

Abstract
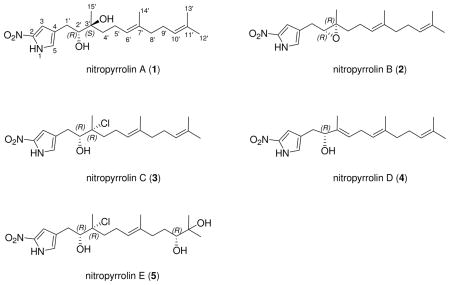
Five new farnesyl-α-nitropyrroles, nitropyrrolins A–E (1–5), were isolated from the saline culture of the marine actinomycete strain CNQ-509. This strain belongs to the “MAR4” group of marine actinomycetes, which have been demonstrated to be a rich source of hybrid isoprenoid secondary metabolites. The structures of the nitropyrrolins are composed of α-nitropyrroles with functionalized farnesyl groups at the C-4 position. These compounds are the first examples of naturally-occurring terpenyl-α-nitropyrroles. Chemical modifications, including one-step acetonide formation from an epoxide, and application of the modified Mosher method, provided the full stereostructures and absolute configurations of these compounds. Several of the nitropyrrolins, nitropyrrolin D in particular, are cytotoxic toward HCT-116 human colon carcinoma cells, but show weak to little antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA).
As part as our continuing interest in the chemistry and biomedical potential of sediment-derived marine actinomycetes, we have isolated a variety of new actinomycete taxa, which show the capacity to produce unique secondary metabolites.1 These taxa include the chemically prolific genera Salinispora2 and “Marinispora”,3 members of which produce new structural classes of secondary metabolites including salinosporamide A,4 a potent proteasome inhibitor that recently completed phase I clinical trial for the treatment of cancer.5 Another group of marine-derived actinomycetes, designated as MAR 4, is proving to be of significant chemical interest.1 To date, at least 15 strains belonging to this group have been isolated from diverse marine sediments. Previous chemical studies of cultured MAR4 strains led to the discovery of a broad range of polyketide-terpenoid secondary metabolites including marinone,6 compounds in the napyradiomycin series7 and azamerone.8 Further chemical analysis of the cultured MAR4 strain CNQ-509, isolated from a marine sediment sample collected off La Jolla, CA, has led to the discovery of a new set of hybrid polyketide-terpenoid metabolites composed of sesquiterpenoid and α-nitropyrrole components. These compounds, nitropyrrolins A–E (1–5), are unusual examples illustrating rare pyrrole nitration in the α position with linear sesquiterpenoid substitution. The structures of these new compounds, including their absolute configurations, were established by combined spectroscopic and chemical methods. The absolute configuration of the epoxide functionality in compound 2 was determined via a one step acetonide formation from the epoxide, a method which when coupled with Mosher ester analysis leads to the assignment of the absolute configuration of trisubstituted epoxides. In this paper we describe this new method as well as the structure elucidation of five new sesquiterpenoid α-nitropyrroles.
Results and Discussion
The MAR 4 strain CNQ-509 was isolated from a marine sediment collected at a depth of 44 m offshore of La Jolla, California. The strain was cultured in 8 × 4 L volumes for seven days and then extracted using Amberlite XAD-7 resin. The resin was filtered, eluted with acetone, and the extraction solvent concentrated under reduced pressure. The extract was subjected to a diversity of HPLC-based purification steps to afford pure samples of nitropyrrolins A–E (1–5).
Nitropyrrolin A (1) was obtained as a light yellow oil that analyzed for the molecular formula C19H30N2O4 by interpretation of HR FAB-MS (obsd [M–H]− at m/z 349.2124) and NMR data. The UV spectrum of 1 displayed absorption bands at 350 nm, indicating the presence of a significant chromophore in the molecule. Two substructures, a linear sesquiterpenoid chain and a pyrrole moiety, were assigned by analyses of 1H, 13C, 1H-1H COSY, HSQC and HMBC NMR spectroscopic data recorded in CDCl3 (Tables 1, 2 and Supporting Information). The linear sesquiterpenoid chain was defined by analysis of NMR data starting from the terminal olefinic gem dimethyl protons (H3-12′ and H3-13′) at δH 1.69 and 1.61 that showed HMBC correlations to one another, to C-10′ (δC 124.1), and to C-11′ (δC 131.6). The proton signal (δH 5.10) derived from H-10′ showed a COSY correlation to an aliphatic methylene proton signal at δH 2.09 (H2-9′), which in turn showed a COSY correlation to another aliphatic methylene proton signal at δH 2.01 (H2-8′). These NMR data established a terminal isoprene unit, which was expanded to a sequential isoprene unit by the observation of HMBC correlations from the methyl singlet of H3-14′ (δH 1.65) to C-8′ (δC 39.7), and from an olefinic proton H-6′ (δH 5.18) to C-14′ (δC 16.1), and by a series of COSY correlations between the H-6′ signal and two aliphatic methylene protons signals at δH 2.20, 2.14, 1.73 and 1.49 (H2-5′ and H2-4′). A remaining isoprenoid fragment was constructed starting from the last methyl proton at δH 1.27 (H3-15′), which showed HMBC correlations to a carbinol carbon (C-2′), an oxygenated quaternary carbon (C-3′) and a methylene carbon (C-4′) at δC 78.3, 74.6 and 36.2, respectively. In addition, the H-2′ proton signal (δH 3.58) showed a COSY correlation to the diastereotopic methylene proton signals H2-1′ observed at δH 2.73 and 2.53. Because the 13C NMR chemical shifts of C-2′ (δC 78.3) and C-3′ (δC 74.6) in 1 (CDCl3) were very similar to those of an anti-α,β-dihydroxylated terpenoid structural unit (δC 78.4 and 74.8, respectively), previously reported in the literature,9 the hydroxy groups were positioned at C-2′ and C-3′. The geometry of C-6′-C-7′ double bond was assigned an E configuration on the basis of a NOESY correlation between H-6′ and H2-8′. This is supported by the 13C chemical shifts of C-14′ and C-8′ (δC 16.1 and 39.7), while their chemical shifts would be approximately δC 23.5 and 40.0, in a Z double bond.9 In addition, the assignment of two terminal methyl groups, C-12′ and C-13′, was confirmed by a NOESY correlation between H-10′ and H3-12′, coupled with a lack of NOESY correlation between H-10′ and H3-13′.
