

Summary
Genomic structural variants (SVs) are abundant in humans, differing from other variation classes in extent, origin, and functional impact. Despite progress in SV characterization, the nucleotide resolution architecture of most SVs remains unknown. We constructed a map of unbalanced SVs (i.e., copy number variants) based on whole genome DNA sequencing data from 185 human genomes, integrating evidence from complementary SV discovery approaches with extensive experimental validations. Our map encompassed 22,025 deletions and 6,000 additional SVs, including insertions and tandem duplications. Most SVs (53%) were mapped to nucleotide resolution, which facilitated analyzing their origin and functional impact. We examined numerous whole and partial gene deletions with a genotyping approach and observed a depletion of gene disruptions amongst high frequency deletions. Furthermore, we observed differences in the size spectra of SVs originating from distinct formation mechanisms, and constructed a map constructed a map of SV hotspots formed by common mechanisms. Our analytical framework and SV map serves as a resource for sequencing-based association studies.
Introduction
Unbalanced structural variants (SVs), or copy number variants (CNVs), involving large-scale deletions, duplications, and insertions form one of the least well studied classes of genetic variation. The fraction of the genome affected by SVs is comparatively larger than that accounted for by single nucleotide polymorphisms1 (SNPs), implying significant consequences of SVs on phenotypic variation. SVs have already been associated with diverse diseases, including autism2,3, schizophrenia4,5 and Crohn’s disease6,7. Furthermore, locus-specific studies suggest that diverse mechanisms may form SVs de novo, with some mechanisms involving complex rearrangements resulting in multiple chromosomal breakpoints8,9.
Initial microarray-based SV surveys focused on large gains and losses10,11,12, with recent advances in array technology widening the accessible size spectrum towards smaller SVs1,13. Microarray-based commonly mapped SVs to approximate genomic locations. However, a detailed SV characterization, including analyses of SV origin and impact, requires knowledge of precise SV sequences. Advances in sequencing technology have enabled applying sequence-based approaches for mapping SVs at fine-scale14,15,16,17,18,19,20,21. These approaches include: (i) paired-end mapping (or read pair ‘RP’ analysis) based on sequencing and analysis of abnormally mapping pairs of clone ends14,22,23,24 or high-throughput sequencing fragments15,17,18; (ii) read-depth (‘RD’) analysis, which detects SVs by analyzing the read depth-of-coverage16,21,25,26,27; (iii) split-read (‘SR’) analysis, which evaluates gapped sequence alignments for SV detection28,29; and (iv) sequence assembly (‘AS’), which enables the fine-scale discovery of SVs, including novel (non-reference) sequence insertions30,31,32. Sequence-based SV discovery approaches have thus far been applied to a limited (<20) number of genomes, leaving the fine-scale architecture of most common SVs unknown.
Sequence data generated by the 1000 Genomes Project (1000GP) provide an unprecedented opportunity to generate a comprehensive SV map. The 1000GP recently generated 4.1 Terabases of raw sequence in pilot projects targeting whole human genomes33 (Supplementary Table 1). These studies comprise a population-scale project, termed ‘low-coverage project’, in which 179 unrelated individuals were sequenced with an average coverage of 3.6X – including 59 Yoruba individuals from Nigeria (YRI), 60 individuals of European ancestry from Utah (CEU), 30 of Han ancestry from Beijing (CHB), and 30 of Japanese ancestry from Tokyo (JPT; the latter two were jointly analyzed as JPT+CHB). In addition, a high-coverage project, termed the ‘trio project’, was carried out, with individuals of a CEU and a YRI parent-offspring trio sequenced to 42X coverage on average.
We report here the results of analyses undertaken by the Structural Variation Analysis Group of the 1000GP. The group’s objectives were to discover, assemble, genotype, and validate SVs of 50 bp and larger in size, and to assess and compare different sequence-based SV detection approaches. The focus of the group was initially on deletions, a variant class often associated with disease9, for which rich control datasets and diverse ascertainment approaches exist1,13,22,28. Less focus was placed on insertions and duplications34 and none on balanced SV forms (such as inversions). Specifically, we applied nineteen methods to generate an SV discovery set. We further generated reference genotypes for most deletions, assessed the SVs’ functional impact, and stratified SV formation mechanism with respect to variant size and genomic context.
