thiocillin I.

(a) BOP-Cl, 35, Et3N, MeCN; (b) LiOH, 1:1 THF-H2O; (c) TFA, CH2Cl2, 4 h; (d) DPPA, Et3N, DMF, 5 h, 12% from 43.
The modular nature of the synthesis just described enables the preparation of natural thiopeptides and their analogues in a straightforward manner, thus, bringing medicinal chemistry research on such antibiotics within the realm of realistic possibility. Results in this area will be reported in due course.
Acknowledgments
We thank the University of British Columbia, the Canada Research Chair program, NSERC, CIHR, and MerckFrosst Canada, for support of our program. We are grateful to Dr. Dan Sorensen, of MerckFrosst Canada, for the HPLC purification and the spectroscopy of synthetic 1, and especially to Dr. Hiroki Satou, of the Shionogi Pharmaceutical Company, Japan, for a gift of authentic thiocillin I.
Supporting Information Available
Full experimental details and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
Novartis, Inc., Cambridge, MA.
Supplementary Material
References
- Reviews: ; a Bagley M. C.; Dale J. W.; Merritt E. A.; Xiong X. Chem. Rev. 2005, 105, 685. [DOI] [PubMed] [Google Scholar]; b Hughes R. A.; Moody C. J. Angew. Chem., Int. Ed. 2007, 46, 7930. [DOI] [PubMed] [Google Scholar]; c Arndt H.-D.; Schoof S.; Lu J.-Y. Angew. Chem., Int. Ed. 2009, 48, 6770. [DOI] [PubMed] [Google Scholar]
- a Shoji J.; Hinoo H.; Wakisaka Y.; Koizumi K.; Mayama M.; Matsuura S.; Matsumoto K. J. Antibiot. 1976, 24, 366. [DOI] [PubMed] [Google Scholar]; b Shoji J.; Kato T.; Yoshimura Y.; Tori K. J. Antibiot. 1981, 29, 1126. [DOI] [PubMed] [Google Scholar]
- a Acker M. G.; Bowers A. A.; Walsh C. J. J. Am. Chem. Soc. 2009, 131, 17563. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bowers A. A.; Walsh C. J.; Acker M. G. J. Am. Chem. Soc. 2010, 132, 12182. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bowers A. A.; Acker M. G.; Koglin A.; Walsh C. J. J. Am. Chem. Soc. 2010, 132, 7519. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Walsh C. J.; Acker M. G.; Bowers A. A. J. Biol. Chem. 2010, 286, 27525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wieland Brown L. C.; Acker M. G.; Clardy J.; Walsh C. T.; Fischbach M. A. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 2559. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liao R.; Duan L.; Lei C.; Pan H.; Ding Y.; Zhang Q.; Chen D.; Shen B.; Yu. Y. Chem. Biol. 2009, 16, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a von Nussbaum F.; Brands M.; Hinzen B.; Weigand S.; Häbich D. Angew. Chem., Int. Ed. 2006, 45, 5072. [DOI] [PubMed] [Google Scholar]; b Isaacs D. Indian Pediat. 2005, 42, 9 and references cited therein. [PubMed] [Google Scholar]; c Hausler T.Viruses vs Superbugs: A Solution to the Antibiotic Crisis?; Palgrave Macmillan: Houndsmill, U.K., 2006. [Google Scholar]
- a Nicolaou K. C.; Nevalainen M.; Safina B. S.; Zak M.; Bulat S. Angew. Chem., Int. Ed. 2002, 41, 1941. [PubMed] [Google Scholar]; b Moody C. J; Hughes R. A.; Thompson S. P.; Alcaraz L. Chem. Commun. 2002, 1760. [DOI] [PubMed] [Google Scholar]; See also ref 1b.