Table 1.
NMR Spectroscopic Data for Nitropyrrolins A-E (1–5) [500 MHz, CDCl3 (δ 7.27), δH mult (J in Hz)]
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| 1 | 9.49, br s | 9.51, br s | 9.48, br s | 9.38, br s | 9.58, br s |
| 3 | 7.06, br s | 7.03, br s | 7.07, br s | 7.01, br s | 7.08, br s |
| 5 | 6.90, br s | 6.87, br s | 6.92, br s | 6.84, br s | 6.92, br |
| 1′ | 2.73, br dd (15.0, 2.0) | 2.71, dd (15.0, 6.0) | 2.93, dd (15.0, 1.5) | 2.72, ma | 2.94, dd (15.0, 2.0) |
| 2.53, dd (15.0, 10.5) | 2.68, dd (15.0, 6.0) | 2.60, dd (15.0, 10.5) | 2.60, dd (15.0, 10.5) | ||
| 2′ | 3.58, br dd (10.5, 2.0) | 2.94, dd (6.0, 6.0) | 3.75, dd (10.5, 1.5) | 4.18, dd (6.5, 6.5) | 3.76, br d (10.5) |
| 4′ | 1.73, br ddd (14.0, 10.5, 6.0) | 1.70, ddd (13.5, 8.0, 6.0) | 2.00, m | 5.38, dd (7.0, 7.0) | 1.98, ddd (14.0, 11.0, 5.0) |
| 1.49, br ddd (14.0, 10.5, 6.0) | 1.49, ddd (13.5, 9.5, 6.5) | 1.75, ddd (14.0, 11.0, 5.0) | 1.72, ddd (14.0, 11.0, 5.0) | ||
| 5′ | 2.20, m | 2.11, m | 2.28, m | 2.73, ma | 2.29, ma |
| 2.14, m | 2.19, m | 2.19, ma | |||
| 6′ | 5.18, br t (7.0) | 5.08, ma | 5.15, td (7.0, 1.0) | 5.08, ma | 5.22, dd (7.0, 7.0) |
| 8′ | 2.01, m | 2.05, m | 1.99, m | 1.99, m | 2.27, ma |
| 2.09, ddd (15.0, 8.0, 8.0) | |||||
| 9′ | 2.09, m | 1.96, br dd (8.0, 7.0) | 2.08, dd (7.5, 7.5) | 2.07, m | 1.60, ma |
| 1.42, dddd (14.0, 10.0. 8.0, 5.0) | |||||
| 10′ | 5.10, tq (7.0, 1.0) | 5.08, ma | 5.09, tq (7.0, 1.5) | 5.09, ma | 3.36, dd (10.0, 1.5) |
| 12′ | 1.69, br d (1.0) | 1.68, br d (1.0) | 1.69, s | 1.69, s | 1.22, s |
| 13′ | 1.61, s | 1.60, s | 1.61, s | 1.61, s | 1.18, s |
| 14′ | 1.65, s | 1.60, s | 1.63, s | 1.63, s | 1.66, s |
| 15′ | 1.27, s | 1.38, s | 1.64, s | 1.71, s | 1.62, s |
The multiplicity of this signal was unresolved due to peak overlapping and the chemical shift was assigned by interpretation of HSQC and HMBC spectroscopic data.
Table 2.
13C NMR Spectroscopic Data for Nitropyrrolins A-E (1–5) [125 MHz, CDCl3 (δ 77.0)]
| C# | A (1) | B (2) | C (3) | D (4) | E (5) |
|---|---|---|---|---|---|
| 2 | 137.5 | 137.4 | 137.5 | 137.5 | 137.4 |
| 3 | 111.2 | 110.9 | 111.3 | 111.5 | 111.5 |
| 4 | 124.3 | 123.0 | 124.0 | 123.7 | 123.9 |
| 5 | 122.2 | 121.6 | 122.2 | 122.1 | 122.2 |
| 1′ | 28.8 | 26.7 | 29.2 | 32.7 | 29.8 |
| 2′ | 78.3 | 63.1 | 78.4 | 77.6 | 78.3 |
| 3′ | 74.6 | 61.5 | 78.0 | 136.0 | 77.9 |
| 4′ | 36.2 | 38.6 | 39.6 | 126.3 | 39.9 |
| 5′ | 22.0 | 23.7 | 23.0 | 26.9 | 23.5 |
| 6′ | 124.1 | 123.3 | 123.0 | 121.9 | 123.9 |
| 7′ | 136.0 | 135.8 | 136.3 | 135.9 | 136.1 |
| 8′ | 39.7 | 26.7 | 39.7 | 39.6 | 36.9 |
| 9′ | 26.6 | 39.7 | 26.6 | 26.7 | 29.9 |
| 10′ | 124.1 | 124.2 | 124.1 | 124.2 | 78.2 |
| 11′ | 131.6 | 131.6 | 131.5 | 131.5 | 73.2 |
| 12′ | 25.7 | 25.8 | 25.7 | 25.7 | 26.2 |
| 13′ | 17.7 | 17.8 | 17.7 | 17.7 | 23.6 |
| 14′ | 16.1 | 16.1 | 16.1 | 16.1 | 16.1 |
| 15′ | 23.4 | 16.8 | 25.6 | 11.8 | 25.9 |
Assignments were made on the basis of DEPT and HMBC/HMQC experimental data.
