

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common genetic kidney condition and is associated with important renal and cardiovascular manifestations in childhood. Renal cystic disease can be documented in some cases as early as in utero. Early intervention is critical if the long-term complications of this condition, including end-stage renal disease, are to be ameliorated. Here we describe our ongoing randomized double-blind placebo-controlled phase III clinical trial to assess the effect of pravastatin treatment on renal and cardiovascular disease progression in 107 children and young adults age 8–22 years with ADPKD who are receiving the angiotensin converting enzyme inhibitor lisinopril. Baseline demographic and laboratory data are provided. Results of this study could markedly impact the standard of care for evaluation and treatment of ADPKD in this population.
Keywords: Pediatric, Autosomal dominant polycystic kidney disease, Statin, Renal volume
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary renal disease, affecting 1 in 400 to 1000 individuals (1). It is responsible for approximately 4% of end-stage renal disease (ESRD) in the United States and 8–10% in Europe (1). ADPKD is caused by mutations in the genes PKD1 or PKD2, which encode polycystin 1 or polycystin 2, respectively. These proteins localize to the primary cilia and appear to interact to detect fluid flow within the renal tubule (2–4). While tubular flow in the normal kidney results in bending of cilia and activation of the polycystin flow sensor that allows Ca2+ influx into the cell (5), inactivation of the polycystin complex due to PKD1 or PKD2 mutations is believed to result in altered Ca2+ homeostasis, thus allowing increased proliferation and apoptosis, altered polarity, and altered secretory properties in ADPKD cells (6). These abnormalities lead to tubular cyst formation. ADPKD is characterized clinically by the progressive development of kidney cysts with renal enlargement and associated loss of renal function, such that approximately half of patients with PKD1 develop ESRD by 60 years of age (1).
Hypertension is a common feature in ADPKD, affecting up to 60–80% of patients. Hypertension occurs prior to the onset of renal insufficiency and is associated with faster progression to ESRD (7–9). There is significant evidence that the renin-angiotensin-aldosterone system (RAAS) is a major contributor to hypertension and cyst growth in both animals and humans with ADPKD (10). Recent studies have focused on the role of 3-hydroxy-3-methylglutaryl coenzyme A (HMG coA) reductase inhibitors (statins) in the amelioration of both progressive nephropathy and cardiovascular disease, and there are data to suggest that statins may have modulatory effects on the RAAS [Reviewed in (11)]. In this randomized double-blind placebo-controlled phase III clinical trial, we are investigating the effects of pravastatin treatment on renal and cardiovascular disease progression in children and young adults with ADPKD who are concurrently treated with the angiotensin converting enzyme (ACE) inhibitor lisinopril. Disease progression will be assessed with a combined primary endpoint of changes in renal volume, left ventricular mass index, and microalbuminuria over the three-year study period. Here we discuss the rationale, study design, and baseline characteristics of study participants.
Scientific rationale
Rationale for use of angiotensin converting enzyme (ACE) inhibition
Activation of the RAAS by cyst expansion and local ischemia has been proposed to play a major role in the pathogenesis of the hypertension and renal cyst growth associated with ADPKD (12;13). Stimulation of RAAS has been observed to occur early in ADPKD, even prior to the development of hypertension (13–15) and may contribute to atherosclerosis, left ventricular hypertrophy, and ESRD (16–18). Angiotensin II is known to increase oxidative stress, leading to vascular inflammation, proliferation, and chemotaxis (19–21) and is an important growth factor for renal proximal tubular cells (22) and renal interstitial fibroblasts (23). Tubular epithelial cell proliferation is of fundamental importance in the pathogenesis of cystic disease (24;25), and interstitial fibrosis appears to be a major contributor to ESRD (25). Hence, it appears that a vicious cycle is created in which renal cysts activate the RAAS, and the increase in angiotensin II enhances cystogenesis, hypertension, and interstitial fibrosis. Our previous studies suggest that ACE inhibition in children with ADPKD and borderline hypertension prevents the decline in glomerular filtration rate and the increase in left ventricular mass index over time (26). Based on these findings, all subjects in this clinical trial are treated with the ACE inhibitor lisinopril.
Rationale for use of statin
Recent clinical studies have demonstrated the efficacy of statins in ameliorating progressive nephropathy (27–31). Statins are known to improve renal blood flow (27;32) and glomerular filtration rate (GFR) by increasing single nephron GFR (33). Statin treatment is also associated with enhanced vascular and glomerular nitric oxide production and attenuation of vascular inflammation [Reviewed in (34)]. Of particular relevance to ADPKD, statins have also been shown to be beneficial in experimental angiotensin II-mediated renal injury. Specifically, rats transgenic for human renin and angiotensinogen develop early hypertension, cardiac hypertrophy, and renal damage, with marked albuminuria and focal cortical necrosis (35). Treatment of such rats with cerivastatin was associated with significant reductions in systolic blood pressure, serum creatinine, and albuminuria. The effect of statin treatment on renal function and structure has been previously assessed in ADPKD. Specifically, van Dijk et al. have demonstrated increases in renal blood flow and GFR in response to a 4-week period of simvastatin treatment in 10 normocholesterolemic normotensive patients with ADPKD (32). It has also been proposed that statins may mediate progression of structural disease in ADPKD. In this regard, lovastatin has been shown in heterozygous male Han:SPRD rats to reduce the severity of PKD as assessed by kidney size, volume density of cysts, and serum urea nitrogen concentration (36). Although the underlying mechanisms are not well understood, it is believed that these renoprotective effects are mediated by statin-related inhibition of G-proteins with resultant decreased cell proliferation (37).
Rationale for study endpoints
Renal volume
With approximately half of patients with PKD1 reaching ESRD by 60 years of age, there has been significant interest in introducing interventions which will slow progression of renal disease. It was observed over two decades ago that renal function remains stable in ADPKD patients for many years, followed by a sharp decrease in glomerular filtration rate once a critical renal size is reached (38). However, it is important to note that the majority of patients with ADPKD manifest renal enlargement prior to loss of renal function, suggesting that distortion of renal architecture is an early clinical feature of ADPKD (39). Renal enlargement is associated with more symptoms including pain and gross hematuria, a greater decrease in renal concentrating ability, elevated blood pressure, increased proteinuria, and faster progression to ESRD (40–44). In addition, overall renal volume, renal volume growth rate, and renal cyst volume have each been shown to correlate inversely with GFR in adults with ADPKD (45;46). Our previous studies have shown an increase in renal volume in hypertensive as compared to normotensive ADPKD children (26). These findings suggest that renal volume and/or cyst volume are appropriate and important endpoints for evaluation of interventions in clinical trials in ADPKD, and these parameters currently form the primary endpoints in a number of clinical trials in ADPKD in adults (47–49). Magnetic resonance imaging (MRI) has now been established as the most accurate and reliable technique for assessment of renal volume in subjects with ADPKD (50).
