

Abstract
In Alzheimer's disease (AD), amyloid-β (Aβ) deposits are frequently surrounded by activated microglia but the precise role of these cells in disease progression remains unclear. The chemokine receptor CX3CR1 is selectively expressed in microglia and is thought to modulate their activity. To study the specific effects of microglia activation on amyloid pathology in vivo, we crossbred mice lacking CX3CR1 with the Alzheimer's mouse model CRND8. Surprisingly, we found that CX3CR1-deficient mice had lower brain levels of Aβ40 and Aβ42 and reduced amyloid deposits. Quantification of Aβ within microglia and time-lapse two-photon microscopy in live mice revealed that these cells were highly effective at the uptake of protofibrillar amyloid but were incapable of phagocytosis of fibrillar congophilic Aβ. CX3CR1 deletion was associated with increased phagocytic ability, which led to greater amyloid content within microglial phagolysosomes. Furthermore, CX3CR1-deficient mice had an increased number of microglia around individual plaques because of higher proliferative rates, which likely contributed to an overall greater phagocytic capacity. CX3CR1 deletion did not affect the degree of neuronal or synaptic damage around plaques despite increased microglia density. Our results demonstrate that microglia can regulate brain Aβ levels and plaque deposition via selective protofibrillar Aβ phagocytosis. Modulation of microglia activity and proliferation by CX3CR1 signaling may represent a therapeutic strategy for AD.
Introduction
Alzheimer's disease (AD) is characterized by the presence of extracellular amyloid-β peptide (Aβ) deposits surrounded by activated glia and dystrophic neurites. Microglia are the resident immune cells of the brain and can become activated by a variety of changes in its structural and biochemical integrity (Davalos et al., 2005; Hanisch and Kettenmann, 2007). Microglia activation and clustering around amyloid deposits tend to occur in early stages of the disease process. Microglia have been shown to be capable of phagocytosis of amyloid fibrils (Frautschy et al., 1991), and deficits in microglia accumulations can accelerate amyloid deposition (El Khoury et al., 2007). However, the role of microglia in AD has not been studied in great detail using in vivo models, and it remains unclear to what extent microglia are capable of removing amyloid from preexisting fibrillar congophilic plaques (Stalder et al., 2001) or of preventing the deposition of smaller protofibrillar Aβ aggregates (Grathwohl et al., 2009). Thus, it is not clear how or to what extent microglia control brain amyloid levels or deposition.
To address these questions, we investigated mice lacking the chemokine receptor CX3CR1 (Jung et al., 2000). In the brain, this receptor is exclusively expressed in microglia and selectively modulates microglial activity in response to its ligand the chemokine fractalkine (Harrison et al., 1998). Thus, CX3CR1 could potentially play a role in modulating microglia function in AD. CX3CR1−/− mice were crossbred with transgenic mice (CRND8) harboring the human amyloid precursor protein gene with the Indiana and Swedish mutations (Chishti et al., 2001). To determine the role of CX3CR1 in microglia phagocytic activity and regulation of amyloid levels, we implemented new in vivo methods for tracking the interactions between microglia and both fibrillar and nonfibrillar amyloid material using longitudinal imaging with high-resolution confocal as well as transcranial two-photon microscopy (TPM).
Previous in vivo results range from showing that microglia are effective at phagocytosis of fibrillar amyloid (Frautschy et al., 1991; Bacskai et al., 2002; Bolmont et al., 2008) to suggesting that microglia play no role in the control of amyloid deposition (Grathwohl et al., 2009). By contrast, in our study, microglia were incapable of phagocytosis of congophilic fibrillar amyloid from plaques but were very effective at the uptake of oligomeric and protofibrillar Aβ. This selective uptake ability was critical for the regulation of total brain amyloid levels and deposition. CX3CR1 deletion increased microglia proliferation and numbers specifically around plaques, which coupled with their increased phagocytic ability, resulted in decreased brain amyloid levels and deposition.
Conflicting evidence in the literature suggests either neurotoxic or neuroprotective effects of CX3CR1 deletion in various disease models (Cardona et al., 2006; Fuhrmann et al., 2010). However, we found no difference in either neuronal loss or synaptic injury around plaques. This occurred despite significant differences in the density of plaque-associated microglia, which in their activated status are thought to have neurotoxic potential (Block et al., 2007). Thus, our data demonstrate that microglia play an important role in selective phagocytosis of oligomeric and protofibrillar Aβ but not of preexisting congophilic amyloid plaques. This function may be critical in controlling Aβ deposition as well as levels of potentially neurotoxic Aβ oligomers (Lambert et al., 1998). Thus, enhancing microglia phagocytosis and proliferation by blocking CX3CR1 signaling could constitute a therapeutic strategy for AD.
Materials and Methods
Mice.
The generation of CX3CR1-deficient mice and TgCRND8 mice has been previously described. Briefly, the CX3CR1 gene locus underwent targeted deletion and direct replacement by a green fluorescent protein (GFP) reporter gene. CX3CR1+/− mice previously backcrossed to C57BL/6 mice for >10 generations were crossbred with TgCRND8 mice to obtain CRND8/CX3CR1−/−, CRND8/CX3CR1+/−, and CRND8/CX3CR1+/+ mice. Experiments in which we quantified amyloid plaque density, Aβ concentrations, and amyloid precursor protein (APP) processing/cleavage were done in male mice. All other experiments including quantification of Aβ within microglia and in vivo imaging experiments were done in mixed gender mice but with equal gender distribution on each experimental group. Experimental protocols were approved by the Northwestern University Feinberg School of Medicine Institutional Animal Care and Use Committee.
In vivo imaging with two-photon microscopy.
GFP-labeled microglia and Methoxy-X04 (MX04)-labeled plaques were imaged through a thinned skull preparation as previously described (Grutzendler et al., 2002). Briefly, transgenic mice were anesthetized with ketamine/xylazine and the skull was exposed with a midline scalp incision. A ∼1-mm-diameter skull region over the somatosensory cortex was thinned with a high speed drill and a microsurgical blade to a final thickness of ∼30 μm. The skull was attached to a custom-made steel plate to stabilize the head while imaging. A mode-locked Ti-sapphire laser (Coherent) was used for two-photon excitation (Prairie Technologies) and tuned to 835 nm for dual imaging of GFP and MX04 or 890 nm for GFP alone. Emission wavelengths for MX04 and GFP were separated from each other with a 490 nm dichroic. Images were taken using a water-immersion objective [Olympus; 40 or 60×; 0.8 and 0.9 numerical aperture (NA), respectively] at z-steps of 1–2 μm and zooms of 1–4×. In most cases, images were taken at depths up to ∼150 μm below the pial surface. The same regions were relocated at different time points using the unchanged pattern of pial blood vessels as reference as well as registration of X–Z coordinates of each scanning area at the initial time point and relocation of the same coordinates using a precision motorized stage at subsequent time points. The precise identity of the areas is confirmed by the presence of unambiguous microvascular shadows or plaque patterns.
In vivo subarachnoid Aβ42 delivery.
A ∼500 μm area of the skull was thinned with a high-speed micro drill at specific cranial stereotaxic coordinates (3.0 mm mediolateral, −8.0 mm anterioposterior from bregma). The tip of a 27 gauge needle was used to make a small cut on the surface of the thinned skull area and into the underlying dura without touching the brain parenchyma. Custom-made polypropylene tubing with a final outer diameter of 90 μm was inserted ∼300 μm into the subarachnoid space and fixed to the skull with cyanoacrylate glue. The tube was connected to a programmable syringe pump through a Hamilton syringe and 10 μl (0.4 μm) of fluorescently labeled Aβ42 monomers (HiLyte Fluor 555-Aβ42; AnaSpec) freshly dissolved in sterile artificial CSF immediately before injection was infused at a rate of 0.2 μl/min. After infusion, tubing was removed and animal scalp was sutured. Animals were imaged in vivo by TPM or in fixed tissue by confocal microscopy at various time intervals after infusion.
Quantification of infused Fluor-Aβ42 phagocytosis.