Prediction of SV candidate loci and assessment of discovery methods
We incorporated the SV discovery methods into a pipeline (Fig. 1AB), with the goal of ascertaining different SV types and assessing each method for its ability to discover SVs. The methods detected SVs by analyzing RD, RP, SR, and AS features, or by combining RP and RD features (abbreviated as ‘PD’). Altogether we generated thirty-six SV callsets by applying the methods on trio and low-coverage data, and by identifying SVs as genomic differences relative to a human reference, corresponding to the reference genome, or to a set of individuals (i.e. population reference; Supplementary Table 2). We initially identified SVs as deletions, tandem duplications, novel sequence insertions, and mobile element insertions (MEIs) relative to the human reference. Subsequent comparative analyses involving primate genomes enabled us to classify SVs as deletions, duplications, or insertions relative to inferred ancestral genomic loci, reflecting mechanisms of SV formation (see below). DNA reads analyzed by SV discovery methods were initially mapped to the human reference genome using a variety of alignment algorithms. Most of these algorithms mapped each read to a single genomic position, although one algorithm (mrFAST16) also considered alternative mapping positions for reads aligning onto repetitive regions (see Supplementary Tables 2-4 for method-specific parameters and full SV callsets). We filtered each callset by excluding SVs <50bp, which are reported elsewhere33. Many SVs exhibited support from distinct SV discovery methods, as exemplified by a common deletion, previously associated with body-mass index35 (BMI), that we identified with RP, RD, and SR methods (Fig. 1C). Nonetheless, we observed notable differences between methods (Fig. 2ABC) in terms of genomic regions ascertained (Supplementary Fig. 1), accessible SV size-range (Fig. 2A), and breakpoint precision (Fig.2C, Supplementary Fig. 2).
Figure 1. SV discovery and genotyping in population scale sequence data.

A. Schematic depicting the different modes (i.e., approaches) of sequence based SV detection we used. The RP approach assesses the orientation and spacing of the mapped reads of paired-end sequences14,15 (reads are denoted by arrows); the RD approach evaluates the read depth-of-coverage25,26; the SR approach maps the boundaries (breakpoints) of SVs by sequence alignment28,29; the AS approach assembles SVs30,31,32. B. Integrated pipeline for SV discovery, validation, and genotyping. Colored circles represent individual SV discovery methods (listed in Supplementary Table 1), with modes indicated by a color scheme: green=RP; yellow=RD; purple=SR; red=AS; green and yellow=methods evaluating RP and RD (abbreviated as ‘PD’). C. Example of a deletion, previously associated with BMI35, identified independently with RP (green), RD (yellow), and SR (red) methods. Grey dots indicate position and mapping quality for individual sequence reads. Targeted assembly confirmed the breakpoints detected by SR.
Figure 2. Comparative assessment of deletion discovery methods.
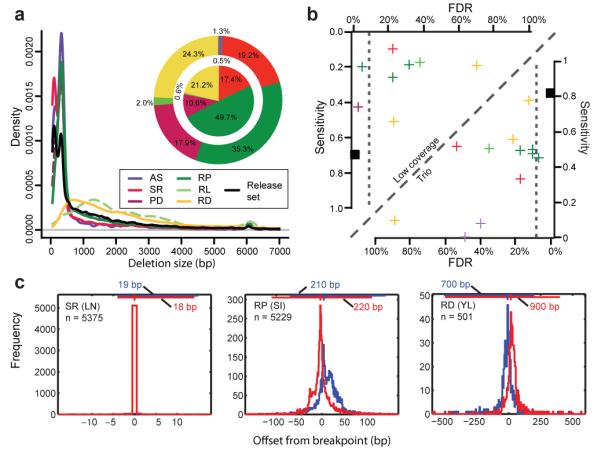
A. Deletion size-range ascertained by different modes of SV discovery. Three groups are visible, with AS and SR, PD and RP, as well as RD and ‘RL’ (RP analysis involving relatively long range (≥1 kb) insert size libraries, resulting in a different deletion detection size range compared to the predominantly used <500kb insert size libraries), respectively, ascertaining similar size-ranges. Pie charts display the contribution of different SV discovery modes to the release set. Outer pie = based on number of SV calls; inner pie = based on total number of variable nucleotides. Of note, not all approaches were applied across all individuals (see Supplementary Table 2). B. Sensitivity and FDR estimates for individual deletion discovery methods based on gold standard sets for individuals sequenced at high (NA12878) and low-coverage (NA12156), respectively. All depicted estimates are summarized in Supplementary Tables 3, 4, 6. Vertical dotted lines correspond to the specificity threshold (FDR≤10%). C. Breakpoint mapping resolution of three deletion discovery methods (the respective method names are in Supplementary Table 2). The blue and red histograms are the breakpoint residuals for predicted deletion start and end coordinates, respectively, relative to assembled coordinates (here assessed in low-coverage data). The horizontal lines at the top of each plot mark the 98% confidence intervals (labeled for each panel), with vertical notches indicating the positions of the most probable breakpoint (the distribution mode).