Left ventricular mass index
With advances in ESRD treatment, the primary cause of death in ADPKD patients is cardiovascular disease (9;51–53). Left ventricular hypertrophy is an important prognostic marker and is known to be associated with systolic and diastolic dysfunction, congestive heart failure, ischemic cardiac disease, arrhythmias, and sudden death. Nearly half of hypertensive adult patients with ADPKD have left ventricular hypertrophy at a mean age of only 44 years (54), and a significant correlation between hypertension and increased left ventricular mass index has been observed in both children and adults with ADPKD (54–56). RAAS has also been shown to contribute to the development of left ventricular hypertrophy in non-ADPKD models (57;58). Consistent with these observations, aggressive control of hypertension (mean arterial pressure 90 mm Hg) has been shown to decrease left ventricular mass index to a greater extent than standard blood pressure control (mean arterial pressure 100 mm Hg) in adult hypertensive subjects with ADPKD (59). Moreover, angiotensin converting enzyme inhibition was shown to be better to reverse left ventricular hypertrophy in ADPKD adults as compared to a calcium channel blocker (59). Studies in rats have shown that simvastatin can decrease left ventricular hypertrophy due to pressure overload (aortic stenosis) in association with a marked statin-mediated reduction in left ventricular angiotensin converting enzyme activity and angiotensin II content (60). The improvement in left ventricular mass appears to occur via a reduction in Ras membrane targeting and activation. We have previously utilized left ventricular mass index as a primary study endpoint in our interventional trials in pediatric ADPKD (26;55;61). Magnetic resonance imaging has now been established as the most accurate and reliable method for serial assessment of left ventricular mass (62;63).
Microalbuminuria
Microalbuminuria is present in nearly 20% of normotensive adults with ADPKD (64), and such patients demonstrate a tendency toward increased systolic blood pressure, plasma renin activity, and left ventricular mass index as compared to those without microalbuminuria. Similar findings have been observed in children with ADPKD. Specifically, overt proteinuria and microalbuminuria have been observed in 23% and 30% of children with ADPKD (65). As anticipated, children with ADPKD demonstrate significantly greater urine protein and microalbumin excretion rates than their unaffected siblings. Also, children with more significant renal cystic disease (> 10 cysts) have increased urine protein excretion rates as compared to those with less severe structural disease. Previous studies have suggested that statin therapy may be of benefit to decrease microalbuminuria and proteinuria in progressive nephropathy (66).
Study design
Hypothesis
Statin therapy will slow progression of renal and cardiovascular disease as assessed by changes in renal volume, left ventricular mass index, and urinary albumin excretion in children and young adults with autosomal dominant polycystic kidney disease treated with lisinopril.
Specific Aims
1) To determine the effect of pravastatin versus placebo on renal and cardiovascular disease progression over a three-year study period in children and young adults with ADPKD who are treated with lisinopril, with disease progression assessed by a combined primary endpoint of 20% or greater change in a) total renal volume as assessed by MRI; b) left ventricular mass index as measured by MRI; c) urinary albumin excretion; 2) To determine the effect of pravastatin versus placebo on change in overall renal volume as assessed by MRI over a three-year study period in children and young adults with ADPKD who are treated with lisinopril; 3) To determine the effect of pravastatin versus placebo on change in left ventricular mass index as assessed by MRI over a three-year study period in children and young adults with ADPKD who are treated with lisinopril; 4) To determine the effect of pravastatin versus placebo on change in microalbuminuria over a three-year study period in children and young adults with ADPKD who are treated with lisinopril.
Summary of Study Design
This is a randomized double-blind placebo-controlled parallel-group phase III clinical trial involving 107 children and young adults age 8–22 years with ADPKD. All subjects are treated with the ACE inhibitor lisinopril. There are 2 treatment arms: pravastatin versus placebo. Pravastatin dose is determined by subject age. The overall study duration is 5 years with individual subject participation of 3 years.
Research design & methods
Organization of the trial
This study is supported by the National Institutes of Health (R01 DK58793) and is conducted at The Clinical Translational Research Center (CTRC) at The Children’s Hospital in Aurora, Colorado, USA. Recruitment began in 2007 and ended in 2009. Subjects were recruited nationally from our ongoing studies of ADPKD, from physician referrals, and from family responses to information from clinicaltrials.gov (NCT00456365) and the Polycystic Kidney Disease Research Foundation. The Colorado Multiple Institutional Review Board reviewed and approved the study protocol. All participants (or parents as appropriate) signed an informed consent for study. Informed assent was obtained from all children aged 7 to 17 years. A data safety monitoring board was established to review the progress of participants and any adverse events on an annual basis.
Inclusion and exclusion criteria
Eligible subjects had ADPKD defined as renal imaging demonstrating bilateral cysts in the setting of a family history of ADPKD or multiple cysts clinically consistent with a new diagnosis of ADPKD. Subjects ranged in age from 8 to 22 years and had normal renal function with Schwartz creatinine clearance > 80 ml/min/1.73 m2 (67). Age 8 years was chosen as the lower limit for study because pravastatin has been FDA-approved for patients beginning at this age as well as anticipated ability to comply with required imaging procedures and to swallow tablets. Exclusion criteria included past allergic history to medications used in study, history of liver or muscle disease, pregnancy or lactation, inability to cooperate with or clinical contraindication for magnetic resonance imaging, identified difficulties interfering with ability to adhere to study regimen, and inability to provide informed consent or assent. Subjects were prohibited from taking both angiotensin converting enzyme inhibitors and angiotensin receptor blockers, mTOR inhibitors, or vasopressin receptor antagonists during the research study.
Study protocol
Once eligibility was confirmed, each subject was seen for a standard two-day admission at the CTRC. All subjects underwent a detailed history and physical examination including growth parameters and Tanner staging (68–70). On the first night of admission, subjects fasted for a 10-hour period. On the following morning, blood was drawn for lipid studies (total, LDL, and HDL cholesterol & triglycerides), routine serum chemistries including complete blood count, creatine kinase, aspartate aminotransferase, alanine aminotransferase, nitrate/nitrite, and in girls of possible childbearing potential (Tanner Stage 2 or higher), a serum pregnancy test (beta human chorionic gonadotropin). Two 24-hour urine collections were obtained for determination of creatinine, volume, protein, microalbumin, transforming growth factor beta, interleukins, vascular endothelial growth factor and angiopoietin excretion rates. Subjects underwent abdominal/cardiac MRI as described below. During the inpatient stay, 12 sitting blood pressure measurements were obtained in the right arm with a programmable blood pressure monitor (Dinamap Pro 300V2, GE Medical Systems Information Technology, Tampa, FL). Blood pressures were related to published data for age-, gender-, and height-matched children (71).
All subjects were given a digital blood pressure monitor (A&D Medical, UA767) with the appropriately sized cuff and subject/parent were instructed in its use. This blood pressure monitor was chosen for use based on its reliability and validity compared to auscultation (72;73). The subject/family was instructed to take the subject’s blood pressure at home on a monthly basis as described in the follow-up section below. Symptoms of orthostatic hypotension were reviewed with parent/subject.