Other characteristic features of the 1H NMR spectrum of 1 were the presence of two aromatic protons at δH 7.06 (1H, br s) and 6.90 (1H, br s), and an exchangeable proton at δH 9.49 (1H, br s). The 1H-1H COSY spectrum showed the cross correlations between these three proton signals, indicating the presence of the NH group in the aromatic ring. HSQC NMR data showed that the aromatic proton signals at δH 7.06 and 6.90 (H-3 and H-5) correlated with 13C NMR bands at δC 111.2 and 122.2, respectively. In the HMBC spectrum of 1, proton H-5′ correlated to two unassigned aromatic quaternary carbons C-2 (δC 137.5) and C-4 (δC 124.3), and the H-3 signal showed an HMBC correlation with C-2. Furthermore, the chemical shift of C-2′, the UV absorption features of 1, and the remaining unaccounted for elements (NO2), suggested that the aromatic ring of 1 was a disubstituted nitropyrrole. Strong IR absorption at 1504 cm−1 also supported the presence of a nitro group in 1. Confirming this assignment, the UV spectrum of 1 showed a maximum absorption at 350 nm characteristic for an α-nitropyrrole, while the UV absorption band for β-nitropyrroles has been recorded at 315 nm.10 HMBC correlations from the diastereotopic methylene protons at δH 2.73 and 2.53 (H2-1′) to the C-3, C-4 and C-5 signals allowed the diol-substituted farnesyl chain of 1 to be connected with the α-nitropyrrole group through C-1′ and C-4. Finally, the γ-sesquiterpenyl-α-nitropyrrole structure of 1 was confirmed by the comparison of NMR and UV data of 1 with those of synthetic γ-farnesyl-α-nitropyrrole and β-farnesyl-α-nitropyrrole. These two isomers were synthesized for comparison to 1. A mixture of pyrrole (7.5 mmol), farnesyl bromide (2.8 mmol), and zinc powder (650 mg) in THF (10 mL) was stirred at room temperature for 10 h to afford 3-farnesyl-pyrrole (6) (270 mg). Nitration of 6 (0.08 mmol) in acetic anhydride (1.0 mL) with a solution of HNO3 (0.08 mmol) in acetic anhydride (0.5 mL), followed by chromatographic purification, yielded γ-farnesyl-α-nitropyrrole (7) (5.0 mg) and β-farnesyl-α-nitropyrrole (8) (8.7 mg) (Scheme 1). The 1H and 13C NMR signals for the nitropyrrole moiety in 1, as well as UV spectroscopic data, were identical to those of synthetic γ-farnesyl-α-nitropyrrole.
Scheme 1.

Formation of 3-farnesyl-pyrrole (6) and the nitration of 6
The absolute configurations of the two asymmetric centers of 1 were first assigned by application of the modified Mosher method11 and by interpretation of the results of a 2D-NOESY NMR experiment. Treatment of 1, in separate experiments, with (R)-(−)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (R-MTPA-Cl) and (S)-(+)-MTPA-Cl, yielded the S-Mosher ester 9a and R-Mosher ester 9b, respectively. Interpretation of the 1H NMR chemical shift differences (ΔδS-R) between 9a and 9b, applying classic Mosher ester analysis, revealed that the absolute configuration of C-2′ is R (Figure 1). The absolute configuration of the tertiary alcohol at C-3′ in 1 was then assigned by defining the relative configuration of C-2′ and C-3′. In the 2D NOESY spectrum, measured in CDCl3, the H-2′ and H2-4′ signals showed consistent NOE correlations with H3-15′ and H2-1′. In addition, the H3-15′ signal showed a NOESY correlation with H2-1′. However, there was no apparent NOE correlation between H-2′ and H2-4′ (Figure 2). These NOE data indicated that the 1,2-diol at C-2′ and C-3′ in 1 was in the erythro configuration. This configuration was further supported by the 13C chemical shifts of C-2′ and C-3′ (δC 78.3 and 74.6).9
Figure 1.

ΔδS-R 1H NMR values in pyridine-d5 for the Mosher esters 9a/9b, the Mosher esters 12a/12b, the Mosher esters 13a/13b, and the bis-Mosher esters 14a/14b.
Figure 2.

Newman Projections along the C-2′-C-3′ bonds illustrating key NOE correlations defining the rotamers of 1 (in CDCl3), 3 (in CDCl3 and DMSO-d6) and 10 (in CDCl3).
Nitropyrrolin B (2) was isolated as a light yellow oil that analyzed for the molecular formula C19H28N2O3 by HR-FAB MS data ([M-H]− at m/z 331.2029) and interpretation of combined NMR data (Tables 1, 2 and Supporting Information). The UV absorption spectrum of 2 showed an absorption band at 351 nm, which was identical with the UV data obtained from nitropyrrolin A (1). The IR spectrum of 2 was also identical to that of 1. Comprehensive analysis of 1D and 2D NMR data for 2, obtained from 1H, 13C, 1H-1H COSY, HSQC and HMBC NMR experiments, allowed complete assignment of the proton and carbon signals, leading to the confident construction of a planar structure of nitropyrrolin B. Unlike 1, the 13C NMR chemical shifts of C-2′ and C-3′ in 2 were observed at δC 63.1 and 61.5. The H-2′ proton signal was also observed at higher field (δH 2.94) than that of 1 (δH 3.58). These data, and considering the unsaturation of 2, indicated an epoxide functionality at C-2′ – C-3′ instead of the diol group as in 1.9
The relative configuration of H-2′ and H2-4′ in nitropyrrolin B (2) was determined as syn by analysis of 1D NOE data. Irradiation of proton H-2′ (δ 2.94) enhanced proton H2-4′ (δ 1.49 and 1.70) signals. The relative configuration of H2-1′ and H3-15′ of 2 was also assigned to be syn on the basis of the NOE correlation between H2-1′ proton signal and H3-15′ methyl signal. Based on these NOE analyses, the relative configuration at C-2′ and C-3′ was assigned as 2′R* 3′R* Next, the absolute configuration of the epoxide in 2 was defined by conversion of the epoxide to the diol via the intermediate acetonide followed by application of the modified Mosher method.11 Treatment of 2 in acetone with BF3-Et2O resulted in a smooth conversion of the epoxide to the acetonide 10.12 In the 2D NOESY spectrum of 10, the H-2′ proton (δH 3.91) showed a strong correlation with H3-15′ (δH 1.23) indicating that the configuration at C-3′ had been inverted to 3′S* after formation of the acetonide. Furthermore, a NOESY correlation between H-2′ and H2-4′ was not observed, supporting the configurations of H-2′ and H3-15′ in 10 (Figure 2). Treatment of 10 in methanol with excess pyridinium p-toluenesulfonate (PPTS) yielded a 2′,3′-diol, whose 1H NMR and NOESY spectra were identical with those of 1. The 1H NMR spectrum of the (S)-Mosher ester of this product was also identical with those of (S)-Mosher ester of 1 (9a). Thus, the absolute configurations at C-2′ and C-3′ in 2 were assigned as R and R, respectively.