Analysis of microglia phagocytosis of Fluor-Aβ42 and presence of thioflavin-S (TS)-positive material within microglia was performed using NIH ImageJ software on confocal images of brain sections obtained from Fluor-Aβ-infused mice. All images were obtained with standardized parameters of laser intensity, gain, and pinhole size. (1) For Fluor-Aβ42 and thioflavin-S content within microglia and astrocytes, microglial or astrocyte cell bodies were identified in z-projections from confocal images. A region of interest (ROI) was drawn around the cell body perimeter as well as a mirror ROI control immediately outside of each microglia or astrocyte. Mean fluorescence intensity values were measured for each cell and control ROI in both red channel for Aβ42 (HiLyte Fluor 555) and blue channel for thioflavin-S-positive fibrillar material. (2) For quantification of Aβ fluorescence outside of microglia, custom-made NIH ImageJ macros were written with the purpose of masking and subtracting areas where microglia were located. Mean fluorescence intensity values were obtained from the resulting area after subtraction of microglia regions. For the purpose of our study, any Fluor-Aβ deposit that was not located inside microglia was considered to be in the extracellular space, given our finding of the limited intake of Fluor-Aβ by astrocytes (supplemental data, available at www.jneurosci.org as supplemental material). Ultimately, however, it is not possible to determine unambiguously if deposits are extracellular given the very small size of this space, which is beyond the resolution of light microscopy.
Quantification of thioflavin-S-labeled amyloid plaques.
Sagittal sections of the entire left hemisphere were obtained with a cryostat at 50 μm thickness. Eight sections for each hemisphere were selected and visually matched between animals to represent the same brain regions. Sections from all genotypes were stained with thioflavin-S with a standardized concentration and time. Whole-brain sections were imaged with a fluorescence microscope (Zeiss Axiovert 200M; objective, 20×; 0.75 NA) using identical parameters of gain and exposure in the CCD camera. All images were stitched together to generate a single image of the entire brain slice. A custom-made thresholding macro based on NIH ImageJ was used to detect all fluorescent amyloid plaques using exactly the same parameters for all sections and genotypes. NIH ImageJ particle counting plug-in was used to calculate the total number of amyloid plaques per brain section. A subset of images was counted manually by an unbiased individual to corroborate the accuracy of our automated counting. The use of automated counting allowed us to count a much larger number of plaques representing the majority of the hemisphere than would have been possible with stereological methods.
Quantification of MXO4-labled plaque area in vivo.
Mice were injected only once with MX04 2 d before the first imaging time point. TPM imaging was done as described above. Image stacks containing plaques were run through a “despeckle” filter to remove noise, and then a maximal projection through a consistent number of slices was taken using NIH ImageJ software. To achieve accurate plaque area measurements regardless of minor brightness differences across time points, images were thresholded for each time point at the average pixel brightness value between a consistent plaque area and the adjacent background.
Quantification of 4G8 and A11 fluorescence inside microglia phagolysosomes.
Cryosections (50 μm thick) underwent sodium citrate antigen retrieval and were immunolabled with the following antibodies: 4G8 (Covance), A11 (BioSource/Invitrogen), IBA1 (Wako Chemicals), and lysosomal-associated membrane protein (LAMP1) (Developmental Studies Hybridoma Bank clone 1D4B) according to manufacturer protocols. Confocal images of plaques selected for their similar size, location and cortical layer were obtained for quantitative imaging using an Olympus FV10i confocal microscope (60× objective; NA, 1.2). Image z-stacks were taken with standardized laser intensity, gain, and pinhole sizes to allow interanimal comparisons. LAMP1-positive lysosomes inside IBA1-labeled microglia have distinct fluorescence intensities compared with LAMP1-positive structures outside microglia. Using NIH ImageJ, we applied a fluorescence threshold to the LAMP1 signal and created a binary image that was then combined with the other fluorescent channels in the z-stack to measure 4G8, A11, and thioflavin-S fluorescence intensities within the microglial LAMP1 structures. Measurements were performed on each optical slice of a given z-stack and then averaged.
Microglia culture and in vitro phagocytosis assay.
One-day-old CX3CR1+/+ and CX3CR1−/− mouse littermates were used for primary cultures of cerebral cortical cells. These cell cultures include abundant microglia, astrocytes, and neurons as confirmed by immunocytochemistry. Briefly, after cell dissociation, a mixture of equal number of brain cells from both genotypes was adjusted to 106 cells/ml in DMEM/10% fetal bovine serum (FBS), followed by plating of the cell suspension on a culture flask. Fifty percent of the medium was replaced at days 3, 10, and 17. At day 21, cells reaching 90% confluence were trypsinized and reseeded to 24-well plates containing poly-lysine-coated coverslips at a density of 5 × 104 cells/ml. The reseeded cells were rinsed in DMEM containing 10% FBS, pretreated with or without 10 μg/ml CX3CR1 neutralizing antibody for 30 min, and then incubated for 2 h in the same medium containing either 2-μm-diameter fluorescent microspheres or 0.05 mg/ml fluorescently conjugated Aβ (HiLyte Fluor 555-Aβ42; AnaSpec) in DMEM. After washing with DMEM, cells were fixed in 2% paraformaldelhyde/PBS for immunocytochemistry. Fixed cells were rinsed in PBS, blocked in 3% normal goat serum/0.3% Triton X-100 for 30 min, and then incubated with primary antibodies (IBA1, rabbit polyclonal, 1:500; GFP, mouse monoclonal, 1:1000) overnight. CX3CR1−/− microglia were differentiated from CX3CR1+/+ microglia because they had positive labeling with both IBA1 and GFP antibodies, whereas CX3CR1+/+ microglia were only labeled by IBA1. We also excluded the possibility of other cell types expressing either IBA1 or GFP in control experiments in which we used dual labeling with antibodies specific for astrocytes (GFAP) or neurons (MAP2). In separate culture preparations containing only CX3CR1−/− microglia, we determined that 100% of IBA1-positive microglia were GFP positive. Microglia of each genotype represent ∼50% of the total microglia population in these mixed cell cultures. Minor differences in the proportion of microglia of each genotype do not affect the final results of the phagocytosis assay given that, in a mixture of cells, there is equal likelihood for each individual cell regardless of its genotype to be exposed to Fluor 555-Aβ42 or microspheres. For quantification of microglia uptake of microspheres or Aβ, we used a custom-made macro with NIH ImageJ software for automated measurement of total fluorescence intensity within individual microglia.
Quantification of microglia, neuron, and synapse density around plaques.
Cryosections (50 μm thick) were immunolabled with any of the following antibodies: anti-IBA1 (Wako Chemicals), Synaptophysin (Millipore Bioscience Research Reagents), and NeuN (Millipore Bioscience Research Reagents) according to manufacturer protocols. Confocal images of plaques selected for their similar size, location, and cortical layer (layers III to VI in somatosensory cortex) were obtained for quantitative imaging using a Zeiss LSM 510 Meta UV microscope (63× objective; NA, 1.4). Images were taken at standardized laser intensity, gain, and pinhole sizes to allow interanimal comparisons. Quantification of microglia and NeuN-positive neuron number was performed using the ImageJ threshold/analyze particle features. Results were validated by manual counting in a subset of stacks. To control for possible differences in microglia density between CX3CR1 genotypes in the absence of AD pathology, we measured the density of microglia in non-AD/CX3CR1 mutant mice that did not differ between genotypes in the brain region analyzed. We have observed full colocalization of IBA1 and F4/80 (an additional microglia marker) demonstrating that all microglia are IBA1 immunoreactive. Furthermore, IBA1 has a 100% colocalization with GFP-labeled cells in the brain parenchyma of CX3CR1 knock-in mice. Thus, IBA1 labels all brain microglia but cannot differentiate them from infiltrating macrophages given that macrophages also label with IBA1. Microglia density around plaques was quantified in blindly selected plaques of the same average size to eliminate size as a variable, which can lead to differences in microglia number unrelated to the CX3CR1 genotype.