Abdominal magnetic resonance imaging (MRI)
All subjects underwent a combined abdominal/cardiac MRI study. For abdominal MRI, a combination of T1, T2, and proton density spin echo sequences was combined with gradient echo sequences with imaging in both axial and coronal planes. Five and ten mm thick sections were acquired depending on subject size and renal volume. Ultrafast single breath-hold sequences were used as appropriate. A phased array surface coil was used for image acquisition. No contrast was administered. Imaging was performed on a 1.5 Tesla Visart System (Toshiba America Medical Systems) with image data transferred to a Vitria 3-D workstation (Vital Images). The liver, spleen, and pancreas were evaluated for cysts. For analysis, DICOM images were deidentified and evaluated by an analyst with no knowledge of the subject’s status using the Analyze software system (Analyze 9.0, Mayo Foundation, Biomedical Imaging Resource, Rochester, MN, USA). Total kidney volume, total cyst volume, and cyst number were obtained using a stereology method. This method has been used extensively for evaluation of kidney and cyst volume in pediatric and adult subjects with ADPKD (50;74–76). Stereology is a simple and fast method of segmenting an object by counting the number of intersections of a randomly oriented and positioned grid over the object (Figure 1). In this technique, the area of the kidney in each image was calculated from the collection of selected points that overlaid the kidney regions and the point count was converted to a pixel count to obtain the area measurements. The total kidney volume was calculated from the set of contiguous images by summing the products of the area measurements and the slice thickness. Renal cyst volume will be calculated from T2-weighted images using the region-based thresholding method (Figure 2) (74). A threshold is selected interactively by the analyst. Cysts are brighter than the renal parenchyma in T2-weighted images and are segmented from voxels with intensity values greater than the threshold. Cyst areas will be calculated in each image, and the total cyst volume will be calculated from each set of contiguous images by summing the products of the area measurements and the slice thickness. Fractional cyst volume will be determined from the ratio of cyst volume to total renal volume. To count the number of cysts, a middle section of the left kidney will be chosen from coronal T2-weighted images, and any cyst with a diameter of ≥ 4 mm will be recorded by the analyst. In our previous studies utilizing this method, reliability coefficients for repeated measurements were 0.972 for kidney volume, 0.987 for cyst volume, and 0.995 for cyst number (77).
Figure 1.

Representative image showing T1-weighted magnetic resonance image of kidneys with overlying grid. The analyst highlights the selected “+” points overlying the each kidney in each magnetic resonance image. This point count is converted to a pixel count to obtain the area measurements. The total kidney volume is then calculated from the set of contiguous images by summing the products of the area measurements and the slice thickness.
Figure 2.
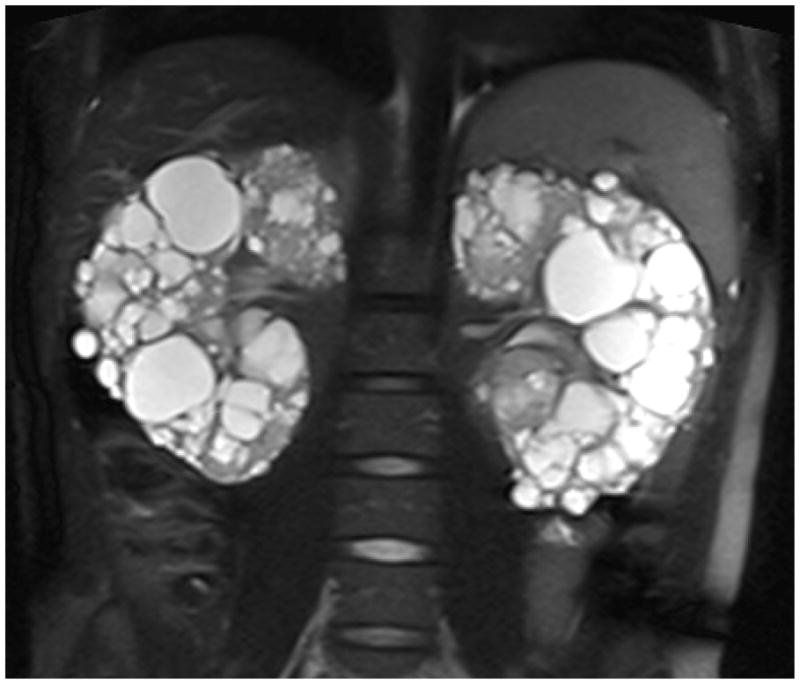
Magnetic resonance image showing highlighted renal cysts on T2-weighted image.
Cardiac MRI
With the same procedure as abdominal MRI, limited cardiac magnetic resonance imaging study was performed for determination of left ventricular mass index, volumes, and ejection fraction. Cardiac electrodes and wires were applied to the chest for cardiac gating during the study. A surface (torso) coil was applied prior to the start of imaging. The imaging protocol consisted of localizers in 3 orthogonal planes (axial, sagittal and coronal). These were followed by steady state free precession (SSFP) breath-hold imaging in the 2-chamber, and 4-chamber planes (total 2 slices). The 4-chamber image was used to prescribe a stack of 8–12, breath-held, SSFP contiguous slices in a short axis orientation to the ventricles. The breath-held SSFP sequences were 4–7 seconds in duration. The entire scan duration was approximately 5–10 minutes, including subject preparation time. After the study was acquired, a Leonardo © (Siemens Medical Systems) workstation was used to manually measure endocardial and epicardial contours of the left ventricle in end-systole and end-diastole. Application of the modified Simpson’s rule was performed to generate left ventricular mass index, left ventricular end-diastolic and end-systolic volume as well as ejection fraction.
Medication treatment
Subjects were randomized to either pravastatin or placebo treatment. Randomization was stratified by gender, age group (8–12 or 13–22 years), and hypertensive status (hypertensive or normotensive). Subjects were numbered consecutively within each stratum. Randomization codes were prepared by the statistician for each gender-, age-, and hypertensive-specific stratum with treatments labeled A and B, with either A or B being the statin treatment and the other being the placebo. The research pharmacist chose which letter represented the statin treatment and placed the code in a sealed envelope. Within each stratum, there were an equal number of A and B treatments, with blocking of 4. The standard dose of medication was 20 mg daily for subjects 8–12 years of age and 40 mg daily for subjects 13 years of age and older. When a child reached the 13th birthday, the dose of pravastatin/placebo was increased from 20 mg daily to 40 mg daily but the subject’s identification code did not change. The initial 3-month supply of pravastatin or placebo was given at the first hospital visit. Potential adverse effects of pravastatin were reviewed with subject and parent, including muscle pain or tenderness, weakness, elevated serum creatine kinase or liver enzymes, neurologic symptoms, headache, abdominal pain, and nausea. Rare serious adverse events could include rhabdomyolysis. Potential side effects were noted in the consent form and were reviewed routinely with subject/parent. Subjects were advised to avoid grapefruit juice.