It can be hypothesized that nitropyrrolin A (1) is biosynthesized by an epoxide hydrolase - induced ring opening of 2. Epoxide hydrolases (EHs) are ubiquitous enzymes that catalyze the hydrolysis of an epoxide to furnish the corresponding vicinal diol. In particular, EHs isolated from actinomycetes such as Rhodococcus and Nocardia sp, were reported as stereoselective biocatalysts for highly substituted epoxides.13 It thus appears that the isolation of compounds 1 and 2 from strain CNQ-509 is suggestive of the presence of an EH, which stereoselectively converts (2R,3R)-epoxides to their corresponding (2R,3S)-diols (Scheme 2). This hypothesis remains to be tested, however, in other epoxide-diol conversions.
Scheme 2.

The formation of the acetonide 10 and the diol 11 from 2.
Nitropyrrolin C (3) was isolated as a dark yellow oil, which was assigned the molecular composition C19H29ClN2O3 based on analysis of HR-FAB MS data (obsd [M-H]− at m/z 367.1791). The isotope ratio (3:1) between the [M-H]− and [M-H+2]− pseudomolecular ion peaks in the mass spectrum clearly indicated that 3 contained one chlorine atom. The UV spectrum of 3 was also identical with those from 1 and 2, indicating the presence of the α-nitropyrrole moiety. The chemical shifts of C-2′, C-3′ and C-4′ in the 13C NMR spectrum of 1 were 78.3, 74.6 and 36.2, while those of 3 were 78.4, 78.0 and 39.6, respectively. The downfield shifts of C-3′ and C-4′ signals in 13C NMR spectrum of 3 were suggestive of the chlorination at C-3′ in 3.14
The absolute configurations of the asymmetric centers in 3 were determined by using the modified Mosher’s method combined with the interpretation of data from a 1D NOE experiment. Treatment of 3 with the R and S-MTPA chlorides as before gave the (S)-MTPA ester 12a and (R)-MTPA ester (12b) via esterification of the 2′ hydroxy group in 3. Analysis of 1H NMR chemical shift differences (ΔδS-R) between 12a and 12b led to the assignment of the R-configuration at C-2′ of 3 (Figure 1). The 1D NOE spectrum of 3 in CDCl3 showed inconclusive NOE correlations (Figure 2), possibly because of the high flexibility of the sesquiterpenoid chain or the eclipsed conformation of chlorine at C-3′ and hydroxy group at C-2′ of 3 in CDCl3. However, the analysis of NOE spectroscopic data using DMSO-d6 instead of CDCl3 clearly revealed a threo configuration of the C-2′ hydroxy and the C-3′ chlorine. Irradiation of the H-2′ signal showed enhancement of the H2-4′ signals and the H3-15′ signal, while irradiation of H2-1′ resulted in only in an observable NOE correlation with H3-15′. These data established threo configurations between the hydroxy and chlorine groups, and between H-2′ and H3-15′. The absolute configurations at C-2′ and C-3′ in 3 were thus assigned as R and R.
Nitropyrrolin D (4) was isolated as a light yellow oil that analyzed for the molecular formula C19H28N2O3 by interpretation of HR-FAB MS (obsd [M-H]− at m/z 331.2023) and NMR data. The UV and IR spectra of 4 were virtually identical to those of 1. Compound 4 was also composed of a linear sesquiterpenoid chain and a nitropyrrole moiety, features that were confidently assigned by analyses of IR, UV and 1D and 2D NMR spectroscopic data. The pyrrole moiety of 4 was identical to those of compounds 1–3, while the sesquiterpenoid chain of 4 contained one additional double bond. In the 1H-1H COSY spectrum of 4, the H-6′ olefinic proton signal at δH 5.08 showed a correlation to the H2-7′ methylene protons signal at δH 2.73, which in turn showed a COSY correlation to another olefinic proton signal at δH 5.38 (H-4′). HMBC NMR correlations from H-4′ to H3-15′ and H2-2′ indicated the presence of the additional double bond in the C-3′ - C-4′ position in 4. The geometry of this olefinic bond, and that of the C-6′ - C-7′ double bond, were assigned as E on the basis of NOESY correlations between H-6′ and H2-8′, and between H-2′ and H-4′. The absolute configuration of C-2′ of 4 was assigned as R based on Mosher analysis, which showed diagnostic 1H NMR chemical shift differences (ΔδS-R) between the MTPA esters of 4 (13a and 13b) (Figure 1).
Nitropyrrolin E (5) was obtained as a light yellow oil that was assigned the molecular formula C19H31ClN2O5, by analysis of HR-FAB MS (obsd [M-H]− at m/z 401.1837) and NMR data. The isotope ratio (3:1) between the [M-H]− and [M-H+2]− pseudomolecular ion peaks in the mass spectrum clearly indicated that 5 also contained one chlorine atom. The 13C NMR chemical shifts of the pyrrole carbons and C-1′ thru C-8′ of 5 were virtually identical with those of 3, while the C-10′ and C-11′ olefinic carbon signals of 1–4 were not observed in the 13C NMR spectrum of 5. The HMBC NMR spectrum of 5 showed correlations from the terminal gem-dimethyl group at δH 1.22 and 1.18 (H3-12′ and H3-13′) to C-10′ (δC 78.2), and to C-11′ (δC 73.2). The H-10′ carbinol proton signal (δH 3.36) showed a COSY correlation to the H2-9′ methylene proton signals at δH 1.60 and 1.42. These 2D NMR spectroscopic data revealed the presence of a diol at C-10′ and C-11′ instead of the terminal olefin found in all of the other nitropyrrolins.
The absolute configurations of the three asymmetric centers in 5 were determined by application of the modified Mosher ester NMR method and interpretation of NOESY data. Treatment of 5 in pyridine with (R)-MTPA-Cl and (S)-MTPA-Cl, in the same manner as before, yielded the bis-(S)-Mosher ester 14a and bis-(R)-Mosher ester 14b, respectively (Figure 1). Analysis of 1H NMR data comparing the shifts of 14a and 14b yielded positive ΔδS-R values for H2-1′ and negative values for H2-4′ and H2-5′ resulting from the anisotropic effects of the Mosher ester group at C-2′. In addition, a negative ΔδS-R value for H3-12′ was observed, while the H2-8′ and H2-9′ proton signals showed positive differences. These data allowed the absolute configurations at C-2′ and C-10′ to be assigned as R and R, respectively. The 2D NOESY spectrum of 5 showed identical NOE correlations between H2-1′, H-2′, H2-4′ and H3-15′ as observed in 3, indicating the threo configuration between hydroxyl and chlorine groups, and the threo configuration of H-2′ and H3-15′. The absolute configurations for all of the asymmetric centers in 5 were thus assigned as R.