- Noteworthy in this regard is the work of Bach: ; a Muller H. M.; Delgado O.; Bach T. Angew. Chem., Int. Ed. 2007, 46, 4771. [DOI] [PubMed] [Google Scholar]; b Delgado O.; Muller H. M.; Bach T. Chem.—Eur. J. 2008, 14, 2322. [DOI] [PubMed] [Google Scholar]; See also: ; c Martin T.; Laguerre C.; Hoarau C.; Marsais F. Org. Lett. 2009, 11, 3690. [DOI] [PubMed] [Google Scholar]; d Martin T.; Verrier C.; Hoarau C.; Marsais F. Org. Lett. 2008, 10, 2909. [DOI] [PubMed] [Google Scholar]
- Ciufolini M. A.; Shen Y. C. J. Org. Chem. 1997, 62, 3804. [DOI] [PubMed] [Google Scholar]
- a Bohlmann F.; Rahtz D. Chem. Ber. 1957, 90, 2265. [Google Scholar]; Review: ; b Bagley M. C.; Glover C.; Merritt E. A. Synlett 2007, 2459. [Google Scholar]; Moody introduced the reaction into current synthetic practice during his pioneering synthesis of promothiocin: ; c Moody C. J.; Bagley M. C. Chem. Commun. 1998, 2049. [Google Scholar]; d Bagley M. C.; Bashford K. E.; Hesketh C. L.; Moody C. J. J. Am. Chem. Soc. 2000, 122, 3301. [Google Scholar]
- a Bagley M. C.; Dale J. W.; Bower J. Chem. Commun. 2002, 1682. [DOI] [PubMed] [Google Scholar]; b Bagley M. C.; Chapaneri K.; Dale J. W.; Xiong X.; Bower J. J. Org. Chem. 2005, 70, 1389. [DOI] [PubMed] [Google Scholar]; See also: ; c Bagley M. C.; Brace C.; Dale J. W.; Ohnesorge M.; Phillips N. G.; Xiong X.; Bower J. J. Chem. Soc., Perkin Trans. 1 2002, 1663. [Google Scholar]
- Aulakh V. S.; Ciufolini M. A. J. Org. Chem. 2009, 74, 5750. [DOI] [PubMed] [Google Scholar]
- Synthetic studies: ; a Yonezawa Y.; Saito H.; Suzuki S.; Shin C.-G. Heterocycles 2002, 57, 903. [Google Scholar]; b Suzuki S.; Yonezawa Y.; Shin C.-G. Chem. Lett. 2004, 33, 814. [Google Scholar]
- Review: ; a Ciufolini M. A.; Lefranc D. Nat. Prod. Rep. 2010, 27, 330. [DOI] [PubMed] [Google Scholar]; Total synthesis and full structural elucidation: ; b Lefranc D.; Ciufolini M. A. Angew. Chem., Int. Ed. 2009, 48, 4198. [DOI] [PubMed] [Google Scholar]
- See refs (viii) and,(xiii) as well as: Ciufolini M. A.; Shen Y.-C. Org. Lett. 1999, 1, 1843. [DOI] [PubMed] [Google Scholar]
- Merritt E. A.; Bagley M. C. Synlett 2007, 954. [Google Scholar]
- This reaction forms a black precipitate of reduced forms of Se. It is certainly possible that said precipitate entrains byproducts formed during the reaction.
- The use of IBX for the oxidation of such carbinols was introduced by Moody (ref (ixc,ixd)). We found that the Dess−Martin periodinane performed better than IBX in the oxidation of 14.