For synaptophysin quantification, multichannel 8 bit images were obtained in which plaques were labeled with thioflavin-S in one channel and synaptophysin in a second channel. Using a custom ImageJ macro, the resultant stacks were analyzed slice by slice to determine the fluorescence of synaptophysin as a function of distance from the plaque edge. The plaque edge was determined by thresholding the thioflavin-S channel at a constant predetermined value of 115. This edge was selected and then enlarged by 2 μm (the selection grows in all directions out from its center for 2 μm) to form an even starting point. Then, 10 donut-shaped selection bands of 3 μm width were created at 3 μm intervals, covering a 30 μm circular area out from the evened plaque edge. This essentially creates concentric circular bands of selection, which were then used to measure average fluorescence intensity in the synaptophysin channel, with the first band nearly adjacent to the plaque (all pixels 2–5 μm from the thresholded plaque edge), and the last band ∼30 μm away from the plaque edge (all pixels 32–35 μm from the thresholded plaque edge).
Quantification of microglia cell body and process dynamics in vivo.
(1) For cell body motility, in vivo TPM images of both microglia and plaques were taken over 4–7 d. For long-term monitoring, a subset of mice was imaged at 1 and 4 month intervals. Microglia cell bodies adjacent to the plaque were counted for each time point, and microglia stability was calculated as the percentage of all adjacent cell bodies in the first time point, which remained in the same location relative to the plaque at the later time point. (2) For cell process dynamics, image stacks of microglia processes at two time points 1 h apart were aligned using ImageJ plug-ins, maximally projected, matched for brightness, and then thresholded. Each time point was subtracted from the other to determine regions that were “lost” (apparent in the first image but not the second) or “gained” (apparent in the second image but not the first). The areas of regions of gains or losses above an arbitrary cutoff of 3.2 μm2 were then measured and counted per image stack.
Microglia proliferation assay.
Mice received daily injections of 5-bromo-2-deoxyuridine (BrdU) (50 mg/kg, i.p.) for 14 d. Brains were removed after 4% paraformaldehyde perfusion and cryosectioned at 50 μm thickness. BrdU was detected after denaturing with 5N HCl followed by incubation with rat anti-BrdU antibody (1:400; Serotec) and rabbit anti-IBA1 antibody for microglia detection. BrdU and IBA1 were detected with Alexa Fluor 555/488-conjugated secondary antibodies and imaged with confocal microscopy. BrdU-labeled microglia were quantified from confocal image stacks in areas in the vicinity (25 μm) and distant from plaques.
ELISA.
Aβ42 and Aβ40 were measured by commercially available ELISA kits (BioSource). Brain hemispheres of CX3CR1+/+, CRND8/CX3CR1+/+, and CRND8/CX3CR1−/− mice were homogenized in cold 5 m guanidine HCl/50 mm Tris-HCl. ELISAs were performed according to the manufacturer's protocol.
Western blot for APP and C-terminal fragments.
Brain tissue lysates were prepared in ice-cold TEEN-Tx [TEEN buffer (50 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm EGTA, and 150 mm NaCl) containing 1% Triton X-100] by sonication. This was followed by centrifugation, protein quantification, addition of buffer, and boiling for 5 min. Proteins were separated using 15% Tris-tricine polyacrylamide gel and incubated with APP C-terminal-specific rabbit anti-APP-CT20 antibody (1:10,000; Calbiochem) and mouse anti-α-tubulin (1:5000; Millipore). Densitometric quantification of band intensity was performed using NIH ImageJ software.
Reverse transcription-PCR.
Mouse presenilin-1 and β-site APP-cleaving enzyme 1 (BACE1) mRNA expression were detected by reverse transcription (RT)-PCR. Briefly, total RNA was extracted and cDNA was obtained by reverse transcription according to manufacturer's instruction. The following pairs of primers were used: 5′-AGC ATG ACA GGC AGA GAC TTG ACA-3′ and 5′-AAC GTA GTC CAC GGC GAC ATT GTA-3′ for presenilin 1, 5′-ATC TAC ACG CCC TTC ACG GAG G-3′ and 5′-TGG GCA GTT TCC ACC AGC ATC-3′ for presenilin 2, 5′-AGG GCT ACT ATG TGG AGA TGA CCG TA-3′ and 5′-TCC CAC TGT CCA CAA TGC TCT TGT-3′ for BACE1, 5′-AGA TGA GAA TGC CAG TCG CTC CTT-3′ and 5′-TGC ACA GTT GAG GTT CCC GAC TAA-3′ for BACE2, and 5′-AAC TTT GGC ATT GTG GAA GGG CTC-3′ and 5′-ACC CTG TTG CTG TAG CCG TAT TCA-3′ for GAPDH (internal control).
Statistical analysis.
Statistical analysis was performed using two-tailed Student's t test or linear correlation analysis. A value of p < 0.05 was considered significant.
Results
CX3CR1 deficiency in microglia leads to reduced brain β-amyloid load and deposition
To examine the role of microglia in the process of brain amyloid buildup, we crossbred CRND8 mice with mice lacking the CX3CR1 receptor (CX3CR1−/−). At 2 months of age, both CRND8/CX3CR1−/− mice and their wild-type littermates (CRND8/CX3CR1+/+) demonstrated low levels of total brain Aβ40 and Aβ42 peptides (Fig. 1A,B). At 5 months, brain Aβ40 and Aβ42 concentrations had risen markedly but were significantly lower in CRND8/CX3CR1−/− mice compared with CRND8/CX3CR1+/+ littermates (Fig. 1A,B). The effect of CX3CR1 deficiency on Aβ deposition was further examined by quantifying the density of TS-labeled fibrillar amyloid deposits. We found that CRND8/CX3CR1−/− mice had a significantly reduced number of amyloid plaques at both 3 and 5 months of age (Fig. 1C,D). The disparity in brain Aβ levels and amyloid plaque number between CX3CR1 genotypes were likely attributable to reduced Aβ accumulation rather than decreased Aβ production given that there was no significant difference in APP concentrations (Fig. 1E), mRNA expression of APP processing enzymes (BACE1 and 2; PS1 and 2) (Fig. 1F), or levels of APP cleavage products (Fig. 1G).
Figure 1.
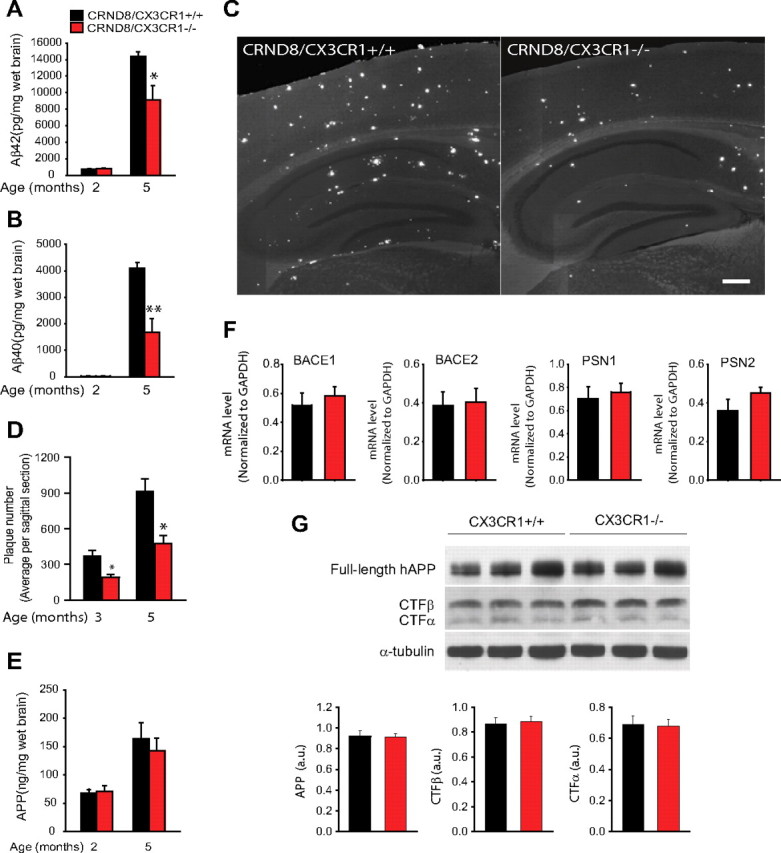
CX3CR1 deficiency leads to reduced β-amyloid load and deposition. A, B, Total Aβ40 and Aβ42 brain concentrations measured by ELISA were very low at 2 months but increased markedly in 5-month-old mice. The 5-month-old CRND8/CX3CR1−/− mice had significantly lower levels of both Aβ40 and Aβ42 than wild-type littermates (*p < 0.05, **p < 0.01, two-tailed Student's t test; n = 6–7 mice per group; values are expressed as mean ± SEM). C, Representative images of neocortex and hippocampus show fewer thioflavin-S-labeled fibrillar amyloid deposits in a 5-month-old CRND8/CX3CR1−/− mouse than its wild-type littermate. Scale bar, 100 μm. D, Quantification of the average plaque number in sagittal brain sections demonstrates significantly fewer plaques in both 3- and 5-month-old CRND8/CX3CR1−/− mice than wild-type littermates (*p < 0.05, two-tailed Student's t test; n = 4 mice per group; values are expressed as mean ± SEM). E, ELISA measurement of APP levels reveals no difference in CRND8/CX3CR1−/− mice and wild-type littermates (n = 6–7 mice per genotype). F, RT-PCR quantification showed no differential effect of CX3CR1 genotype on mRNA levels of BACE1, BACE2, PSN1, and PSN2 in 2-month-old mice (n = 4 mice per group; values are expressed as mean ± SEM). G, Western blot analysis using a C terminus anti-APP antibody reveals no significant difference in the concentration of APP cleavage products between CX3CR1 genotypes (APP, p = 0.92; CTFβ, p = 0.67; CTFβ, p = 0.78, two-tailed Student's t test; n = 11 mice of average age of 4 months per genotype).