All subjects were also treated with the angiotensin converting enzyme inhibitor lisinopril. In normotensive subjects (blood pressure < 95th percentile for height, age, and sex), the initial dose of lisinopril was 2.5 mg/day. Higher doses could be started in hypertensive subjects at the discretion of the supervising physician based on the subject’s blood pressure, history of hypertension, prior treatment with angiotensin converting enzyme inhibitors, and/or size. Adjustments of the lisinopril dose were made once per month based on home blood pressure reports at the discretion of the physician at an increase of 2.5 mg/dose until at least 4 of 6 systolic and at least 4 of 6 diastolic blood pressures on home blood pressure monitoring were between the 50th and the 75th percentile, or the maximum dose of 0.5 mg per kg of body weight up to 20 mg BID was reached. For subjects whose blood pressure was below the 25th percentile, lisinopril was not started but home blood pressure monitoring was instituted and lisinopril started if blood pressure rose above the 50th percentile. For subjects who did not tolerate lisinopril, the study protocol included treatment with losartan. However, no subject required such treatment. Subjects were advised to avoid grapefruit juice.
If the maximum dose of lisinopril was reached before the blood pressure goal for hypertensive children was achieved, amlodipine was added and adjusted every month. The starting dose was 2.5 mg/day (0.1 mg/kg/day), with dose increases of 2.5 mg/day each month to a maximum dose of 20 mg/day. Medication doses could be raised more quickly at the discretion of the supervising physician based on the subject’s blood pressure and size. If additional or alternative antihypertensive medications were required to reach the assigned blood pressure goal, these could be prescribed at the discretion of the supervising physician based on current standard of care. Any normotensive subject who developed hypertension (blood pressure ≥ 95th percentile) during the course of the study was treated as outlined above for hypertensive subjects.
Follow up
All participants were asked to submit home blood pressure reports on a monthly basis. The blood pressure was to be measured 6 times 3 minutes apart in the right arm after 5 minutes of quiet sitting, 24 hours after the last medication dose or 12 hours after the last medication dose for twice a day dosing. The report was relayed to us by phone, mail, or secure on-line internet submission, along with a report of any adverse effects, concurrent illnesses, any other medications taken, and for females Tanner stage 2 and above, results of home urine pregnancy testing. We have previously used this approach to home blood pressure monitoring with good compliance.
Potential side effects of pravastatin, lisinopril, and other antihypertensive medications were discussed at initiation of treatment and were reviewed monthly. Subjects were also instructed to contact us if any adverse effects developed or for any symptoms suggestive of orthostatic hypotension. Along with the monthly blood pressure report, subjects were asked to describe any potential adverse drug effects as well as intercurrent illnesses. Potential adverse drug effects were reviewed on a case by case basis by the principal investigators. The decision was made in each case to adjust, continue, or discontinue the medication.
Medication compliance was addressed by frequent contact with subjects and by monitoring of medication supply for all study medications.
Follow up laboratory studies were obtained at one month and six months after the initial visit. These studies included creatine kinase, aspartate aminotransferase, and alanine aminotransferase. These could be obtained at the local contracted laboratory or a kit could be sent to the subject/parent to have blood drawn and sent to a central laboratory in Denver for analysis. Our previous experience utilizing angiotensin converting enzyme inhibitors in children and young adults with ADPKD and normal renal function suggested that the incidence of hyperkalemia or increased serum creatinine was extremely rare, and these parameters were not routinely monitored.
All girls of possible childbearing potential (Tanner stage 2 or higher) were required to perform urine pregnancy tests every month at home using a commercial kit that was provided to them with results reported to us as a component of drug safety monitoring. At the initial visit, these subjects received individual counseling regarding pregnancy and birth control, side effects of medications during pregnancy, and utilization of the home urine pregnancy kit, as appropriate. Confidentiality concerns were reviewed with both subject and parent (for subjects under 18 years of age) and subjects were given several options for reporting of results. If a positive pregnancy test is documented, the principal investigators will contact the subject to discuss the results. Subjects with positive pregnancy tests will be encouraged to discuss the results with their parents with the assistance of the principal investigators if desired. Medications will be stopped immediately as deemed safe by the principal investigators (alternate medications known to be safe in pregnancy may be recommended to those subjects with severe hypertension) and the girl will be referred urgently to an obstetrician.
Blood pressure reports are reviewed on a monthly basis and medications adjusted as needed. Medications and home urine pregnancy tests as appropriate are mailed to the subject at least every 3 months.
At 1–1/2 years and 3 years following the initial visit, each subject returns for a standard two-day admission. Thus, each subject will have a total of 3 inpatient visits over a three-year chronologic period. Upon admission to the CTRC, a detailed history is obtained and physical examination including growth parameters and Tanner staging is performed. On the first night of admission, subjects fast for a 10-hour period. On the following morning, blood is drawn for lipid studies (total, LDL, and HDL cholesterol & triglycerides), routine serum chemistries including complete blood count, creatine kinase, aspartate aminotransferase, alanine aminotransferase, nitrate/nitrite, and in girls of possible childbearing potential (Tanner Stage 2 or higher), a serum pregnancy test (beta human chorionic gonadotropin). Two 24-hour urine collections are obtained for determination of creatinine, volume, protein, microalbumin, transforming growth factor beta, interleukins, vascular endothelial growth factor and angiopoietin excretion rates. Subjects undergo abdominal/cardiac MRI. Blood pressure monitoring is performed as previously noted. Counseling regarding home urine pregnancy tests as described above is provided for any subjects who have progressed from Tanner stage 1 to 2 since the previous visit.
Data analysis
The primary outcome variable is a dichotomous variable which is positive if a subject has a 20% or greater change over a three-year interval in any or all of the following three endpoints: 1) total renal volume as assessed by MRI; 2) left ventricular mass index as assessed by MRI; and 3) urinary albumin excretion. The composite endpoint is negative if all three measures fail to show a 20% or greater increase. The composite endpoint will be compared between two randomized study groups: the treatment group, who receive pravastatin and lisinopril, and the control group, who receive placebo and lisinopril. The primary analysis will be the comparison of the percentage of children reaching the combined endpoint between the two study groups, by Chi-square analysis (Fisher’s exact test will be substituted if warranted by small cell sizes), with a p value of less than 0.05 considered significant. The secondary endpoints are percent change in renal volume and cystic volume as assessed by MRI, percent change in left ventricular mass index as assessed by MRI, and percent change in microalbuminuria. Since the primary endpoint is a composite of the secondary endpoints, ANCOVA will be used to test for differences between groups on each of the secondary endpoints after adjusting for sex, age, body mass index, and other relevant covariates. In addition, mixed model, longitudinal data analysis will be employed to examine the change in secondary endpoints over time after adjustment for covariates. These analyses will allow us to model a random slope and intercept for each subject while taking into consideration the correlation among repeated measurements. In addition, the mixed model is robust to missing data. All analyses will be intent-to-treat and data for all available time points will be included. Given the modest sample size, these analyses will be considered secondary; thus no correction for multiple tests will be used. The correlation between each of the three endpoints and LDL cholesterol concentration or blood pressure will also be assessed using Pearson correlation coefficients.