We evaluated the cancer cell cytotoxicities and the antibacterial activities of the nitropyrrolins A–E (1–5), as well as several synthetic derivatives, against the human colon carcinoma cell line HCT-116 and against methicillin-resistant Staphylococcus aureus (MRSA). Against HCT-116 colon carcinoma, nitropyrrolin D (4) was the most active showing IC50 = 5.7 μM. The other nitropyrrolins were far less active, while some of the synthetic derivatives showed activities between 10–30 μM (see Table 3). Curiously, the synthetic analogs 3-farnesylpyrrole and γ-farnesyl-α-nitropyrrole showed modest, but significant cytotoxicities. This trend was also observed for activity against MRSA. The nitropyrrolins showed little to no activity, but the synthetic compound 3-farnesylpyrrole was active with a modest MIC of 2.8 μg/mL.
Table 3.
Cytotoxicities and Antibacterial Activities of the Nitropyrrolins A–E (1–5) and Some Synthetic Derivatives.
| Compound | HCT-116 | MRSA Inhibition |
|---|---|---|
| IC50 (μM) | MIC (μg/mL) | |
| nitropyrrolin A (1) | 31.1 | NSA |
| nitropyrrolin B (2) | 31.0 | NSA |
| nitropyrrolin C (3) | NSA | NSA |
| nitropyrrolin D (4) | 5.7 | NSA |
| nitropyrrolin E (5) | NSA | NSA |
| 3-farnesylpyrrole | 13.7 | 2.8 |
| γ-farnesyl-α-nitropyrrole | 9.2 | NSA |
| α-nitropyrrole | NSA | NSA |
| β-nitropyrrole | NSA | NSA |
| 2-farnesylpyrrole | 24.4 | NSA |
NSA = Not Significantly Active (Inhibition observed only at >20 μg/mL)
The nitropyrrolins are hybrid isoprenoids composed of two distinct biosynthetic units, an α-nitropyrrole and a linear sesquiterpenoid chain. Secondary metabolites possessing this specific structural composition have not been previously reported. Although there are many natural products composed of terpenoid substructures joined to aromatic ring systems,14 farnesyl pyrroles are rare.15 Only two examples of pyrroloterpene natural products, the glaciapyrroles16 and pyrrolostatin,17 are known to possess a linear terpenoid chain attached to a pyrrole ring or a pyrrole-2-carboxylic acid.
In summary, the structures of the nitropyrrolins were elucidated based on the analysis of spectroscopic data, application of the modified Mosher method, and direct acetonide formation from an epoxide. The rigidity of the acetonide ring allowed the relative configuration of the trisubstituted epoxide to be easily defined by NOE measurements. Hydrolysis to the diol then allowed the absolute configurations at both carbons to be defined by the classical Mosher ester NMR method. The nitropyrrolins appear to be derived from a mixed biosynthesis involving terpenoid precursors and either amino acids or polyketides. In addition, this strain produces terpenoid compounds incorporating phenazine and naphterpin scaffolds (unpublished data) thus making it a remarkable source of diverse hybrid isoprenoid secondary metabolites. The discovery of the nitropyrrolins from a MAR 4 strain provides additional evidence that this subgroup of actinomycetes in the family Streptomycetaceae is a rich source of secondary metabolites produced by mixed terpenoid biosynthetic pathways.
Experimental Section
General Experimental Procedures
Optical rotations were measured on PerkinElmer model 343 polarimeter. UV and IR spectra were recorded using PerkinElmer Lambda 35 UV/Vis spectrophotometer and Thermo Scientific Nicolet iS10 spectrometer, respectively. 1H, 13C and 2D NMR spectral data were obtained in CDCl3 or DMSO-d6 on a Varian Unity 500 MHz NMR spectrometer. Low resolution ESIMS were measured on a Waters Alliance 2695/Quattro micro API system. High-resolution mass spectral data were acquired on JEOL/JMS-AX505WA instrument. Lichroprep RP-18 (Merch, 40–63 um) was used for column chromatography. Semi-preparative HPLC separations were performed using a Gilson 321 HPLC system with a Phenomenex Luna C18(2) 10 μm column (10 × 250 mm) at a flow rate 4 mL/min. A Waters 1525 HPLC-PDA system with a Phenomenex Luna C18(2) 5 μm column (4.6 × 100 mm) was used for the analysis of extracts and fractions.
Isolation of Strain CNQ-509, Cultivation, Extraction
Strain CNQ-509 was isolated on A1 agar medium (10 g of starch, 4 g of peptone, 2 g of yeast extract and 18 g of agar in 1L of seawater) from a marine sediment sample collected at a depth of 44.2 m one mile Northwest of the Scripps Institution of Oceanography Pier (La Jolla, California). The 16S rRNA gene sequence for this strain (1451 base pairs) has been deposited with GenBank (accession number EF581384). It shares 99.2% sequence identity with the type strain for Streptomyces aculeolatus (AB184624), which interestingly is the only non-marine strain reported to date that falls within the MAR4 group. Strain CNQ-509 was cultured in 8 replicate 7 L fermentors (LiFlus GR, BioTron Inc.) each containing 4 L of fermentation medium A1 without agar for 7 days at 27 °C while stirring at 400 rpm. At the end of the fermentation period, 20 g/L Amberlite XAD-7 adsorbent resin was added into each fermentor and stirring was continued for one additional hour at 100 rpm. The resin was then collected by filtration through cheesecloth, washed with deionized water, and eluted twice with acetone. The acetone solution was concentrated to afford 3.5 g of organic extract.
Isolation and Purification of Nitropyrrolins A-E (1–5)
The extract (3.5 g) was absorbed on Celite (5 g) and subjected to C-18 (50 g) reversed-phase column chromatography eluting with a step gradient of MeOH-H2O solvent mixtures (increasing the MeOH by 20% per 500 mL from 10% to 90% MeOH-H2O). The combined 70% and 90% MeOH fractions (360 mg) was then further fractionated by C-18 reversed-phase HPLC with a MeCN-H2O gradient solvent system (Phenomenex Luna C-18(2) 21.2 × 250 mm, 10 mL/min, 50–100% MeCN-H2O for 40 min) to obtain five subfractions containing the nitropyrrolins (A, 16 mg; B, 30 mg; C, 27 mg; D, 11 mg and E, 5 mg ). Each fraction was subsequently purified by C-18 reversed-phase HPLC using Phenomenex Luna C18(2) column (10 × 250 mm, 10 μm) with 75 % MeCN-H2O as an eluting solvent (flow rate: 4 mL/min) to yield four pure compounds, nitropyrrolin A (1, 6.0 mg), nitropyrrolin B (2, 16 mg), nitropyrrolin C (3, 12 mg), nitropyrrolin D (4, 4 mg) and nitropyrrolin E (5, 2 mg).