To estimate callset specificity, we carried out extensive validations (Methods), including PCRs for over 3,000 candidate loci, and microarray data analyses for 50,000 candidate loci (Supplementary Tables 3, 4; Supplementary Fig. 3). We combined PCR and array-based analysis results to estimate false discovery rates (FDRs), and found that eight callsets (three deletion, four insertion, and one tandem duplication callset) met the pre-specified specificity threshold33 (FDR≤10%), whereas the other callsets yielded lower specificity (FDRs of 13%-89%).
We further assessed the sensitivity of deletion discovery methods by collating data from four earlier surveys1,13,22,28 into a gold standard (Methods, Supplementary Tables 5, 6, and Supplementary Fig. 4A), and specifically assessing the detection sensitivity for an individual sequenced at high-coverage (NA12878) as well as for an individual sequenced at low-coverage (NA12156). Unsurprisingly, given the typical trade-off between sensitivity and specificity, in the trios the highest sensitivities were achieved by RD and RP methods with FDR>10% (Fig. 2B). By comparison, in the low-coverage data, the individual method with the greatest accuracy (FDR=3.7%) was the second most sensitive based on our gold standard (Fig. 2B), and the most sensitive when expanding the gold standard to a larger set of individuals (Supplementary Fig. 4B). This method, Genome STRiP (to be described elsewhere36), integrated both RP and RD features (PD), implying that considering different evidence types can improve SV discovery.
Construction of a high-confidence SV discovery set
To construct our SV discovery set (“release set”), we joined calls from different discovery methods corresponding to the same SV with a merging approach that was aware of each callset’s precision in SV breakpoint detection (Supplementary Fig. 5 and Methods). Most SVs in the release set (61%) were contributed by individual methods meeting the pre-defined specificity threshold (FDR≤10%). The remaining 39% of calls were contributed by lower specificity methods following experimental validation. Altogether, the release set comprised 22,025 deletions, 501 tandem duplications, 5,371 MEIs, and 128 non-reference insertions (Table 1, Supplementary Table 7). With our gold standard we estimated an overall sensitivity of deletion discovery of 82% in the trios, and 69% in low-coverage sequence (Fig. 2B) using a 1 bp overlap criterion. When instead applying a stringent 50% reciprocal overlap criterion for sensitivity assessment (which required SV sizes inferred on different experimental platforms to be in close agreement) our sensitivity estimates decreased by 12% and 18%, respectively, in trio and low-coverage sequence (Supplementary Table 8). We further examined an alternative approach that involved the pairwise integration of deletion discovery methods, and tested its ability to discover SVs without relying on the inclusion of lower specificity calls following experimental validation (“algorithm-centric set”; Fig. 1B). While this alternative approach resulted in an increased number (by ~13%) of high-specificity (FDR<10%) calls compared to the release set (Supplementary Text), it overall resulted in fewer SV calls owing to its decreased sensitivity at the lower (<200bp) SV size range. In the following analyses we thus focused on the release set.
Table 1.
Summary of discovered structural variation
| Deletions | Tandem Duplications |
Mobile element insertions |
Novel sequence insertions |
Total | |
|---|---|---|---|---|---|
| Individual Callsets <10% FDR | 11215 | 501 | 5371 | - | 17087 |
| Validated Experimentally * | 10810 | - | - | 128 | 10938 |
|
| |||||
| Release set | 22025 | 501 | 5371 | 128 | 28025 |
Only tabulates validated calls which were not already present in the individual callsets with <10% FDR
Extent and impact of our SV discovery set
We next assessed the extent and impact of our SV discovery (release) set. The median SV size was 729 bp (mean=8 kb), approximately four times smaller than in a recent tiling CGH based study1, reflecting the high resolution of DNA sequence based SV discovery. We also compared our set to a recent survey of SVs in an individual genome37 based on capillary sequencing and array-based analyses24, and observed a similar size distribution for deletions, but differences in the size distributions of other SV classes, reflecting underlying differences in SV ascertainment (Supplementary Fig. 6). By comparing our SVs to databases of structural variation and to additional personal genome datasets, we classified 15,556 SVs in our set as novel, with an enrichment of low frequency SVs and small SVs amongst the novel variants (Methods and Supplementary Text).