Given that the expression of CX3CR1 in the brain is microglia specific, the reduction in Aβ levels and plaque density in CRND8/CX3CR1−/− mice suggests a critical role of microglia and CX3CR1 in controlling Aβ accumulation.
Microglia are unable to phagocytose fibrillar congophilic amyloid in vivo
Although microglia appear to be capable of soluble (Mandrekar et al., 2009) and fibrillar (Frautschy et al., 1992) Aβ uptake, their phagocytic properties in vivo are not well characterized. To determine the mechanisms by which microglia control the accumulation of Aβ in the brain, we examined their phagocytic capabilities in living mice by time-lapse transcranial TPM. We first labeled plaques by intraperitoneal injection of the fluorescent amyloid-binding dye methoxy-XO4 (Klunk et al., 2002) 48 h before the first imaging session and without relabeling on subsequent sessions. We reasoned that, if microglia phagocytosis of fibrillar amyloid were a robust phenomenon in vivo, the size and shape of the initial fluorescent plaque label would change significantly over time as portions of amyloid deposits are removed by microglia. However, time-lapse TPM images over intervals of up to 1 month showed virtually no change in the size or shape of the original fluorescent plaque label (Fig. 2A,B). In addition, we labeled fixed brain slices with the fluorescent fibrillar amyloid binding dye TS and with IBA1 antibody to visualize fibrillar amyloid contained within microglia using high-resolution confocal microscopy. Quantification of TS fluorescence within IBA1-immunolabeled cells revealed absence of any intracellular TS labeling (Fig. 2C,D) regardless of the CX3CR1 genotype (Fig. 2D).
Figure 2.

Microglia are unable to phagocytose fibrillar congophilic amyloid in vivo. A, Time-lapse in vivo two-photon microscopy of fibrillar amyloid plaques in the mouse cortex. Plaques were labeled with the amyloid binding dye methoxy-XO4 48 h before initial imaging and without relabeling in subsequent sessions. The shape and size of the amyloid plaque at day 3 remains virtually unchanged at days 7 and 30 after labeling. Scale bar, 15 μm. B, Quantification of changes in amyloid plaque size over time from in vivo images shows significant stability (p = 0.92, two-tailed t test; n = 2–4 mice per group and 30 plaques; values are expressed as mean ± SEM). C, Confocal image of IBA1-labeled microglia (green) surrounding a thioflavin-S-labeled amyloid plaque (blue). The intensity of thioflavin-S fluorescence was measured within microglia cell bodies and equivalent areas immediately adjacent but devoid of microglia (dotted circles). D, Fluorescence quantification shows that microglia contain the same negligible levels of blue fluorescence as the immediately adjacent background areas. CX3CR1 genotype had no differential effect on fluorescence within microglia (n = 3 mice, 120 plaques, and >400 microglia per genotype; p > 0.75 between CX3CR1 genotypes and background fluorescence).
To further establish whether fibrillar amyloid within plaques is stable or is constantly being removed, we developed a method for subarachnoid infusion of fluorescently tagged Aβ peptides. We obtained time-lapse images with two-photon microscopy while Aβ42(HiLyte)-555 was being infused. During imaging, we observed very rapid binding of Aβ42 (within minutes) to preexisting amyloid plaques (Fig. 3A), suggesting a very high affinity of soluble Aβ42 for fibrillar amyloid within plaques. However, nonplaque areas immediately adjacent demonstrated very low fluorescence levels (Fig. 3A), suggesting that unbound Aβ42 was rapidly cleared possibly through the blood–brain barrier (BBB) (DeMattos et al., 2002). Once bound to plaques, we found by time-lapse imaging that Aβ42 was highly stable over many weeks like we observed with methoxy-XO4. This was formally quantified from fixed tissue confocal images of amyloid plaques obtained from animals infused with Aβ42(HiLyte)-555 and killed either 1 or 90 d after infusion. We reasoned that, if there were any significant phagocytosis of plaque-associated Aβ, we would observe a reduction in the average Aβ42(HiLyte)-555 fluorescence intensity and/or plaque diameter over time. However, we found complete stability of the label at least over this 90 d interval (Fig. 3B,C).
Figure 3.
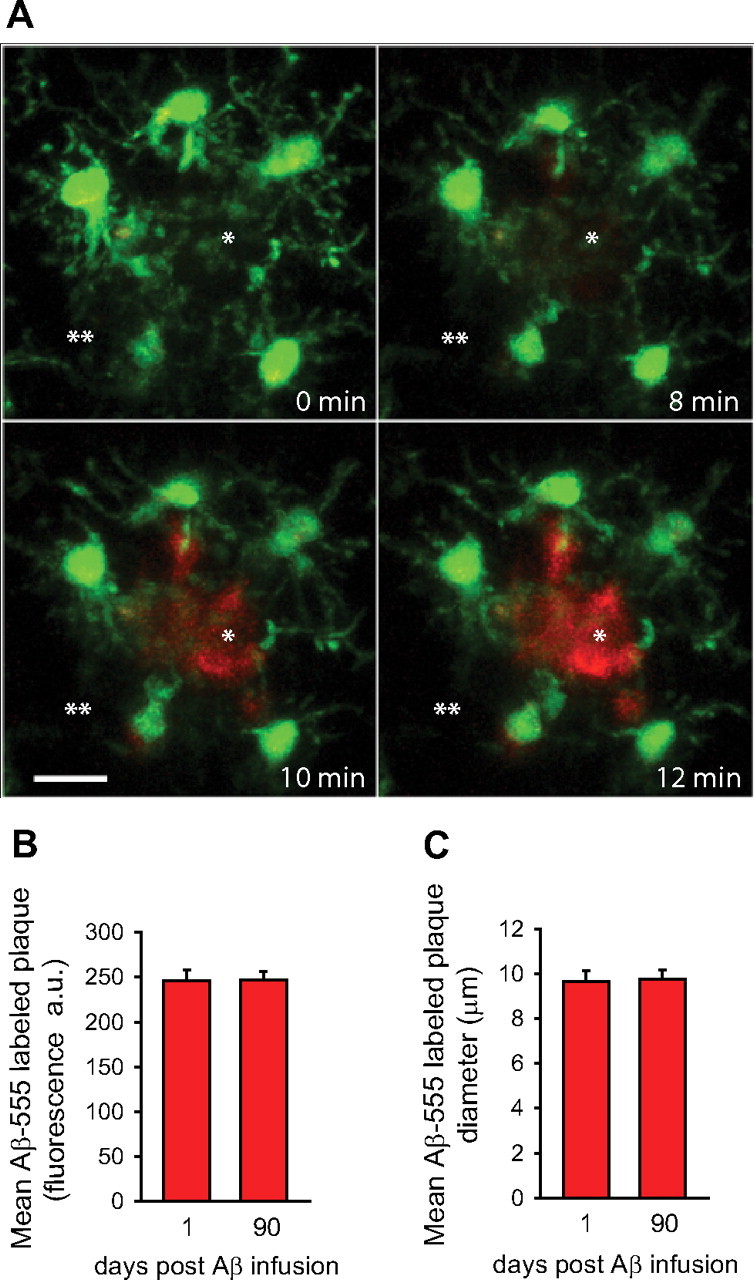
Exogenous fluorescent Aβ42 binds to plaques with high affinity and remains stably bound for several months. A, HiLyte-555-conjugated soluble Aβ42 was infused through the subarachnoid space, and time-lapse two-photon imaging was obtained during the infusion. A fibrillar plaque (asterisk) was identified by the stereotypical arrangement of surrounding microglia (GFP, green) in a CRND8/CX3CR1+/− mouse and imaged every 2 min. The image shows very rapid binding of Aβ42 to a fibrillar plaque surrounded by microglia (green). The fluorescence intensity outside of the plaque area is very low, suggesting rapid clearance of unbound Aβ42 through the BBB. Quantification of confocal images obtained from brain slices in HiLyte-555 Aβ42-infused mice shows that the average fluorescence intensity (B) and average diameter (C) of labeled plaques in age-matched mice infused 1 or 90 d before killing were virtually identical, demonstrating the lack of any significant removal of recently fibrillized Aβ (n = 3 mice and >200 plaques per time point; p = 0.1678 and 0.8526 for fluorescence intensity and diameter, respectively).