Nitropyrrolin A (1)
a light yellow oil, [α]D20 +8 (c 0.05, CH2Cl2); UV (MeOH) λmax (log ε) : 350 nm (3.90) nm; IR (KBr) νmax: 3360, 2967, 2926, 1504, 1452, 1384, 1367, 1293, 1272 cm−1; See Table 1 for 1H NMR data and Table 2 for 13C NMR data. ESI MS: m/z 373 [M+Na]+, 349 [M-H] −; HR-FAB MS: m/z 349.2124 [M-H]− (calcd for C19H29N2O4,349.2127).
Nitropyrrolin B (2)
a light yellow oil, [α]D20 +3 (c 0.05, CH2Cl2); UV (MeOH) λmax (log ε): 351 nm (3.86); IR (KBr) νmax: 3259, 3127, 2963, 2926, 2873, 1510, 1455, 1383, 1368, 1302, 1272, 1251 cm−1; See Table 1 for 1H NMR data and Table 2 for 13C NMR data. ESI MS: m/z 355 [M+Na]+, 331 [M-H] −; HR-FAB MS: m/z 331.2029 [M-H]− (calcd for C19H27N2O3, 331.2022).
Nitropyrrolin C (3)
a light yellow oil, [α]D20 -2 (c 0.07, CH2Cl2); UV (MeOH) λmax (log ε): 348 nm (3.82); IR (KBr) νmax: 3303, 3128, 2968, 2927, 2856, 2731, 1709, 1632, 1591, 1504, 1450, 1384, 1370, 1295, 1273 cm−1; See Table 1 for 1H NMR data and Table 2 for 13C NMR data. ESI MS (rel. int.): m/z 391 [M+Na]+, 369 (40) [M-H+2]−, 367 (100) [M-H] −; HR-FAB MS: m/z 367.1791 [M-H]− (calcd for C19H28ClN2O3, 367.1788).
Nitropyrrolin D (4)
a light yellow oil, [α] D20 -9 (c 0.003, MeOH); UV (MeOH) λmax (log ε): 351 nm (3.90); IR (KBr) νmax: 3301, 2968, 2926, 1503, 1452, 1383, 1366, 1276 cm−1; See Table 1 for 1H NMR data and Table 2 for 13C NMR data. ESI MS: m/z 355 [M+Na]+, 331 [M-H] −; HR-FAB MS: m/z 331.2023 [M-H]− (calcd for C19H27N2O3, 331.2021).
Nitropyrrolin E (5)
a light yellow oil, [α]D20 +15 (c 0.002, MeOH); UV (MeOH) λmax (log ε): 350 nm (3.85); IR (KBr) νmax 3403, 2973, 2931, 1503, 1451, 1384, 1366, 1293, 1271 cm−1; See Table 1 for 1H NMR data and Table 2 for 13C NMR data. ESI MS (rel. int.): m/z 403 [M-H+2]− (40), 401 [M-H]− (100); HR-FAB MS: m/z 401.1837 [M-H]− (calcd for C19H30ClN2O5, 401.1843).
Preparation of the 3-farnesylpyrrole by zinc-mediated farnesylation of pyrrole
A mixture of pyrrole (7.5 mmol), farnesyl bromide (2.8 mmol), zinc powder (650 mg) in THF (10 mL) was stirred at room temperature for 10 hours. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with saturated NH4Cl (20 mL) and diluted with water (10 mL), and extracted with ethyl acetate (2 × 40 mL). Evaporation of the solvent followed by purification on normal phase HPLC [Luna Silica (2), 250 × 21.2 mm, flow rate 10 mL/min] as an isocratic condition [hexane-EtOAc (20:1)] afforded pure 3-farnesyl-pyrrole (6) (270 mg). For 3-Farnesylpyrrole (6): UV (MeOH) λmax (log ε): 227 nm (4.49); ESI MS: m/z 270 [M-H]−. 1H NMR (500 MHz, CDCl3) δ 1H NMR (500 MHz, CDCl3) δ 8.02 (1H, NH), 6.75 (1H, br m, H-5), 6.58 (1H, br m, H-2), 6.12 (1H, br m, H-4), 5.41 (1H, br tq, J = 7.5, 1.0 Hz, C-2′), 5.17 (1H, br tq, J = 7.0, 1.0 Hz, H-6′), 5.13 (1H, br tqq, J = 7.0, 1.0, 1.0 Hz, H-10′), 3.25 (2H, d, J = 7.5 Hz, H2-2′), 2.15 (2H, m, H2-5′), 2.07–2.09 (4H, m, H2-4′ and H2-9′), 2.01 (2H, dd, J = 8.5, 7.0 Hz, H2-8′), 1.72 (3H, br d, J = 1.0 Hz, H3-15′), 1.71 (3H, br d, J = 1.0 Hz, H3-12′), 1.63 (6H, br s, H3-13′ and H3-14′).