Preliminary data are available on each of the 3 measurements used in the combined endpoint. Previous data on children with ADPKD measured at least 2–1/2 years apart, and defining “increase” as a 20% or greater change over 3 years, showed that 75% had an increase in renal volume as measured by ultrasound, 80% had an increase in renal volume as measured by MRI (the sample size for MRI was small), 22% had an increase in left ventricular mass index, 53% had an increase in urinary microalbumin, and 83% had an increase in either renal volume by ultrasound, left ventricular mass index, or urinary microalbumin. None of these children were taking HMG coA reductase inhibitors. From the preliminary data we predict that approximately 80% of the control group will reach the combined endpoint. With α = 0.05, we would have 80% power to detect a difference of 30% in the percentage of subjects reaching the combined endpoint (i.e., 80% of control and 50% of treatment subjects will reach the combined endpoint) with 40 subjects finishing the study in each group. A continuity correction was not utilized due to anticipated cell sizes for results. With an expected drop-out rate of 25%, we planned to recruit 50 subjects for each group, for a total sample size of 100 subjects. The 25% drop-out rate was based on the observed drop-out rate in our previous clinical trial in pediatric ADPKD with a five-year follow up period (26).
Baseline characteristics of subjects
One hundred seven subjects were enrolled and randomized to either pravastatin or placebo in a double-blind manner, including 55 subjects in group A and 52 subjects in group B. No significant differences were detected in the baseline characteristics of the study groups, including age, gender, prevalence of hypertension, systolic or diastolic blood pressure, 24-hour urine creatinine clearance, hematocrit, left ventricular mass index, ejection fraction, or renal volume (Table 1). Serum and urine chemistries were similar between groups except for a statistically significant but clinically insignificant difference in serum chloride concentration (Table 2).
Table 1.
Demographics and Baseline Characteristics of Subjects
| Parameter | Group A Mean | Group B Mean | p |
|---|---|---|---|
| N | 55 | 52 | |
| Age | 15.5 ± 3.9 | 15.7 ± 3.4 | 0.41 |
| Gender (Male/Female) | 21/34 (38%) | 19/33 (37%) | 0.86 |
| Hypertension (%) | 18 (33%) | 19 (36%) | 0.68 |
| Systolic blood pressure (mm Hg) | 122 ± 2 | 123 ± 2 | 0.83 |
| Diastolic blood pressure (mm Hg) | 72 ± 1 | 71 ± 1 | 0.60 |
| 24-h urine creatinine clearance (ml/min/1.73m2) | 138 (132–147) | 133 (125–141) | 0.36 |
| Hematocrit (%) | 41.4 ± 0.50 | 40.4 ± 0.53 | 0.20 |
| Left ventricular mass index (gm/m2) | 55 ± 2 | 53 ± 2 | 0.38 |
| Left ventricle ejection fraction (%) | 57 ± 1 | 58 ± 1 | 0.45 |
| Mean renal volume corrected by height (cm2) | 1.00 ± 0.07 | 1.05 ± 0.07 | 0.58 |
| Renal volume (ml) | 213 (192–252) | 233 (202–265) | 0.57 |
Note. All analyses adjusted for sex and height in ANOVA. Data are presented as least square means ± SE or geometric mean and 95% CI. p < 0.05 considered significant.
Table 2.
Baseline Urine and Serum Chemistries
| Parameter | Group A Mean | Group B Mean | p |
|---|---|---|---|
| Urine | |||
| Microalbumin (mcg/min) | 20 (15–28) | 24 (17–33) | 0.50 |
| Protein (mg/24h) | 112 (95–132) | 122 (102–144) | 0.51 |
| Serum | |||
| Creatinine (mg/dl) | 0.65 (0.62–0.69) | 0.65 (0.61–0.69) | 0.79 |
| Sodium (mEq/L) | 138 ± 1 | 133 ± 1 | 0.36 |
| Potassium (mEq/L) | 4.0 ± 0.1 | 3.9 ± 0.1 | 0.32 |
| Chloride (mEq/L) | 104 ± 1 | 103 ± 1 | 0.02 |
| Urea Nitrogen (mg/dl) | 13 ± 1 | 13 ± 1 | 0.39 |
| Glucose (mg/dl) | 90 ± 1 | 91 ± 1 | 0.21 |
| Total Cholesterol (mg/dl) | 142 ± 4 | 149 ± 4 | 0.25 |
| LDL Cholesterol (mg/dl) | 83 ± 3 | 87 ± 3 | 0.36 |
| HDL Cholesterol (mg/dl) | 47 ± 2 | 47 ± 2 | 0.99 |
| Triglycerides (mg/dl) | 87 ± 7 | 99 ± 8 | 0.28 |
| Aspartate aminotransferase (AST) (IU/L) | 23 ± 1 | 22 ± 1 | 0.65 |
| Alanine aminotransferase (ALT) (IU/L) | 19 ± 1 | 22 ± 2 | 0.06 |
| Creatine Kinase (CK) (IU/L) | 93 ± 7 | 97 ± 8 | 0.69 |
All analyses adjusted for sex and height in ANOVA. Data are presented as least square means ± SE or geometric mean and 95% CI. p < 0.05 considered significant.
Significance
ADPKD is the most common life-threatening hereditary renal disease. Over the past few decades, it has become apparent that this condition can significantly affect the health of children as young as in utero. Because renal cyst formation and hypertension begin very early in life in many patients, it is imperative that interventions be initiated early in childhood if complications of this disease are to be ameliorated. From previous studies, it is apparent that renal volume and blood pressure are major variables that influence the course of ADPKD. Moreover, with advancements in renal replacement therapy, cardiovascular disease has become a major contributor to morbidity and mortality in patients with ADPKD. If renal and cardiovascular disease progression can be altered with the interventions described in this protocol, medical management of this disease will be drastically altered. Specifically, such results will lead to earlier routine screening in children from affected families and initiation of treatment specific to the condition in early life. The results of this study could potentially markedly decrease the rate of progression to end-stage renal disease and the prevalence of cardiovascular disease in this population.
Acknowledgments
This research study was supported by NIH NIDDK Grant R01 DK058793, NIH NCRR Grant MO1 RR00069, and the Zell Family Foundation.