A major advantage of sequence-based SV discovery is the nucleotide resolution mapping of SVs. We initially mapped the breakpoints of 7,066 deletions and 3,299 MEIs using SR and AS features. Using the TIGRA-targeted assembly approach38 we further identified the breakpoints of an additional 4,188 deletions and 160 tandem duplications, initially discovered by RD, RP, and PD methods (Methods, Supplementary Table 2). Altogether, we mapped ~15,000 SVs at nucleotide resolution, 48% of which were novel. Few deletion loci (4.4%) displayed different SV breakpoints in different samples, which is explainable by rare TIGRA misassemblies, or alternatively, by recurrently formed, multi-allelic SVs (Supplementary Text). TIGRA further enabled us to validate an additional 7,359 SVs discovered with RP or RD features by identifying the SVs’ breakpoints (Methods), and to evaluate the mapping precision of SV discovery methods (Fig. 2C, Supplementary Figure 2).
We further assessed the putative functional impact of SVs in our set by relating them to genomic annotation. Seventeen hundred SVs affected coding sequences, resulting in full gene overlaps or exon disruptions (Table 2), many of which led to out-of-frame exons (Supplementary Table 9). We related gene disruptions to gene functions, and observed significant enrichments for several functional categories including cell defense and sensory perception (Supplementary Table 10). High levels of structural variation, including copy-number variation, were previously described for both processes15,22,39. These SVs might be maintained in the population by selection for the purpose of functional redundancy. While most SVs intersecting with genes were deletions, several validated tandem duplications and MEIs also intersected with coding sequences (Table 2).
Table 2. Functional impact of our fine resolution SV set.
Figures in parentheses indicate numbers of validated SVs per category. We inferred gene overlap with Gencode gene annotation47.
| SV class | Gene Overlap |
Total Gene overlap |
Total Inter- genic |
|||
|---|---|---|---|---|---|---|
| Full gene overlap |
Coding exon affected (partial) |
UTR overlap | Intron overlap |
|||
| Deletions | 654 (631) |
1093 (1031) |
315 (290) |
7319 (6481) |
9381 (8433) |
12644 (10386) |
|
Tandem
duplications |
2 (2) |
7 (6) |
9 (5) |
197 (62) |
215 (75) |
286 (76) |
|
Mobile element
insertions |
- | 3 (-) |
36 (-) |
1304 (97) |
1348 (112) |
4023 (758) |
|
Novel sequence
insertions |
- | - | 2 (2) |
49 (49) |
51 (51) |
77 (77) |
|
| ||||||
| Sum | 656 (633) |
1119 (1040) |
351 (309) |
8869 (6689) |
10995 (8671) |
17030 (11280) |
Population genetic properties of deletions
We next sought to generate genotypes for deletions discovered in the 1000GP data, both to facilitate population genetics analyses and to make our SV set amenable to association studies in the form of a reference genotype set. In this regard, the Genome STRiP36 genotyping method was developed, a method combining information from RD, RP, SR and haplotype features of population-scale sequence data for genotyping (Methods, Supplementary Text). Using this approach we generated genotypes for 13,826 autosomal deletions in 156 individuals. The genotypes displayed 99.1% concordance with CGH array1 based genotypes (available for 1,970 of the deletions), suggesting high genotyping accuracy.
Fig. 3 presents allele frequency analyses based on these genotypes. As expected, common polymorphisms (minor allele frequency (MAF) >5%) were generally shared across populations, while rare alleles were frequently observed in only one population (Figs. 3ABC). We observed several candidates for monomorphic deletions (i.e., genomic segments putatively deleted in all individuals), explainable by rare insertions present in the reference genome or by remaining genotyping inaccuracies (Supplementary Text).
Figure 3. Analysis of deletion presence and absence in two populations.
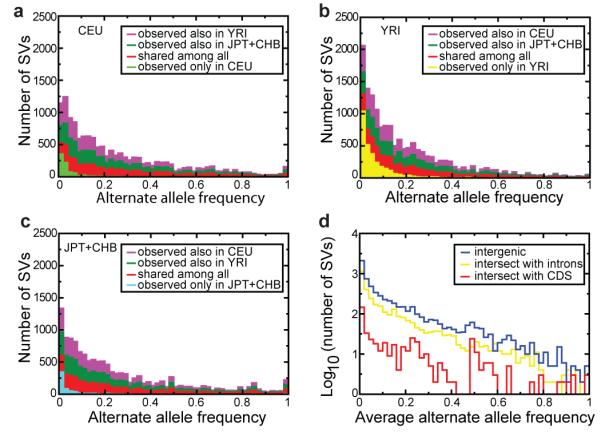
A-C. Deletion allele frequencies and observed sharing of alleles across populations, displayed for deletions discovered in the CEU, YRI, and JPT+CHB population samples in terms of stacked bars. D. Allele frequency spectra for deletions intersecting with intergenic (blue), intronic (yellow), and protein-coding sequences (red).