Preparation of the γ-farnesyl-α-nitropyrrole (7) and β-farnesyl-α-nitropyrrole (8) by nitration of 3-farnesylpyrrole (6)
A solution of HNO3 (0.08 mmol) in Ac2O (0.5 mL) was added dropwise to a solution of 3-farnesylpyrrole (6) (0.08 mmol) in Ac2O (1 mL), and the reaction was stirred at − 50°C for 2 h and then at room temperature for 1 h. After the reaction was complete (as indicated by TLC analysis), the reaction mixture was diluted with H2O(5 mL), and extracted with Et2O (2 × 10 mL). The organic layer was washed with aqueous NaHCO3 solution (2%), concentrated under reduced pressure, then purified by normal phase HPLC [Luna silica (2), 250 × 10 mm, flow rate 4 mL/min] under isocratic conditions using hexane-EtOAc (92:8) to afford γ-farnesyl-α-nitropyrrole (7) (5.0 mg) and β-farnesyl-α-nitropyrrole (8) (8.7 mg). For γ-farnesyl-α-nitropyrrole (7) a light yellowish oil; UV (PDA detector using MeCN-H2O solvent mixture) λmax: 353 nm; ESI MS: m/z 315 [M-H]−; 1H NMR (500 MHz, CDCl3); δ 9.24 (1H, br s, NH), 6.96 (1H, br s, H-3), 6.74 (1H, br s, H-1), 5.30 (1H, br tq, J = 7.5, 1.0 Hz, H-2′), 5.11 (1H, m, H-6′), 5.10 (1H, m, H-10′), 3.19 (2H, d, J = 7.5 Hz, H2-1′), 2.13 (2H, m, H2-5′), 2.06–2.08 (4H, m, H2-4′ and H2-9′), 1.99 (2H, m, H2-8′), 1.69 (6H, s, H3-12′ and H3-15′), 1.61 (3H, s, H3-13′), 1.60 (3H, s, H3-14′); 13C NMR (125 MHz, CDCl3) δ 137.3a (C-2), 137.2 (C-3′), 135.3 (C-7′), 131.4 (C-11′), 127.1 (C-4), 124.3 (C-6′), 123.9 (C-10′), 121.5 (C-2′), 121.0 (C-5), 110.8 (C-3), 39.7 (C-8′), 39.6 (C-4′), 26.7 (C-9′), 26.4 (C-5′), 25.7 (C-12′), 25.3 (C-1′), 17.7 (C-13′), 16.1 (C-15′), 16.0 (C-14′). aThis chemical shift was assigned by the analysis of HSQC and HMBC spectra because it was not observed in the 13C NMR spectrum. For β-farnesyl-α-nitropyrrole (8): a light yellowish oil; UV (PDA detector using MeCN-H2O solvent mixture) λmax: 340 nm; ESI MS: m/z 315 [M-H]−; 1H NMR (500 MHz, CDCl3); δ 9.35 (1H, br s, NH). 6.86 (1H, br dd, J= 2.5, 2.0 Hz, H-5), 6.20 (1H, br dd, J= 2.5, 2.0 Hz, H-4), 5.35 (1H, br tq, J = 7.0, 1.0 Hz, H-2′), 5.12 (1H, m, H-6′), 5.10 (1H, m, H-10′), 3.62 (2H, d, J= 7.0 Hz, H2-1′), 2.13 (2H, m, H2-5′), 2.06–2.07 (4H, m, H2-4′ and H2-9′), 1.99 (2H, m, H2-8′), 1.71 (3H, s, H3-15′), 1.68 (3H, s, H3-12′), 1.61 (6H, s, H3-13′ and H3-14′); 13C NMR (125 MHz, CDCl3) δ 137.7 (C-3′), 135.2 (C-7′), 134.1a (C-2), 131.3 (C-11′), 129.3 (C-3), 124.3 (C-10′), 124.0 (C-6′), 121.9 (C-5), 120.2 (C-2′), 112.4 (C-4), 39.7 (C-8′), 39.6 (C-4′), 26.7 (C-9′), 26.5 (C-5′), 25.7 (C-1′ and C-12′), 17.7 (C-13′), 16.2 (C-15′), 16.0 (C-14′). aThis chemical shift was assigned by the analysis of HSQC and HMBC spectra because it was not detected in the 1D 13C NMR spectrum.
Preparation of MTPA Esters
Compounds 1, 3, 4, 5 and 11 were divided into two portions, and each was dissolved in 600 μL of pyridine-d5 in a 5 mm NMR tube. To each NMR tube were added a slight excess dimethylaminopyridine (DMAP). The samples were then treated with 5 μL of (R)-α-methoxy-a-(trifluoromethyl) phenylacetyl chloride (MTPA-Cl) and 5 μL of (S)-MTPA-Cl at room temperature. After 12 h, the reaction was complete, and 1H NMR and 1H-1H gCOSY spectra for (S)-mosher esters (9a, 12a, 13a, 14a and the (S)-mosher ester of 11) and (R)-mosher esters (9b, 12b, 13b and 14b) were recorded (see Supporting Information for NMR data). Because of the difficulty in interpreting some crude ester mixtures, compounds 13a, 13b, 14a and 14b were purified by RP-HPLC with a Luna C-8 column (250 × 10 mm) using 50% MeCN-H2O to 100% MeCN as a linear gradient for 40 min (flow rate 4 mL/min),
Preparation of the Acetonide 10
Nitropyrrolin B (2) (2 mg) was dissolved in dry Me2CO (1 mL), and BF3 · Et2O (20 μL) was added at 0 °C. The reaction was allowed to stir for 3 h at 0 °C, the reaction was then quenched with saturated aqueous NaHCO3, and the aqueous phase was extracted twice with EtOAc. The combined EtOAc solution was concentrated under reduced pressure, and the residue was purified by RP HPLC with Luna C-8 column (250 × 10 mm) using a linear gradient solvent system of 50 % MeCN-H2O to 100 % MeCN for 20 min to provide compound 10 (1.2 mg). For acetonide 10: 1H NMR (500 MHz, CDCl3) δ 9.30 (1H, br s, NH), 7.07 (1H, br s, H-3), 6.90 (1H, br s, H-5), 5.16 (1H, dd, J = 7.0, 7.0 Hz, H-6′), 5.11 (1H, dd, J = 7.0, 7.0 Hz, H-10′), 3.91 (1H, dd, J = 10.0, 3.5 Hz, H-2′), 2.76 (1H, J = 15.0, 10.0 Hz, H2-1′a), 2.58 (1H, J = 15.0, 3.5 Hz, H2-1′b), 2.23 (1H, m, H2-5′a), 2.08 (1H, m, H2-5′b), 2.08 (2H, m, H2-9′), 2.00 (2H, m, H2-8′), 1.70 (3H, br s, H3-12′), 1.65 (1H, m, H2-4′a), 1.64 (3H, br s, H3-14′), 1.62 (3H, br s, H3-13′), 1.46 (3H, s, acetonide methyl), 1.37 (3H, s, acetonide methyl), 1.27 (1H, m, H2-4′b), 1.23 (3H, s, H3-15′); ESI MS: m/z 413 [M+Na]+.