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- ACE
angiotensin converting enzyme
- ESRD
end-stage renal disease
- GFR
glomerular filtration rate
- MRI
magnetic resonance imaging
- RAAS
renin-angiotensin-aldosterone system
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ecder T, Fick-Brosnahan G, Schrier RW. Polycystic Kidney Disease. In: Schrier RW, editor. Diseases of the Kidney and Urinary Tract. 8. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 502–39. [Google Scholar]
- 2.Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol. 2004 Oct;15(10):2528–36. doi: 10.1097/01.ASN.0000141055.57643.E0. [DOI] [PubMed] [Google Scholar]
- 3.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002 Oct;13(10):2508–16. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 4.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003 Feb;33(2):129–37. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 5.Praetorius HA, Frokiaer J, Nielsen S, Spring KR. Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J Membr Biol. 2003 Feb 1;191(3):193–200. doi: 10.1007/s00232-002-1055-z. [DOI] [PubMed] [Google Scholar]
- 6.Torres VE, Harris PC. Mechanisms of Disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006 Jan;2(1):40–55. doi: 10.1038/ncpneph0070. [DOI] [PubMed] [Google Scholar]
- 7.Ecder T, Schrier RW. Hypertension in autosomal-dominant polycystic kidney disease: Early occurrence and unique aspects. J Am Soc Nephrol. 2001 Jan;12(1):194–200. doi: 10.1681/ASN.V121194. [DOI] [PubMed] [Google Scholar]
- 8.Schrier RW, Mcfann KK, Johnson AM. Epidemiological study of kidney survival in autosomal dominant polycystic kidney disease. Kidney Int. 2003 Feb;63(2):678–85. doi: 10.1046/j.1523-1755.2003.00776.x. [DOI] [PubMed] [Google Scholar]
- 9.Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1995;5:2048–56. doi: 10.1681/ASN.V5122048. [DOI] [PubMed] [Google Scholar]
- 10.Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009 Sep;20(9):1888–93. doi: 10.1681/ASN.2008080882. [DOI] [PubMed] [Google Scholar]
- 11.Dechend R, Fiebler A, Lindschau C, Bischoff H, Muller D, Park JK, Dietz R, Haller H, Luft FC. Modulating angiotensin II-induced inflammation by HMG Co-A reductase inhibition. Am J Hypertens. 2001 Jun;14(6 Pt 2):55S–61S. doi: 10.1016/s0895-7061(01)02070-2. [DOI] [PubMed] [Google Scholar]
- 12.Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med. 1990 Oct 18;323(16):1091–6. doi: 10.1056/NEJM199010183231602. [DOI] [PubMed] [Google Scholar]
- 13.Barrett BJ, Foley R, Morgan J, Hefferton D, Parfrey P. Differences in hormonal and renal vascular responses between normotensive patients with autosomal dominant polycystic kidney disease and unaffected family members. Kidney Int. 1994;46(4):1118–23. doi: 10.1038/ki.1994.374. [DOI] [PubMed] [Google Scholar]
- 14.Harrap SB, Davies DL, Macnicol AM, Dominiczak AF, Fraser R, Wright AF, Watson ML, Briggs JD. Renal, cardiovascular and hormonal characteristics of young adults with autosomal dominant polycystic kidney disease. Kidney Int. 1991;40(3):501–8. doi: 10.1038/ki.1991.238. [DOI] [PubMed] [Google Scholar]
- 15.Bell PE, Hossack KF, Gabow PA, Durr JA, Johnson AM, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease. Kidney Int. 1988 Nov;34(5):683–90. doi: 10.1038/ki.1988.233. [DOI] [PubMed] [Google Scholar]
- 16.Brunner HR. Experimental and clinical evidence that angiotensin II is an independent risk factor for cardiovascular disease. Am J Cardiol. 2001;87:3C–9C. doi: 10.1016/s0002-9149(01)01538-7. [DOI] [PubMed] [Google Scholar]
- 17.Hirsch AT, Pinto YM, Schunkert H, Dzau VJ. Potential role of the tissue renin-angiotensin system in the pathophysiology of congestive heart failure. Am J Cardiol. 1990;66:22D–30D. doi: 10.1016/0002-9149(90)90473-e. [DOI] [PubMed] [Google Scholar]
- 18.Wolf G. Angiotensin II: A pivotal factor in the progression of renal diseases. Nephrol Dial Transplant. 1999;14( Suppl 1):S42–S44. doi: 10.1093/ndt/14.suppl_1.42. [DOI] [PubMed] [Google Scholar]
- 19.Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–93. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- 20.Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK. P22phox is a critical component of the superoxide-generating NADH/NADPH system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem. 1996;271:23317–21. doi: 10.1074/jbc.271.38.23317. [DOI] [PubMed] [Google Scholar]
- 21.Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle: New concepts. Hypertension. 1997;29:366–73. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 22.Chatterjee PK, Weerackody RP, Mistry SK, Hawksworth GM, McLay JS. Selective antagonism of the AT1 receptor inhibits angiotensin II stimulated DNA and protein synthesis in primary cultures of human proximal tubular cells. Kidney Int. 1997;3:699–705. doi: 10.1038/ki.1997.385. [DOI] [PubMed] [Google Scholar]
- 23.Ruiz-Ortega M, Egido J. Angiotensin II modulates cell growth-related events and synthesis of matrix proteins in renal interstitial fibroblasts. Kidney Int. 1997;6:1497–510. doi: 10.1038/ki.1997.480. [DOI] [PubMed] [Google Scholar]
- 24.Bernstein J, Evan AP, Gardner KD., Jr Epithelial hyperplasia in human polycystic kidney diseases. Its role in pathogenesis and risk of neoplasia. Am J Pathol. 1987;1:92–101. [PMC free article] [PubMed] [Google Scholar]
- 25.Grantham JJ. Mechanisms of progression in autosomal dominant polycystic kidney disease. Kidney Int Suppl. 1997;63:S93–S97. [PubMed] [Google Scholar]
- 26.Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol. 2009 Apr;4(4):820–9. doi: 10.2215/CJN.02810608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hafez KS, Inman SR, Stowe NT, Novick AC. Renal hemodynamic effects of lovastatin in a renal ablation model. Urology. 1996 Dec;48(6):862–7. doi: 10.1016/s0090-4295(96)00314-7. [DOI] [PubMed] [Google Scholar]
- 28.Glazer AA, Inman SR, Stowe NT, Novick AC. Renal microcirculatory effects of lovastatin in a rat model of reduced renal mass. Urology. 1997 Nov;50(5):812–7. doi: 10.1016/S0090-4295(97)00338-5. [DOI] [PubMed] [Google Scholar]