Thus, the long-term persistence of methoxy-XO4 and Aβ42(HiLyte)-555 plaque labeling coupled with the absence of TS label inside microglia strongly suggests that little phagocytosis of fibrillar amyloid takes place in vivo.
Microglia are effective at phagocytosis of protofibrillar amyloid in vivo
To test whether microglia were capable of phagocytosis of soluble or “protofibrillar” Aβ (broadly defined in our study as Aβ deposits that do not colabel with thioflavin-S), we infused Aβ42(HiLyte)-555 into the brain via the subarachnoid space and obtained brain slices at different time intervals for confocal microscopy. Within hours of infusion, we observed strong binding of Aβ42(HiLyte)-555 to preexisting methoxy-XO4-labeled fibrillar amyloid plaques as seen by in vivo imaging. However, little Aβ42(HiLyte)-555 was observed in areas outside of plaques in the first 12 h after infusion, likely because of rapid Aβ efflux through the BBB. Beginning at ∼24 h, however, scattered fluorescent Aβ deposits became evident (Fig. 4C) possibly because of aggregation of Aβ42(HiLyte)-555 that failed to be cleared through the BBB or because of binding to preexisting protofibrillar aggregates. Many of these early Aβ deposits outside of microglia were likely to be located in the extracellular space given that they did not colocalize with astrocytes either (Fig. 4F; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, at this early time point, a substantial number of deposits were already observed inside microglia within vesicular compartments consistent with phagolysosomes (Frackowiak et al., 1992) (Fig. 4A,C; supplemental Movie 1, available at www.jneurosci.org as supplemental material).
Figure 4.
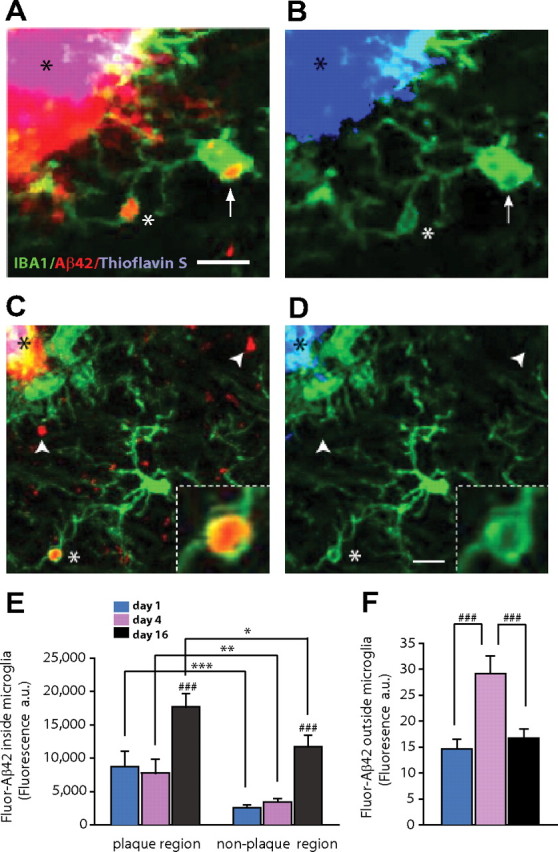
Microglia are effective at phagocytosis of noncongophilic amyloid aggregates in vivo. A, Confocal images of brain slices obtained 4 d after in vivo subarachnoid injection of fluorescently labeled-Aβ42 (red) in CRND8/CX3CR1+/− mice. IBA-1-labeled microglia (green) near amyloid plaques have prominent vesicular structures inside their soma (arrow) and processes (white asterisk) that contain fluorescent Aβ42 (red). Also notice the strong binding of Aβ42 to amyloid plaques (red, black asterisk). B, Thioflavin-S strongly labels the same amyloid plaque (black asterisk). However, despite maximally increasing the blue channel intensity, no thioflavin-S fluorescence can be detected within microglia vesicles that contained Aβ42 fluorescent deposits. C, At 4 d after Aβ42 infusion, most Aβ deposits (white arrowheads) appeared outside microglia, but a substantial number of Fluor-Aβ42 aggregates were already engulfed and found within microglia processes and cell bodies (white asterisk; inset). D, Increasing the blue channel brightness to saturation demonstrates that neither engulfed nor extracellular Fluor-Aβ42 deposits colabeled with thioflavin-S, suggesting that these are newly formed protofibrillar Aβ deposits. Scale bar, 5 μm. E, Quantification of Aβ42 fluorescence (red) shows a greater Aβ42 content within microglia located near the plaque edge (within a 25 μm radius) regardless of the postinfusion interval (***p < 0.001, **p < 0.01, *p < 0.05, two-tailed Student's t test). Sixteen days after infusion, an approximately twofold increase in Aβ42 was observed inside microglia (###p < 0.001, two-tailed Student's t test; n = 39–83 microglia from 3 mice per postinfusion time point; values are expressed as mean ± SEM). F, Fluorescence accumulation outside of microglia undergoes an approximately twofold increase 4 d after infusion but declines again at 16 d (###p < 0.001, two-tailed Student's t test; n = 3 mice per postinfusion time point; values are expressed as mean ± SEM).
Quantification of Aβ42(HiLyte)-555 fluorescence within individual microglia showed an approximately threefold rise 16 d after subarachnoid infusion (Fig. 4E). Concurrent with the increase in fluorescence inside microglia, at day 16 there was a large decrease in the number and intensity of fluorescent aggregates outside microglia (Fig. 4F), suggesting that these newly formed Aβ deposits were being phagocytosed. Virtually none of these Aβ42(HiLyte)-555 accumulations colabeled with TS (Fig. 4B,D), suggesting that these deposits were composed of material in a protofibrillar Aβ conformation. Together, these data indicate that microglia exhibit an increasing capacity for Aβ phagocytosis as it gradually adopts protofibrillar conformations but fail to phagocytose fibrillar deposits that have adopted a β-pleated sheet conformation.