- Bagley M. C.; Dale J. W.; Jenkins R. L.; Bower J. Chem. Commun. 2004, 102. [DOI] [PubMed] [Google Scholar]
- Godet T.; Bonvin Y.; Vincent G.; Ciufolini M. A. Org. Lett. 2004, 6, 3281. [DOI] [PubMed] [Google Scholar]
- a Ishihara K.; Yamamoto H. J. Am. Chem. Soc. 1994, 116, 1561. [Google Scholar]; b Ishihara K.; Kaneeda M.; Yamamoto H. J. Am. Chem. Soc. 1994, 116, 11179. [Google Scholar]; Review: ; c Yamamoto H.; Cheon C. H. In Catalytic Asymmetric Synthesis, 3rd ed.; Ojima I., Ed.; John Wiley & Sons: Hoboken, NJ, 2010; Chapter 3, pp 119ff. Early examples: [Google Scholar]; d Deaton M. V.; Ciufolini M. A. Tetrahedron Lett. 1993, 34, 2409. [Google Scholar]; See also: ; e Ciufolini M. A.; Zhu S.; Deaton M. V. J. Org. Chem. 1997, 62, 7806. [Google Scholar]
- a Bagley M. C.; Chapaneri K.; Dale J. W.; Xiong X.; Bower J. J. Org. Chem. 2005, 70, 1389. [DOI] [PubMed] [Google Scholar]; See also: ; b Bagley M. C.; Brace C.; Dale J. W.; Ohnesorge M.; Phillips N. G.; Xiong X.; Bower J. J. Chem. Soc., Perkin Trans. 1 2002, 1663. [Google Scholar]
- Eiden F.; Herdeis C. Arch. Pharm. 1977, 310, 744. [DOI] [PubMed] [Google Scholar]; A similar reaction may be carried out in ionic liquids: Karthikeyan G.; Perumal P. T. Can. J. Chem. 2005, 83, 1746. [Google Scholar]
- Nagai K.; Kamigiri K.; Arao N.; Suzumura K.-I.; Kawano Y.; Yamaoka M.; Zhang H.; Watanabe M.; Suzuki K. J. Antibiot. 2003, 56, 123. [DOI] [PubMed] [Google Scholar]
- Ref (xi) as well as Huang L.; Quada J. C. Jr.; Lown J. W. Het. Commun. 1995, 1, 335. [Google Scholar]
- Ciufolini M. A; Valognes D.; Xi N. Angew. Chem., Int. Ed. 2000, 39, 2493 and references cited therein. [DOI] [PubMed] [Google Scholar]
- a Hamada Y.; Kohda K.; Shioiri T. Tetrahedron Lett. 1984, 25, 5303. [Google Scholar]; b Hamada Y.; Shibata M.; Shioiri T. Tetrahedron Lett. 1985, 26, 5155. [Google Scholar]; c Hamada Y.; Shibata M.; Shioiri T. Tetrahedron Lett. 1985, 26, 5159. [Google Scholar]; d Sugiyama H.; Yokokawa F.; Shioiri T. Org. Lett. 2000, 2, 2149. [DOI] [PubMed] [Google Scholar]
- For a related construction see ref (xiia).
- This special grade of MnO2 is available at modest cost from the Wako Chemical Co., Japan.
- Nakamura Y.; Shin C.-G.; Umemura K.; Yoshimura J. Chem. Lett. 1992, 1005. [Google Scholar]
- a Shioiri T.; Ninomiya K.; Yamada S. J. Am. Chem. Soc. 1972, 94, 6203. [DOI] [PubMed] [Google Scholar]; Use in macrolactamization reactions: ; b Hamada Y.; Shioiri T. Chem. Rev. 2005, 105, 4441. [DOI] [PubMed] [Google Scholar]; Reviews: ; d Shioiri T. TCIMail 2008, 134, 1 (http://www.tci-asiapacific.com/tcimail/backnumber/article/134drE.pdf). [Google Scholar]; c Liang H. Synlett 2008, 2554. [Google Scholar]
- One of the referees recommended that an NMR spectrum of a mixture of natural and synthetic thiocillin I be provided. Unfortunately, the sample of synthetic 1 had deteriorated by the time that the suggestion was made, preventing us from obtaining said spectrum. On the other hand, stacked plots of the 1H NMR spectrum of natural and synthetic thiocillin I (provided as Supporting Information) reveal that all chemical shifts of nonexchangeable protons match to within ±0.01 ppm and coupling constants likewise match. This leaves no doubt that synthetic 1 is structurally identical to authentic thiocilin I.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.