- 29.Inman SR, Stowe NT, Cressman MD, Brouhard BH, Nally JV, Jr, Satoh S, Satodate R, Vidt DG. Lovastatin preserves renal function in experimental diabetes. Am J Med Sci. 1999 Apr;317(4):215–21. doi: 10.1097/00000441-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Joyce M, Kelly C, Winter D, Chen G, Leahy A, Bouchier-Hayes D. Pravastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, attenuates renal injury in an experimental model of ischemia-reperfusion. J Surg Res. 2001 Nov;101(1):79–84. doi: 10.1006/jsre.2001.6256. [DOI] [PubMed] [Google Scholar]
- 31.Bianchi S, Bigazzi R, Caiazza A, Campese VM. A controlled, prospective study of the effects of atorvastatin on proteinuria and progression of kidney disease. Am J Kidney Dis. 2003 Mar;41(3):565–70. doi: 10.1053/ajkd.2003.50140. [DOI] [PubMed] [Google Scholar]
- 32.van Dijk MA, Kamper AM, van Veen S, Souverijn JH, Blauw GJ. Effect of simvastatin on renal function in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2001 Nov;16(11):2152–7. doi: 10.1093/ndt/16.11.2152. [DOI] [PubMed] [Google Scholar]
- 33.Stowe NT, Inman SR, Tapolyai M, Brouhard BH, Hodge EE, Novick AC. Lovastatin has direct renal hemodynamic effects in a rodent model. J Urol. 1996 Jul;156(1):249–52. [PubMed] [Google Scholar]
- 34.McFarlane SI, Muniyappa R, Francisco R, Sowers JR. Pleiotropic effects of statins: Lipid reduction and beyond. J Clin Endocrinol Metab. 2002;87:1451–8. doi: 10.1210/jcem.87.4.8412. [DOI] [PubMed] [Google Scholar]
- 35.Park JK, Muller DN, Mervaala EM, Dechend R, Fiebeler A, Schmidt F, Bieringer M, Schafer O, Lindschau C, Schneider W, et al. Cerivastatin prevents angiotensin II-induced renal injury independent of blood pressure- and cholesterol-lowering effects. Kidney Int. 2000 Oct;58(4):1420–30. doi: 10.1046/j.1523-1755.2000.00304.x. [DOI] [PubMed] [Google Scholar]
- 36.Gile RD, Cowley BD, Gattone VH, O’Donnell MP, Swan SK, Grantham JJ. Effect of lovastatin on the development of polycystic kidney disease in the Han:SPRD rat. Am J Kidney Dis. 1995;26:501–7. doi: 10.1016/0272-6386(95)90497-2. [DOI] [PubMed] [Google Scholar]
- 37.Zafar I, Tao Y, Falk S, McFann K, Schrier RW, Edelstein CL. Effect of statin and angiotensin-converting enzyme inhibition on structural and hemodynamic alterations in autosomal dominant polycystic kidney disease model. Am J Physiol Renal Physiol. 2007 Sep;293(3):F854–F859. doi: 10.1152/ajprenal.00059.2007. [DOI] [PubMed] [Google Scholar]
- 38.Franz KA, Reubi FC. Rate of functional deterioration in polycystic kidney disease. Kidney Int. 1983 Mar;23(3):526–9. doi: 10.1038/ki.1983.51. [DOI] [PubMed] [Google Scholar]
- 39.Gabow PA, Chapman AB, Johnson AM, Tangel DJ, Duley IT, Kaehny WD, Manco-Johnson M, Schrier RW. Renal structure and hypertension in autosomal dominant polycystic kidney disease. Kidney Int. 1990 Dec;38(6):1177–80. doi: 10.1038/ki.1990.330. [DOI] [PubMed] [Google Scholar]
- 40.Gabow PA, Johnson AM, Kaehny WD, Kimberling WJ, Lezotte DC, Duley IT, Jones RH. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int. 1992;41(5):1311–9. doi: 10.1038/ki.1992.195. [DOI] [PubMed] [Google Scholar]
- 41.Fick GM, Duley IT, Johnson AM, Strain JD, Manco-Johnson ML, Gabow PA. The spectrum of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol. 1994;4(9):1654–60. doi: 10.1681/ASN.V491654. [DOI] [PubMed] [Google Scholar]
- 42.Gabow PA, Duley IT, Johnson AM. Clinical profiles of gross hematuria in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1992;20(2):140–3. doi: 10.1016/s0272-6386(12)80541-5. [DOI] [PubMed] [Google Scholar]
- 43.Gabow PA, Kaehny WD, Johnson AM, Duley IT, Manco-Johnson ML, Lezotte DC, Schrier RW. The clinical utility of renal concentrating capacity in polycystic kidney disease. Kidney Int. 1989 Feb;35(2):675–80. doi: 10.1038/ki.1989.38. [DOI] [PubMed] [Google Scholar]
- 44.Chapman AB, Johnson AM, Gabow PA, Schrier RW. Overt proteinuria and microalbuminuria in autosomal-dominant polycystic kidney disease. J Am Soc Nephrol. 1994 Dec;5(6):1349–54. doi: 10.1681/ASN.V561349. [DOI] [PubMed] [Google Scholar]
- 45.Fick-Brosnahan GM, Belz MM, McFann K, Johnson AM, Schrier RW. Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease: A longitudinal study. Am J Kidney Dis. 2002 Jun;39(6):1127–34. doi: 10.1053/ajkd.2002.33379. [DOI] [PubMed] [Google Scholar]
- 46.King BF, Reed JE, Bergstralh EJ, Sheedy PF, Torres VE. Quantification and longitudinal trends of kidney, renal cyst, and renal parenchyma volumes in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2000 Aug;11(8):1505–11. doi: 10.1681/ASN.V1181505. [DOI] [PubMed] [Google Scholar]
- 47.Chapman AB, Torres VE, Perrone RD, Steinman TI, Bae KT, Miller JP, Miskulin DC, Rahbari OF, Masoumi A, Hogan MC, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol. 2010 Jan;5(1):102–9. doi: 10.2215/CJN.04310709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Horl WH, Obermuller N, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010 Aug 26;363(9):830–40. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- 49.Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010 Aug 26;363(9):820–9. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 50.Chapman AB, Guay-Woodford LM, Grantham JJ, Torres VE, Bae KT, Baumgarten DA, Kenney PJ, King BF, Jr, Glockner JF, Wetzel LH, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int. 2003 Sep;64(3):1035–45. doi: 10.1046/j.1523-1755.2003.00185.x. [DOI] [PubMed] [Google Scholar]
- 51.Iglesias CG, Torres VE, Offord KP, Holley KE, Beard CM, Kurland LT. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am J Kidney Dis. 1983 May;2(6):630–9. doi: 10.1016/s0272-6386(83)80044-4. [DOI] [PubMed] [Google Scholar]
- 52.Roscoe JM, Brissenden JE, Williams EA, Chery AL, Silverman M. Autosomal dominant polycystic kidney disease in Toronto. Kidney Int. 1993 Nov;44(5):1101–8. doi: 10.1038/ki.1993.355. [DOI] [PubMed] [Google Scholar]
- 53.Hateboer N, van Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999 Jan 9;353(9147):103–7. doi: 10.1016/s0140-6736(98)03495-3. [DOI] [PubMed] [Google Scholar]
- 54.Chapman AB, Johnson AM, Rainguet S, Hossack K, Gabow P, Schrier RW. Left ventricular hypertrophy in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1997 Aug;8(8):1292–7. doi: 10.1681/ASN.V881292. [DOI] [PubMed] [Google Scholar]