We then aimed at determining whether microglia, in addition to exogenously administered Aβ42, can also uptake endogenously produced amyloid. We obtained high-resolution confocal images of brain slices from CRND8 mice stained with A11 and 4G8 antibodies, which detect oligomeric and other protofibrillar conformations, respectively (Lesné et al., 2006; Kayed et al., 2007). We found bright clusters of A11- and 4G8-immunoreactive material that did not colabel with TS, mostly inside LAMP1-immunoreactive vesicles inside microglia (Fig. 5A–D; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Thus, endogenously produced oligomeric and protofibrillar amyloid but not thioflavin-S-positive fibrillar material are efficiently phagocytosed by microglia and may be targeted for degradation given their presence within phagolysosomes.
Figure 5.
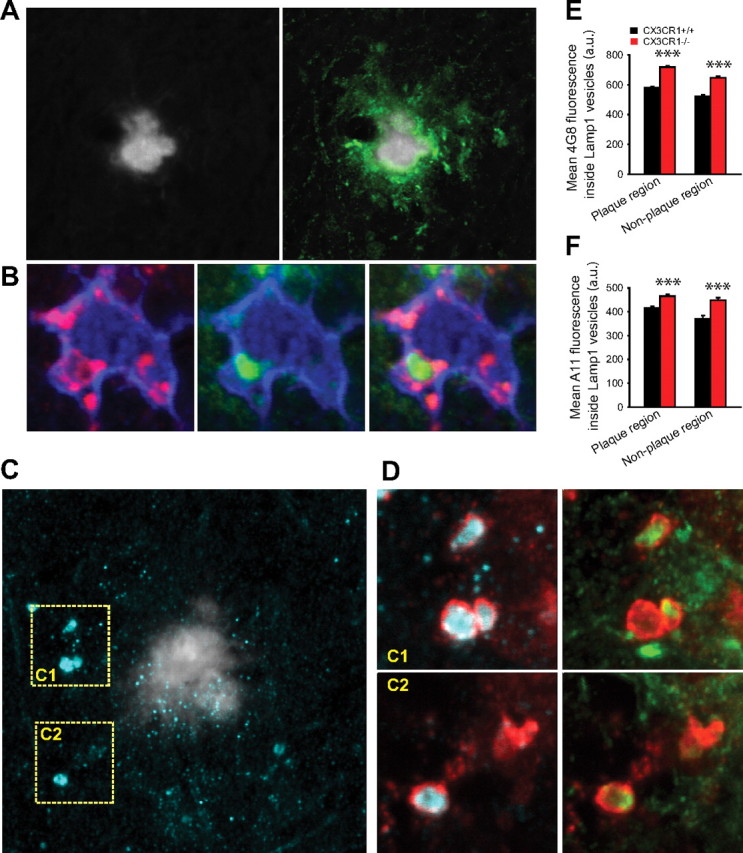
CX3CR1-deficient microglia have an enhanced capacity for protofibrillar Aβ phagocytosis in vivo. A, Confocal image shows a thioflavin-S fibrillar plaque (gray) surrounded by a large halo of 4G8-immunoreactive protofibrillar Aβ (green). B, High-resolution confocal image of a plaque-associated IBA1-labeled microglia (blue) containing a 4G8-immunoreactive Aβ aggregate (green) within a LAMP1-immunoreactive phagolysosome (red). C, Left panel, Thioflavin-S-labeled fibrillar plaque (gray) surrounded by abundant A11-immunoreactive oligomeric Aβ (cyan). D, Insets C1 and C2 correspond to dotted squares in Figure 4C. Large LAMP1-labeled phagolysosomes (red) (which are exclusively present in microglia) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) contain abundant A11-immunoreactive oligomeric (cyan) and 4G8-immunoreactive protofibrillar Aβ. E, F, Quantification of 4G8 and A11 fluorescence inside LAMP1-immunoreactive phagolysosomes near (<25 μm) and away from fibrillar plaques. Microglial phagolysosomes in CRND8/CX3CR1−/− mice exhibit a significant increase in 4G8 and A11 content in both plaque and nonplaque regions compared with CRND8/CX3CR1+/+ mice (***p < 0.001; values are expressed as mean ± SEM; n = 3 mice, 180 plaques, and >4000 lysosomes per genotype).
CX3CR1-deficient microglia have an enhanced capacity for Aβ phagocytosis
To explore the possibility that the disparities we observed in brain amyloid accumulation between CX3CR1 genotypes could be explained by differential microglial phagocytic capacity, we quantified A11 and 4G8 immunofluorescence within LAMP1-labeled phagolysosomes in microglia (Fig. 5A–F). We found a significantly increased A11 and 4G8 immunofluorescence in phagolysosomes from CRND8/CX3CR1−/− mice compared with wild-type littermates, suggesting that CX3CR1−/− microglia have an increased capacity to phagocytose endogenously produced amyloid.
To further prove that the increase in protofibrillar Aβ observed within phagolysosomes in vivo in CX3CR1-deficient mice was the result of increased phagocytosis rather than decreased degradation, we performed an in vitro phagocytosis assay. We generated dissociated cortical cell cultures containing a mixture of CX3CR1−/− and CX3CR1+/+ microglia and incubated them for 2 h with either Aβ42(HiLyte)-555 or 2 μm fluorescent microspheres. After incubation, we performed quantitative imaging and compared the average fluorescence intensity within microglia of both genotypes (Fig. 6A–D). We found that CX3CR1−/− microglia had a greater capacity to uptake both Aβ peptides and microspheres, suggesting an overall enhanced phagocytic activity. Preincubation with anti-CX3CR1 neutralizing antibody 2 h before the phagocytosis assay led to increased microsphere phagocytosis in CX3CR1+/+ microglia but not those lacking CX3CR1 (Fig. 6E), demonstrating rapid and specific modulation of phagocytosis by the reduction of CX3CR1 activity. The fact that microsphere phagocytosis was increased demonstrates that differences in Aβ within microglia are not likely attributable to intracellular degradation but rather uptake differences. It also indicates that the increased phagocytic capacity is not specific to amyloid but a general property of CX3CR1−/− microglia.
Figure 6.

CX3CR1 deficiency enhances microglia phagocytosis of Aβ peptides and microspheres in vitro. A, B, Dissociated brain cell cultures containing a mixture of CX3CR1+/+ microglia (recognized by lack of GFP label and presence of IBA-1 immunoreactivity; yellow arrow) and CX3CR1−/− microglia (both IBA1 and GFP labeling is present; white arrowhead). After a 2 h incubation with either 2 μm fluorescent microspheres (A) or fluorescence-conjugated Aβ42 (B), cells were imaged and the total amount of fluorescence within microglia of both genotypes was quantified. Scale bar, 10 μm. C, D, Fluorescence intensity within individual microglia showed a 57.7% (microsphere) and 34.7% (Aβ42 peptide) greater uptake, respectively, in CX3CR1−/− than in CX3CR1+/+ microglia (***p < 0.001, two-tailed Student's t test; values are expressed as mean ± SEM; n = 600–967 cells pooled from 3–4 postnatal day 1 pups per group). E, Pretreatment with anti-CX3CR1 neutralizing antibody 2 h before assay significantly increased microsphere uptake in CX3CR1+/+ but not in CX3CR1−/− microglia (***p < 0.001, two-tailed Student's t test; values are expressed as mean ± SEM; n = 706–908 cells per group).
CX3CR1 deficiency is associated with increased microglia proliferation and clustering around fibrillar amyloid plaques
To examine additional cellular mechanisms that might explain differences in Aβ accumulation between CX3CR1 genotypes, we analyzed the spatial relationship between amyloid plaques of similar size and their surrounding microglia. We found a greater average number of microglia within a 25 μm radius of the plaque edge in 5-month-old CRND8/CX3CR1−/− mice than in wild-type littermates (Fig. 7A,B). Several mechanisms could explain the observed difference in microglia number such as differential proliferation, migration, and/or retention rates.
Figure 7.