We next assessed the allele frequencies of gene deletions (Fig. 3D). Similar to a recent array-based study1, we observed a depletion of high frequency alleles among deletions intersecting with protein-coding sequence compared to other deletions (P=1.1×10−11; KS test), consistent with purifying selection keeping most gene deletions at low frequency. Nonetheless, several coding sequence deletions were observed with high allele frequency (>80%). Most of these occurred in regions annotated as segmental duplications, consistent with lessened evolutionary constraintin functionally redundant gene categories22. Intriguingly, common gene deletions also affected many unique genes with no obvious paralogs. We further analyzed the abundance of gene deletions in different populations and observed highly differentiated loci, albeit with no statistically significant relationship between differentiation and particular categories of gene overlap, i.e., intronic vs. exonic (Supplementary Text).
By comparing deletion genotypes with genotypes of nearby SNPs, we found, consistent with earlier studies1,13,40, that deletions in genomic regions accessible to short read sequencing display extensive linkage disequilibrium (LD) with SNPs. 81% of common deletions had one or more SNPs with which they are strongly correlated (r2>0.8; Supplementary Fig. 7). This suggests that many deletions mapped in our study will be identifiable through tagging SNPs in future studies (Supplementary Text). On the other hand, a fifth of the genotyped deletions were not tagged by HapMap SNPs (a figure similar to the fraction of SNPs that are not tagged by HapMap SNPs41), implying that these SVs should be genotyped directly in association studies. Furthermore, the LD properties of complex SVs (e.g., multiallelic SV) have not yet been fully ascertained as methods for genotyping such SVs with similar accuracy still being developed.
SV formation mechanism analysis
Nucleotide resolution breakpoint information enables inference of SV formation mechanisms15,22. Recent studies broadly distinguished between several germline rearrangement classes, some of which may comprise more than one SV formation mechanism15,22,42,43: non-allelic homologous recombination (NAHR), associated with long sequence similarity stretches around the breakpoints; rearrangements in the absence of extended sequence similarity (abbreviated as “non-homologous” or NH), associated with DNA repair by non-homologous end-joining (NHEJ) or with microhomology-mediated break-induced replication (MMBIR); the shrinking or expansion of variable number of tandem repeats (VNTRs), frequently involving simple sequences, by slippage; and MEIs. We distinguished among the classes NAHR, NH, VNTR, and MEI by examining the breakpoint junction sequence of SVs initially discovered as deletions or tandem duplications relative to a human reference.
We first compared the SVs to orthologous primate genomic regions to distinguish deletions from insertions/duplications with respect reconstructed ancestral loci using the BreakSeq classification approach43. This analysis showed that of the 11,254 nucleotide-resolution SVs discovered as deletions relative to a human reference, 21% actually represented insertion and 2% represented tandem duplications relative to the putative ancestral genome. Of the remaining SVs, 60% were classified as deletions relative to ancestral sequence, whereas the ancestral state of 17% was undetermined. By comparison, out of 160 nucleotide-resolution SVs identified as tandem duplications relative to the reference genome, 91.6% were classified as duplications relative to the ancestral genome, whereas the ancestral state of 8.4% remained undetermined (Supplementary Text). Our breakpoint analysis revealed that 70.8% of the deletions and 89.6% of the insertions exhibited breakpoint microhomology/homology ranging from 2-376 bp in size, with distribution modes of 2 bp (attributable to NH) and 15 bp (attributable to MEI), respectively (Fig. 4A, Supplementary Text). As expected42, a small portion of the deletions (16.1%) displayed non-template inserted sequences at their breakpoint junctions. By comparison, the tandem duplications showed extensive stretches displaying ≤95% sequence identity at the breakpoint linearly correlating in lenght with SV size (Fig. 4A). In addition, most tandem duplications displayed 2-17 bp of microhomology at the breakpoint junctions (Supplementary Text).
Figure 4. Contribution of SV formation mechanisms to the SV size spectrum.