- 55.Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Increased left ventricular mass in children with autosomal dominant polycystic kidney disease and borderline hypertension. Kidney Int. 2008 Nov;74(9):1192–6. doi: 10.1038/ki.2008.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ivy DD, Shaffer EM, Johnson AM, Kimberling WJ, Dobin A, Gabow PA. Cardiovascular abnormalities in children with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1995;5( 12):2032–6. doi: 10.1681/ASN.V5122032. [DOI] [PubMed] [Google Scholar]
- 57.Schunkert H, Jackson B, Tang SS, Schoen FJ, Smits JF, Apstein CS, Lorell BH. Distribution and functional significance of cardiac angiotensin converting enzyme in hypertrophied rat hearts. Circulation. 1993 Apr;87(4):1328–39. doi: 10.1161/01.cir.87.4.1328. [DOI] [PubMed] [Google Scholar]
- 58.Morgan HE, Baker KM. Cardiac hypertrophy. Mechanical, neural, and endocrine dependence. Circulation. 1991 Jan;83(1):13–25. doi: 10.1161/01.cir.83.1.13. [DOI] [PubMed] [Google Scholar]
- 59.Schrier R, McFann K, Johnson A, Chapman A, Edelstein C, Brosnahan G, Ecder T, Tison L. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol. 2002 Jul;13(7):1733–9. doi: 10.1097/01.asn.0000018407.60002.b9. [DOI] [PubMed] [Google Scholar]
- 60.Luo JD, Zhang WW, Zhang GP, Guan JX, Chen X. Simvastatin inhibits cardiac hypertrophy and angiotensin-converting enzyme activity in rats with aortic stenosis. Clin Exp Pharmacol Physiol. 1999 Nov;26(11):903–8. doi: 10.1046/j.1440-1681.1999.03165.x. [DOI] [PubMed] [Google Scholar]
- 61.Cadnapaphornchai MA, Fick-Brosnahan GM, Duley I, Johnson AM, Strain JD, DeGroff CG, Schrier RW. Design and baseline characteristics of participants in the study of antihypertensive therapy in children and adolescents with autosomal dominant polycystic kidney disease (ADPKD) Contemp Clin Trials. 2005 Apr;26(2):211–22. doi: 10.1016/j.cct.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 62.Grothues F, Smith GC, Moon JC, Bellenger NG, Collins P, Klein HU, Pennell DJ. Comparison of interstudy reproducibility of cardiovascular magnetic resonance with two-dimensional echocardiography in normal subjects and in patients with heart failure or left ventricular hypertrophy. Am J Cardiol. 2002 Jul 1;90(1):29–34. doi: 10.1016/s0002-9149(02)02381-0. [DOI] [PubMed] [Google Scholar]
- 63.Rajappan K, Bellenger NG, Melina G, Di Terlizzi M, Yacoub MH, Sheridan DJ, Pennell DJ. Assessment of left ventricular mass regression after aortic valve replacement--cardiovascular magnetic resonance versus M-mode echocardiography. Eur J Cardiothorac Surg. 2003 Jul;24(1):59–65. doi: 10.1016/s1010-7940(03)00183-0. [DOI] [PubMed] [Google Scholar]
- 64.Martinez-Vea A, Gutierrez C, Bardaji A, Pastor R, Garcia C, Peralta C, Richart C, Oliver JA. Microalbuminuria in normotensive patients with autosomal-dominant polycystic kidney disease. Scand J Urol Nephrol. 1998 Sep;32(5):356–9. doi: 10.1080/003655998750015331. [DOI] [PubMed] [Google Scholar]
- 65.Sharp C, Johnson A, Gabow P. Factors relating to urinary protein excretion in children with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1998;9:1908–14. doi: 10.1681/ASN.V9101908. [DOI] [PubMed] [Google Scholar]
- 66.Nakamura T, Ushiyama C, Hirokawa K, Osada S, Shimada N, Koide H. Effect of cerivastatin on urinary albumin excretion and plasma endothelin-1 concentrations in type 2 diabetes patients with microalbuminuria and dyslipidemia. Am J Nephrol. 2001 Nov;21(6):449–54. doi: 10.1159/000046648. [DOI] [PubMed] [Google Scholar]
- 67.Schwartz GJ, Haycock GB, Edelmann CM, Jr, Spitzer A. A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics. 1976;2:259–63. [PubMed] [Google Scholar]
- 68.Marshall WA, Tanner JM. Growth and physiological development during adolescence. Annu Rev Med. 1968;19:283–300. doi: 10.1146/annurev.me.19.020168.001435. [DOI] [PubMed] [Google Scholar]
- 69.Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. 1969;44(235):291–303. doi: 10.1136/adc.44.235.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. 1970 Feb;45(239):13–23. doi: 10.1136/adc.45.239.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.National high blood pressure education program working group on high blood pressure in children and adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114:555–76. [PubMed] [Google Scholar]
- 72.Artigao LM, Llavador JJ, Puras A, Lopez Abril J, Rubio MM, Torres C, Vidal A, Sanchis C, Divison JA, Naharro F, et al. Evaluation and validation of Omron Hem 705CP and Hem 706/711 monitors for self-measurement of blood pressure. Aten Primaria. 2000;25(2):96–102. doi: 10.1016/S0212-6567(00)78470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Brien E, Waeber B, Parati G, Staessen J, Myers MG. Blood pressure measuring devices: recommendations of the European Society of Hypertension. BMJ. 2001 March;322(3):531–6. doi: 10.1136/bmj.322.7285.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bae KT, Commean PK, Lee J. Volumetric measurement of renal cysts and parenchyma using MRI: phantoms and patients with polycystic kidney disease. J Comput Assist Tomogr. 2000 Jul;24(4):614–9. doi: 10.1097/00004728-200007000-00019. [DOI] [PubMed] [Google Scholar]
- 75.O’Neill WC, Robbin ML, Bae KT, Grantham JJ, Chapman AB, Guay-Woodford LM, Torres VE, King BF, Wetzel LH, Thompson PA, et al. Sonographic assessment of the severity and progression of autosomal dominant polycystic kidney disease: the Consortium of Renal Imaging Studies in Polycystic Kidney Disease (CRISP) Am J Kidney Dis. 2005 Dec;46(6):1058–64. doi: 10.1053/j.ajkd.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 76.Torres VE, King BF, Chapman AB, Brummer ME, Bae KT, Glockner JF, Arya K, Risk D, Felmlee JP, Grantham JJ, et al. Magnetic resonance measurements of renal blood flow and disease progression in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2007 Jan;2(1):112–20. doi: 10.2215/CJN.00910306. [DOI] [PubMed] [Google Scholar]
- 77.Cadnapaphornchai MA, Masoumi A, Strain JD, McFann K, Schrier RW. Magnetic Resonance Imaging of Kidney and Cyst Volume in Children with ADPKD. Clin J Am Soc Nephrol. 2010 Nov 29; doi: 10.2215/CJN.03780410. [DOI] [PMC free article] [PubMed] [Google Scholar]