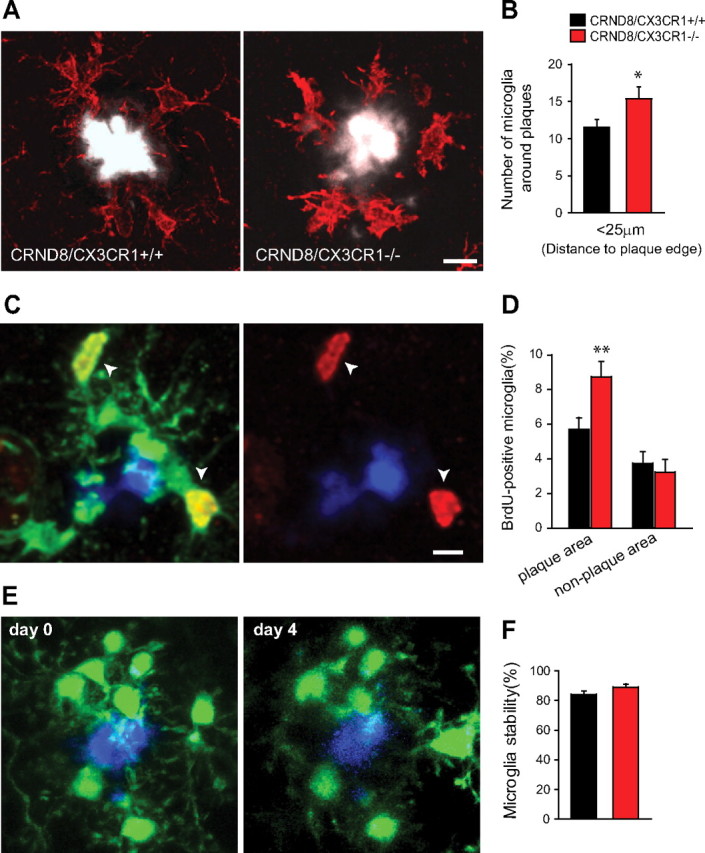
Microglia are more abundant around plaques in CX3CR1-deficient mice because of increased proliferation. A, Confocal images show greater number of IBA1-labeled microglia (red) around plaques of similar size (white) in 5-month-old CRND8/CX3CR1−/− mice than in wild-type littermate. Scale bar, 10 μm. B, Quantification of microglia cell bodies within a 25 μm radius of the plaque edge shows a greater number in CRND8/CX3CR1−/− mice than wild-type littermates (*p < 0.05, two-tailed Student's t test; n = 5 animals per genotype; values are expressed as mean ± SEM). C, Confocal image shows several BrdU-labeled (white arrows) IBA1-immunoreactive microglia (green) around an amyloid plaque (blue). Scale bar, 10 μm. D, The proportion of BrdU-labeled microglia within a 25 μm radius from plaques was significantly greater in CRND8/CX3CR1−/− than wild-type littermates (p < 0.01, two-tailed Student's t test; n = 300 and 329 plaques from 3 mice per genotype). There was no significant difference in the percentage of BrdU-labeled microglia in areas distant from plaques (p = 0.64, two-tailed Student's t test; n = 3 mice and ∼1500 microglia per genotype). E, Time-lapse two-photon image of microglia (green) and a methoxy-XO4-labeled amyloid plaque (blue) over a 4 d interval. The majority of microglia in the immediate vicinity of plaques appears stable. F, Quantification of changes in microglia number/position demonstrates no difference between CX3CR1 genotypes at least over this short time interval of 4 d (p = 0.11 for both groups, two-tailed t test; n = 6–7 mice and 67–69 plaques for CRND8/CX3CR1+/− and CRND8/CX3CR1−/−, respectively; values are expressed as mean ± SEM).
To examine the extent of microglia proliferation, we administered daily injections of BrdU for 2 weeks in 5-month-old mice. We quantified the number of BrdU-labeled microglia located in the immediate vicinity of plaques in mice of both CX3CR1 genotypes. Interestingly, we found that the overall proportion of microglia that were BrdU-labeled was significantly greater in CRND8/CX3CR1−/− mice than in CRND8/CX3CR1+/+ littermates (Fig. 7C,D), whereas nonplaque areas showed substantially lower overall proliferation rates and no significant differences between genotypes (Fig. 7D). Thus, increased proliferation rates around plaques are likely to explain the greater number of plaque-associated microglia in CX3CR1-deficient mice.
To determine whether migration/retention also plays a role, we used time-lapse two-photon microscopy in live mice to track the dynamics of GFP-labeled microglia around plaques. We found that CX3CR1-deficient microglia display enhanced process dynamism over short intervals (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Microglia cell bodies, however, can remain stable over months (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), but no significant difference in stability between mice of different CX3CR1 genotypes was observed (Fig. 7E,F). However, technical limitations of in vivo imaging make it unfeasible to quantify microglia cell body dynamics over longer intervals with sufficient precision. Thus, we cannot rule out that subtle variations in microglia recruitment/retention could contribute to the observed disparities in microglia number around plaques.
CX3CR1 deletion does not affect the degree of neuronal or synaptic injury around amyloid plaques
Results of studies in a variety of disease models have yielded conflicting results regarding secondary neurotoxicity in CX3CR1 mutants. For example, mice deficient in CX3CR1 were found to have enhanced neuronal death in models of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) toxicity and amyotrophic lateral sclerosis (Cardona et al., 2006), whereas CX3CR1 deficiency was neuroprotective in models of cerebral ischemia (Dénes et al., 2008) and mutant tau/APP-associated neurodegeneration (Fuhrmann et al., 2010). Since our results demonstrated that CX3CR1-deficient mice had increased numbers of microglia around amyloid plaques, we wondered whether this would lead to increased neuronal injury given that activated microglia can secrete potentially neurotoxic substances (Block et al., 2007). We thus investigated whether CX3CR1 deficiency had any effect on morphological correlates of neurotoxicity as assessed by neuronal and synaptic density in the immediate surroundings of amyloid plaques. Quantification of cells with the neuronal nuclear marker NeuN and of synaptophysin-immunoreactive puncta demonstrated no statistically significant difference between wild-type and CX3CR1-deficient mice (Fig. 8). Thus, it appears that CX3CR1 deficiency has neither neuroprotective nor neurotoxic effects around individual plaques at least in this model of Alzheimer's-like pathology. However, given that CX3CR1-deficient mice have fewer plaques, it is likely that the overall pathological burden is diminished.
Figure 8.

CX3CR1 deficiency does not alter the degree of neuronal or synaptic damage around plaques. A, Confocal images of synaptophysin-immunoreactive puncta (gray) around thioflavin-S-labeled amyloid plaques (cyan) were quantified by measuring their density at different distances from the plaque edge. B, No statistically significant difference was observed in synaptophysin density at any point up to ∼30 μm away from the plaque edge in CX3CR1-deficient mice compared with wild-type littermates (p > 0.05, two-tailed t test for all distances; n = 30 plaques from 3 5-month-old male mice per genotype; values are expressed as mean ± SEM). C, Confocal images of NeuN-immunoreactive neurons (gray) around thioflavin-S-labeled amyloid plaques (cyan) were quantified by counting the number of neuronal cell bodies present within a standardized three-dimensional volume around plaques. Scale bar, 10 μm. D, No statistically significant difference was observed in the density of NeuN-immunoreactive neurons in CX3CR1-deficient mice compared with wild-type littermates (p > 0.05, two-tailed t test; 3 mice per genotype and 23 plaques; values are expressed as mean ± SEM).
Discussion
Our results provide new insights into the role of microglia in the pathogenesis of Alzheimer's disease in vivo. We show that deletion of the fractalkine receptor CX3CR1, which in the brain is specifically expressed in microglia, leads to a significant reduction in the total levels of brain Aβ and in the number of amyloid plaques in a mouse model of AD. This demonstrates that microglia play an important role in regulating brain amyloid levels and deposition.
The effect of CX3CR1 deletion on brain amyloid was mediated by a general enhancement of microglia phagocytic ability as demonstrated both in vitro and in vivo. In addition, we also found that, with CX3CR1 deficiency, microglia proliferation near plaques was increased leading to a greater density. This may further enhance the global brain capacity for Aβ clearance.