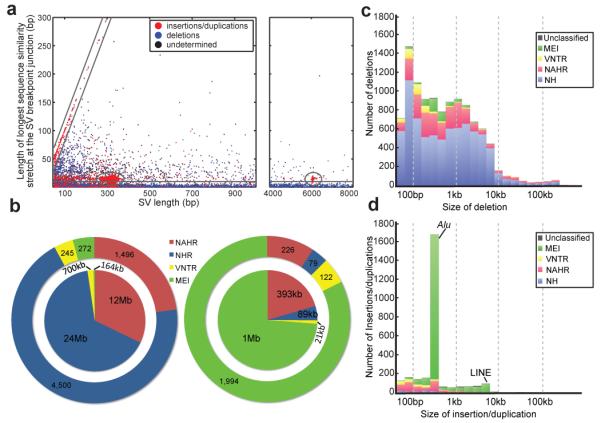
A. Breakpoint junction homology/microhomology length plotted as a function of SV size for SVs originally identified as deletions compared to a human reference. Dots are colored according to the SVs’ classification as deletions, insertions/duplications, or “undetermined” relative to inferred ancestral genomic loci. Gray lines mark groups of SVs likely formed by a common formation mechanism. The diagonal highlights tandem duplications (and few reciprocal deletion events), in which the length of the duplicated sequence correlates linearly with the length of the longest breakpoint junction sequence identity stretch. The ellipses indicate MEIs, i.e., Alu (~300 bp) and L1 (~6 kb) insertions, associated with target site duplications of up to 28 bp in size at the breakpoints. The horizontal group corresponds mostly to NH-associated deletions with <10 bp microhomology at the breakpoints. The remaining (ungrouped) SVs comprise truncated MEIs, VNTR expansion and shrinkage events, as well as NAHR-associated deletions and duplications. B. Relative contributions of SV formation mechanisms in the genome. Numbers of SVs are displayed on the outer pie chart and affected base pairs on the inner. Left panel: SVs classified as deletions relative to ancestral loci. Right panel: SVs classified as insertions/duplications. C. Size spectra of deletions classified relative to ancestral loci. D. Size spectra of insertions/duplications.
We subsequently applied BreakSeq43 to infer formation mechanisms for all SVs classified with regard to ancestral state. Using BreakSeq, we inferred NH as the dominating deletion mechanism, and MEI as the dominating insertion mechanism (Fig. 4BC, Supplementary Table 11). Furthermore, an abundance of microhomology at tandem duplication breakpoints suggested frequent formation of this SV class by a rearrangement process acting in the absence of homology (NH). When relating SV formation to the variant size spectrum, we observed marked insertion peaks for MEIs at 300 bp, corresponding to Alu elements, and at 6 kb, corresponding to L1/LINEs (Fig. 4C). By comparison, NH and NAHR based mechanisms occurred across a wide size-range, whereas VNTR expansion/shrinkage, consistent with earlier findings1, led to relatively small SV sizes (Figs. 4C,D).
Furthermore, when displaying the genomic distribution of SVs (Fig. 5A), we observed a notable clustering of SVs into ’SV hotspots’. We analyzed this clustering in detail by examining the distribution of non-overlapping, adjacent SVs, and observed a marked clustering of SVs formed by NAHR, VNTR, and NH, respectively, a signal extending to hundreds of kilobases (Fig.5B). The clustering was influenced by an abundance of VNTR near the centromeres43 and NAHR near the telomeres (Fig.5A). A significant enrichment of NAHR near recombination hotspots (P=1.3e-15) and segmental duplications (P=3.1e-17) further contributed to the clustering (Supplementary Table 13).
Figure 5. Mapping hotspots of SV formation in the genome.
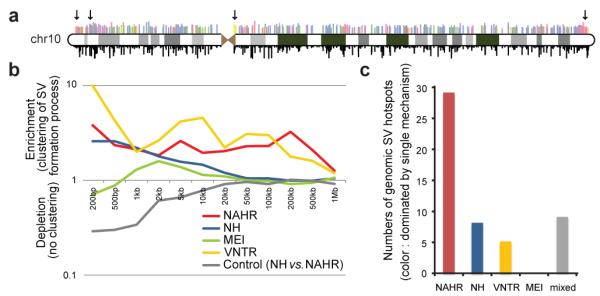
A. Distribution of SVs on chromosome 10 (“chr10”). Above the ideogram, colored bars indicate SV formation mechanisms (same color scheme as in B and C); bar lengths relate to the logarithm of SV size. Below the ideogram, bar lengths are directly proportional to allele frequencies. Arrows indicate an SV hotspot near the centromere underlying mainly VNTR, and several hotspots near the telomeres underlying mainly NAHR events. B. Enrichment of SVs inferred to be formed by the same formation mechanism for different genomic window sizes. Displayed is an enrichment of nearby, non-overlapping SVs formed by the same mechanism relative to an SV set where mechanism assignments are shuffled randomly. C. SV hotspots are mostly dominated by a single formation mechanism. Colored bars depict numbers of SV hotspots in which at least 50% of the variants were inferred to be formed by a single formation mechanism. The average abundance of NAHR-classified SVs in NAHR hotspots was 70% (compared with 77% for VNTR-hotspots; 69% for NH). The gray bar (“mixed”) corresponds to SV hotspots with no single mechanism dominating.