Preparation of the Diol 11 from 10
To a solution of 10 in anhydrous MeOH (2 mL) was added pyridinium-p-toluensulfonate (2 mg), and the reaction was stirred for 24 h. The reaction was then quenched with saturated aqueous NaHCO3, and the aqueous phase was extracted twice with EtOAc. The EtOAc solution was concentrated under reduced pressure, and the residue was purified by RP HPLC [Luna C-8 column (250 × 10 mm), flow rate 4 mL/min, 45 % MeCN-H2O to 95 % MeCN as a linear gradient for 40 min] to afford diol 11. For diol (11): 1H NMR (500 MHz, CDCl3) δ 9.40 (1H, br s, NH), 7.06 (1H, br s, H-3), 6.90 (1H, br s, H-5), 5.18 (1H, br t, J = 7.0 Hz, H-6′), 5.10 (1H, tq, J = 7.0, 1.0 Hz, H-10′), 3.58 (1H, ddd, J = 11.0, 4.0, 2.0 Hz, H-2′), 2.73 (1H, dd, J = 15.0, 2.0 Hz, H2-1′a), 2.53 (1H, dd, J = 15.0, 11.0 Hz, H2-1′b), 2.20 (1H, m, H2-5′a), 2.14 (1H, m, H2-5′b), 2.14 (1H, d, J = 4.0 Hz, 2′-OH), 2.19 (2H, m, H2-9′), 2.01 (2H, m, H2-8′), 1.97 (1H, br s, 3′-OH), 1.73 (1H, m, H2-4′a), 1.69 (3H, br s, H3-12′), 1.66 (3H, br s, H3-14′), 1.61 (3H, br s, H3-13′), 1.49 (1H, m, H2-4′b), 1.27 (3H, br s, H3-15′); ESI MS: m/z 373 [M+Na]+.
Supplementary Material
Acknowledgments
This research is a result of financial support from the NIH, National Cancer Institute under grant R37 CA044848 (to WF) and the NOAA California Sea Grant College Program Project R/NMP-100 (NA100AR41700360 to PRJ) through NOAA’S National Sea Grant College Program, U.S. Dept. of Commerce. The statements, findings, conclusions and recommendations are those of the author(s) and do not necessarily reflect the views of California Sea Grant or the U.S. Dept. of Commerce. Fellowship support for APE from the CNPq (Conselho Nacional de Desenvolvimento Cientifico e Tecnológico, Brasil) is gratefully acknowledged.
Footnotes
Supporting Information Available: Spectroscopic data sets (1D and 2D NMR, ESI MS, UV/VIS, HRMS, etc) for 1–5, 1H NMR spectra for 6 and (S)-Mosher ester of 11, 1H NMR and 1H-1H COSY spectra for 9a, 9b, 12a, 12b, 13a, 13b, 14a and 14b, 1H NMR and 2D NOESY spectra for 10 and 11, and 1H and 13C NMR spectra for 7 and 8. This material is available free of charge via the Internet at http://pubs.acs.org
References and Notes
- 1.Fenical W, Jensen PR. Nat Chem Biol. 2006;2:666–673. doi: 10.1038/nchembio841. [DOI] [PubMed] [Google Scholar]
- 2.(a) Mincer TJ, Jensen PR, Kauffman CA, Fenical W. Appl Environ Microbiol. 2002;68:5005–5011. doi: 10.1128/AEM.68.10.5005-5011.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maldonado LA, Fenical W, Jensen PR, Kauffman CA, Mincer TJ, Ward AC, Bull AT, Goodfellow M. Int J Sys Evol Microbiol. 2005;55:1759–1766. doi: 10.1099/ijs.0.63625-0. [DOI] [PubMed] [Google Scholar]
- 3.(a) Jensen PR, Mincer TJ, Williams PG, Fenical W. Antonie Van Leeuwenhoek. 2005;87:43–48. doi: 10.1007/s10482-004-6540-1. [DOI] [PubMed] [Google Scholar]; (b) Kwon HC, Kauffman CA, Jensen PR, Fenical W. J Am Chem Soc. 2006;128:1622–1632. doi: 10.1021/ja0558948. [DOI] [PubMed] [Google Scholar]
- 4.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Angew Chem Int Edit. 2003;42:355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 5.Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC. Bioorg Med Chem. 2009;17:2175–2180. doi: 10.1016/j.bmc.2008.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pathirana C, Jensen PR, Fenical W. Tetrahedron Lett. 1992;33:7663–7666. [Google Scholar]
- 7.Soria-Mercardo IE, Prieto-Davó A, Jensen PR, Fenical W. J Nat Prod. 2005;68:904–910. doi: 10.1021/np058011z. [DOI] [PubMed] [Google Scholar]
- 8.Cho JY, Kwon HC, Williams PG, Jensen PR, Fenical W. Org Lett. 2006;8:2471–2474. doi: 10.1021/ol060630r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deouet D, Cauret L, Brosse JC. Eur Polym J. 2003;39:671–686. [Google Scholar]
- 10.Morgan KJ, Morrey DP. Tetrahedron. 1966;22:57–62. [Google Scholar]
- 11.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 12.Yadav JS, Pratap TV, Rajender V. J Org Chem. 2007;72:5882–5885. doi: 10.1021/jo0704762. [DOI] [PubMed] [Google Scholar]
- 13.Steinreiber A, Farber K. Curr Opin Biotechnol. 2001;12:552–558. doi: 10.1016/s0958-1669(01)00262-2. [DOI] [PubMed] [Google Scholar]
- 14.Nii K, Tagami K, Kijima M, Munakata T, Ooi T, Kusumi T. Bull Chem Soc Jpn. 2008;81:562–57. [Google Scholar]
- 15.(a) Chemical Database, Version 13.1. Chapman & Hall; 2004. Dictionary of Natural Products on CD-ROM. [Google Scholar]; (b) Harborne JB, Williams CA. Nat Prod Rep. 1998;15:631–651. [Google Scholar]
- 16.Macherla VR, Liu J, Bellows C, Teisan S, Nicholson B, Lam KS, Potts BCM. J Nat Prod. 2005;68:780–783. doi: 10.1021/np049597c. [DOI] [PubMed] [Google Scholar]
- 17.Kato S, Shindo K, Kawai H, Odagawa A, Matsuoka M, Mochizuki J. J Antibiot. 1993;46:892–899. doi: 10.7164/antibiotics.46.892. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.