However, in contrast to recent imaging studies (Bolmont et al., 2008), we found that microglia, although highly effective at phagocytosis of oligomeric and protofibrillar Aβ, were incapable of removing fibrillar amyloid from preexisting plaques in vivo. Thus, microglia may only be able to remove fibrillar amyloid from plaques during heightened levels of activation such as in injury models (DiCarlo et al., 2001) or after Aβ immunization (Bacskai et al., 2002). Our study also contrasts with recent studies of microglia ablation showing no role for these cells in brain amyloid plaque regulation (Grathwohl et al., 2009). Ablation is likely to induce marked astrocyte activation and could turn these cells into much more effective phagocytes, which could compensate for the loss of microglia and thus explain the apparent lack of effect of microglia ablation on Aβ levels.
Although we define microglia uptake of Aβ as “phagocytosis,” the precise mechanism of Aβ uptake is unclear and it has been proposed that microglia can also use fluid-phase macropinocytosis to clear Aβ (Mandrekar et al., 2009). However, given that CX3CR1 deletion is able to modulate phagocytosis of polystyrene microspheres, we assume that the disparity in Aβ uptake between genotypes is mostly attributable to differential phagocytic capacity rather than macropinocytosis. Ongoing microglial phagocytosis around growing fibrillar deposits could reduce levels of oligomeric and protofibrillar Aβ, which may change the equilibrium between monomers, oligomers, and fibrils, altering the process of nucleation-dependent polymerization that leads to fibrillar plaque formation (Harper and Lansbury, 1997; Walsh et al., 1999). This not only may reduce the number of amyloid deposits but the overall concentration of oligomeric and protofibrillar Aβ, which could have important implications in the progression of AD given the enhanced toxicity of these smaller Aβ conformations (Lambert et al., 1998; Walsh et al., 1999). Thus, it is likely that the main role played by microglia is to ameliorate the formation of new fibrillar plaques rather than to remove preexisting ones, with the most critical effect likely being at the earliest stages of plaque deposition.
In addition to microglia, peripheral macrophages have been shown to be capable of Aβ phagocytosis in AD models (Simard et al., 2006). Although currently it is thought that macrophages may normally not enter the brain in significant numbers in the absence of blood–brain barrier disruption (Ajami et al., 2007; Mildner et al., 2007), fractalkine/CX3CR1 signaling may also have chemotactic effects on macrophages (Imai et al., 1997). Thus, we cannot rule out that some of the effects we observed are at least partly attributable to peripheral macrophages entering the brain.
Despite increased clustering of activated microglia in CX3CR1-deficient mice, we did not observe any difference in neuronal or synaptic density around plaques. This suggests that plaque-associated microglia do not exert a significant neuroprotective or neurotoxic “bystander” effect. Microglia have been shown to exacerbate neuronal death mainly in acute and subacute injury models in which they are fully activated (Wu et al., 2000; Cardona et al., 2006; Block et al., 2007). However, in slowly progressive neurodegenerative disorders, evidence of neurotoxicity or neuroprotection has been more conflicting. Thus, it appears that microglia activation is not an all-or-none process and its net effect may depend on the particular pathological scenario (Hanisch and Kettenmann, 2007). In the case of CX3CR1, our results indicate that reducing microglia activation or phagocytosis could have deleterious effects in AD. This may at least partly explain why recent attempts at prevention of AD with nonsteroidal antiinflammatory therapy have been unsuccessful or even counterproductive (Martin et al., 2008).
Our findings support the concept that fractalkine through its receptor CX3CR1 (Harrison et al., 1998) acts as a repressor of microglia activation (Cardona et al., 2006). This would explain the increased proliferation and clustering around plaques with CX3CR1 deletion. Although the increased number of microglia around amyloid deposits would likely lead to an overall greater phagocytic capacity, the precise mechanism through which CX3CR1 affects phagocytosis at the single microglia level is less clear. A large number of pattern recognition cell surface receptors in microglia such as the Toll-like family, RAGE (receptor for advanced glycation end-products), and scavenger receptors have been implicated in the recognition of amyloid oligomers and fibrils (Salminen et al., 2009). Signal transduction pathways associated with these receptors are known to modulate phagocytosis (Tahara et al., 2006; Richard et al., 2008) and cytokine production (Reed-Geaghan et al., 2009) after amyloid exposure. Furthermore, cross talk can occur between chemokine and pattern recognition receptor signaling pathways leading to functional changes in phagocytes (Kishore et al., 2005; Alves-Filho et al., 2009). This receptor transactivation could partly explain the effect of CX3CR1 on phagocytosis. In addition, CX3CR1 deletion has been shown to directly affect cytokine production (Cardona et al., 2006). Thus, CX3CR1 modulation of phagocytosis may involve the indirect effect of some proinflammatory cytokines that can attenuate phagocytosis (Koenigsknecht-Talboo and Landreth, 2005). However, our experiment showing a rapid increase in phagocytic ability after incubation with anti-CX3CR1 neutralizing antibody suggests that CX3CR1 may also exert direct effects on microglia phagocytosis.
Expression of various chemokines has been shown to be increased in the aging brain (Mo et al., 2003) and plasma levels of fractalkine can be increased in AD patients (Kim et al., 2008). Increased fractalkine signaling in aging could in theory restrain microglia and predispose individuals to amyloid deposition and toxicity. Although our results were obtained in a model in which CX3CR1 was developmentally rather than conditionally depleted and should thus be cautiously extrapolated, attempts at treating a variety of conditions with fractalkine/CX3CR1 agonists or other microglia inhibitors should take into account their potential for exacerbating AD. However, selective inhibition of fractalkine or CX3CR1 in the brain, which could modulate microglia proliferation and phagocytosis, may represent a novel therapeutic approach for AD.
Footnotes
This work was supported by National Institutes of Health–National Institute on Aging Grant R01 AG027855 (J.G.), Ellison Medical Foundation New Scholar Award in Aging (J.G.), and Dana Foundation Brain and Immunoimaging Award (J.G.).
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Alves-Filho JC, Freitas A, Souto FO, Spiller F, Paula-Neto H, Silva JS, Gazzinelli RT, Teixeira MM, Ferreira SH, Cunha FQ. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci U S A. 2009;106:4018–4023. doi: 10.1073/pnas.0900196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT. Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J Neurosci. 2002;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- Dénes A, Ferenczi S, Halász J, Környei Z, Kovács KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–1012. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Baird A, Cole GM. Effects of injected Alzheimer beta-amyloid cores in rat brain. Proc Natl Acad Sci U S A. 1991;88:8362–8366. doi: 10.1073/pnas.88.19.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frautschy SA, Cole GM, Baird A. Phagocytosis and deposition of vascular beta-amyloid in rat brains injected with Alzheimer beta-amyloid. Am J Pathol. 1992;140:1389–1399. [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TS, Lim HK, Lee JY, Kim DJ, Park S, Lee C, Lee CU. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer's disease. Neurosci Lett. 2008;436:196–200. doi: 10.1016/j.neulet.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Kishore SP, Bungum MK, Platt JL, Brunn GJ. Selective suppression of Toll-like receptor 4 activation by chemokine receptor 4. FEBS Lett. 2005;579:699–704. doi: 10.1016/j.febslet.2004.12.047. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE. Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J Neurosci. 2009;29:4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, Craft S, Evans D, Green R, Mullan M. Cognitive function over time in the Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65:896–905. doi: 10.1001/archneur.2008.65.7.nct70006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Mo R, Chen J, Han Y, Bueno-Cannizares C, Misek DE, Lescure PA, Hanash S, Yung RL. T cell chemokine receptor expression in aging. J Immunol. 2003;170:895–904. doi: 10.4049/jimmunol.170.2.895. [DOI] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and Toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard KL, Filali M, Préfontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β1–42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci. 2008;28:5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer's disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol. 2009;87:181–194. doi: 10.1016/j.pneurobio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Stalder M, Deller T, Staufenbiel M, Jucker M. 3D-reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging. 2001;22:427–434. doi: 10.1016/s0197-4580(01)00209-3. [DOI] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of Toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- Wu Q, Combs C, Cannady SB, Geldmacher DS, Herrup K. Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging. 2000;21:797–806. doi: 10.1016/s0197-4580(00)00219-0. [DOI] [PubMed] [Google Scholar]