To further explore this clustering we devised a segmentation approach for predicting SV hotspots (Methods), which yielded a map of 51 putative SV hotspots (Supplementary Table 14). 80% of the hotspots mainly comprised SVs originating from a single formation mechanism (Fig. 5C). Most of these corresponded to NAHR hotspots, although hotspots dominated by NH and VNTR also were evident. These observations suggest that SV formation is frequently associated with the locus-specific propensity for genomic rearrangement.
Conclusions and discussion
By generating an SV set of unprecedented size along with breakpoint assemblies and reference genotypes, we demonstrate the suitability of population-scale sequencing for SV analysis. Nucleotide resolution data allow the construction of reference datasets and make SVs readily assessable across different experimental platforms using genotyping approaches. Our fine-scale map enabled us to examine the functional impact of SVs, as exemplified by our analysis of gene disruption variants, which will be of value for genome and exome sequencing studies.
Our map further enabled us to examine size spectra of SV formation mechanisms and led us to identify genomic SV hotspots that are commonly dominated by a single formation mechanism. Recurrent rearrangements, implicated in genomic disorders, are hypothesized to be associated with local genome architecture44, e.g., with segmental duplications that facilitate NAHR. Also, DNA rearrangement in the absence of homology, i.e., MMBIR, has been implicated in recurrent SV formation8,45. In this regard, we noticed that out of the hotspots we report, six fall into critical regions of known genetic disorders associated with recurrent de novo deletions, including Miller-Dieker syndrome and Leri-Weill dyschondrosteosis (Supplementary Table 14). Irrespective of potential disease relevance, or inferred mechanism of formation, our analysis revealed a map of SV hotspots that may constitute local centers of de novo SV formation, consistent with the concept that local genome architecture contributes to genomic instability44.
Our study focused on characterizing deletions, which are often associated with disease9. Facilitated by ancestral analyses of SV loci, we also characterized insertions and tandem duplications, albeit in less detail than deletions. Companion papers with more detailed analyses of MEIs, and copy-number variation within segmental duplications are published elsewhere34,46. Of note, most SV discovery methods depend on mapping reads onto their genomic locus of origin, i.e., the ‘accessible’ fraction of the genome, a fraction lessened in segmental duplications that are of high interest to SV analysis. Nonetheless, owing to the abilities of RP and RD methods in detecting SVs in these regions and in interpreting reads with multiple mapping positions, the ‘accessible’ fraction of the genome is higher for SVs than for SNPs16. In the future, sequencing technologies generating longer DNA reads will increase the accessible genome, and will enable the assessment of SVs embedded in long repeat structures, such as balanced inversions.
Our SV resource will enable the discovery, genotyping, and imputation of SVs in larger cohorts. Numerous genomes will be sequenced in the coming months to facilitate disease association studies. Systematic characterization of SVs in these genomes will benefit from the concepts and datasets presented here.
Methods Summary
Samples
Sequence data for 179 unrelated individuals and six individuals from parent-offspring trios were obtained as part of the 1000GP. These data were generated with Illumina/Solexa, Roche/454, and Life Technologies/SOLiD sequencing technology platforms.
SV discovery and breakpoint assembly
The SV discovery methods we applied comprised six RP, four RD, three SR, four AS, and two PD based methods. TIGRA38 was used for targeted breakpoint assembly.
Experimental validation
We validated SV calls by PCR, array CGH and SNP microarrays, targeted assembly, and custom microarray-based sequence capture. PCR was performed in various different laboratories33, CGH analysis was performed based on tiling array data provided by the Genome Structural Variation Consortium (ArrayExpress: E-MTAB-40), and SNP array analysis based on data obtained from the International HapMap Consortium (http://hapmap.ncbi.nlm.nih.gov).
Genotyping
Genome STRiP36 was used for deletion genotyping in low coverage sequence data. Initial genotype likelihoods were derived with a Bayesian model and imputation into a SNP genotype reference panel from the HapMap41 (Hapmap3r2) was achieved with Beagle (v3.1; http://faculty.washington.edu/browning/beagle/beagle.html).
SV formation mechanism analysis
SV breakpoints mapped at nucleotide resolution were analyzed with BreakSeq43 to classify SVs relative to putative ancestral loci and to infer SV formation mechanisms. SV hotspots were mapped with custom Perl and R scripts.
Supplementary Material
Acknowledgements
We would like to acknowledge Claire Hardy, Richard Smith, Anniek De Witte, and Shane Giles for their assistance with validation. M.A.B’s group was supported by grants from the National Institutes of Health (RO1 GM59290) and G.T.M’s group by grants R01 HG004719 and RC2 HG005552, also from the NIH. J.O.K.’s group was supported by an Emmy Noether Fellowship of the German Research Foundation (Deutsche Forschungsgemeinschaft). J.W.’s group was supported by the National Basic Research Program of China (973 program no. 2011CB809200), the National Natural Science Foundation of China (30725008; 30890032; 30811130531; 30221004), the Chinese 863 program (2006AA02Z177; 2006AA02Z334; 2006AA02A302; 2009AA022707), the Shenzhen Municipal Government of China (grants JC200903190767A; JC200903190772A; ZYC200903240076A; CXB200903110066A; ZYC200903240077A; ZYC200903240076A and ZYC200903240080A), and the Ole RØmer grant from the Danish Natural Science Research Council. C.L.’s group was supported by grants from the National Institutes of Health: P41 HG004221, RO1 GM081533, and UO1 HG005209 and X.S. was supported by a T32 fellowship award from the NIH. We thank the Genome Structural Variation Consortium (http://www.sanger.ac.uk/humgen/cnv/42mio/) and the International HapMap Consortium for making available microarray data. The authors acknowledge the individuals participating in the 1000 Genomes Project by providing samples, including The Yoruba people of Ibadan, Nigeria, the community at Beijing Normal University, the people of Tokyo, Japan, and the people of the Utah CEPH community. Furthermore, we thank Richard Durbin and Lars Steinmetz for comments on the manuscript.
Footnotes
Competing interests statement: The authors declare competing financial interests. H.E.P. and Y.F. are employees of Life Technologies, the manufactures of the SOLiD sequencing platform. R.K.C. is an employee of Illumina Cambridge Ltd., the manufacturer of the Illumina sequencing platform.
Data retrieval: The data sets described here can be obtained from the 1000 Genomes Project website at www.1000genomes.org (July 2010 Data Release). Individual SV discovery methods can be obtained from sources mentioned in Supplementary Table 1, or upon request from the authors. Abbreviations used in this paper are summarized in the Supplementary Text.
References
- 1.Conrad DF, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCarthy SE, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craddock N, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarroll SA, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–12. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 10.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 11.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 12.Sharp AJ, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCarroll SA, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 14.Tuzun E, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 15.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alkan C, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009;6:S13–20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- 20.McKernan KJ, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiang DY, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S, Cheran E, Brudno M. A robust framework for detecting structural variations in a genome. Bioinformatics. 2008;24:i59–67. doi: 10.1093/bioinformatics/btn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pang AW, et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 2010;11:R52. doi: 10.1186/gb-2010-11-5-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey JA, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 26.Campbell PJ, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mills RE, et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simpson JT, et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hajirasouliha I, et al. Detection and characterization of novel sequence insertions using paired-end next-generation sequencing. Bioinformatics. 2010;26:1277–1283. doi: 10.1093/bioinformatics/btq152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li R, et al. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463:311–317. doi: 10.1038/nature08696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The-1000-Genomes-Project-Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sudmant PH, et al. Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willer CJ, Willer CJ. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Handsaker RE, Korn JM, Nemesh J, McCarroll SA. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. submitted. [DOI] [PMC free article] [PubMed]
- 37.Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, et al. TIGRA local targeted assembly of structural variants. 2010. submitted.
- 39.Hasin-Brumshtein Y, Lancet D, Olender T. Human olfaction: from genomic variation to phenotypic diversity. Trends Genet. 2009;25:178–184. doi: 10.1016/j.tig.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet. 2006;38:82–85. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]
- 41.Altshuler DM, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conrad DF, et al. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lam HY, et al. Nucleotide-resolution analysis of structural variants using BreakSeq and a breakpoint library. Nat Biotechnol. 2010;28:47–55. doi: 10.1038/nbt.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 45.Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 46.Stewart C, et al. A comprehensive map of mobile element insertion polymorphisms in humans. in preparation. [DOI] [PMC free article] [PubMed]
- 47.Harrow J, et al. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006;7(Suppl 1):S4, 1–9. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.