

Abstract
Colorectal cancer (CRC) is a complex disease related to environmental and genetic risk factors. Several studies have shown that susceptibility to complex diseases can be mediated by ancestral alleles. Using RNAi screening, CTNNBL1 was identified as a putative regulator of the Wnt signaling pathway, which plays a key role in colorectal carcinogenesis. Recently, single nucleotide polymorphisms (SNPs) in CTNNBL1 have been associated with obesity, a known risk factor for CRC. We investigated whether genetic variation in CTNNBL1 affects susceptibility to CRC and tested for signals of recent selection. We applied a tagging SNP approach that cover all known common variation in CTNNBL1 (allele frequency >5%; r2>0.8). A case-control study was carried out using two well-characterized study populations: a hospital-based Czech population composed of 751 sporadic cases and 755 controls and a family/early onset-based German population (697 cases and 644 controls). Genotyping was performed using allele specific PCR based TaqMan® assays (Applied Biosystems, Weiterstadt, Germany). In the Czech cohort, containing sporadic cases, the ancestral alleles of three SNPs showed evidence of association with CRC: rs2344481 (OR 1.44, 95%CI 1.06-1.95, dominant model), rs2281148 (OR 0.59, 95%CI 0.36-0.96, dominant model) and rs2235460 (OR 1.38, 95%CI 1.01-1.89, AA vs. GG). The associations were less prominent in the family/early onset-based German cohort. Data derived from several databases and statistical tests consistently pointed to a likely shaping of CTNNBL1 by positive selection. Further studies are needed to identify the actual function of CTNNBL1 and to validate the association results in other populations.
Keywords: Colorectal cancer, case-control study, ancestral-susceptibility model, selective pressure, CTNNBL1
Introduction
Colorectal cancer (CRC) is one of the most common cancers in industrialized countries and it represents one of the leading causes of cancer-related morbidity and mortality. The incidence rates for CRC vary among different groups and populations depending on race, gender and age. The highest incidence rate can be found in New Zealand, Australia, North America, Europe and more recently in Japan, whereas lower rates are reported in Asia and Africa [1, 2]. Several non-genetic factors, such as nutrition, life style and environment [3], as well as genetic variation, reflected by familial aggregation and identified high- and low-penetrance mutations [4, 5], contribute to the risk of developing CRC. Inherited susceptibility underlies ∼35% of variance in colorectal cancer risk [6]. Yet, high-penetrance germline mutations, such as mismatch repair genes and adenomatous polyposis coli (APC) mutations, only account for less than 5% of all CRC cases but they contribute to the heritability of CRC in general [4, 5, 7, 8]. In comparison, low-penetrance mutations contribute to a small proportion of familial cases and to a larger proportion of sporadic cases [4, 5, 8].
The CTNNBL1 [catenin (cadherin-associated protein) b-like 1] (GeneID: 56259) gene was newly identified using RNAi screening as a putative regulator of the canonical Wnt signaling pathway, acting upstream of, or in parallel to β-catenin (D.I. and M.B., unpublished data). Liu et al. have also connected CTNNBL1 to Wnt signaling through structural homologies of CTNNBL1 to β-catenin, the main mediator in the Wnt pathway [9]. Wnt signaling plays an important role in cell proliferation, differentiation and stem cell maintenance during embryonic development and tissue renewal in adult organisms. Importantly, dysregulation of Wnt signaling, mainly through mutations of the pathway components, plays a key role in colorectal carcinogenesis [10]. Moreover, nutrition related diseases, such as obesity and type 2 diabetes, are affected by genetic and functional variations in the Wnt signaling pathway [11]. Single nucleotide polymorphisms (SNPs) in CTNNBL1 have been associated with increased body mass index (BMI) and fat mass in case-control studies of different populations [9, 12, 13]. Obesity, high BMI and high fat mass are also known risk factors for the development of CRC. Taken together, these facts suggest that CTNNBL1 may be involved in CRC and other nutrition related diseases if mutated or dysregulated.
Nutrition related traits are common targets for selection and local adaptation [14, 15]. Several diseases, such as obesity, type 2 diabetes and hypertension, have been linked to polymorphisms in which the ancestral allele increases the risk of the disease [16]. This kind of allelic effect is described as the ancestral-susceptibility model [16]. Such polymorphisms frequently show significant differences in the worldwide allele distribution. In the case of non-synonymous polymorphisms, it is usually straightforward to describe the cause and the direction of selective pressure due to the altered function, whereas in the case of non-coding, low-penetrance polymorphisms, complex processes and interactions may cause the final phenotype. Especially in complex diseases, the detected effect of a low-penetrance polymorphism may point an undiscovered intermediate phenotype or it might have developed due to selection of another trait linked to the analyzed one [17].
We considered CTNNBL1 as a promising candidate gene to be associated with CRC. We investigated whether genetic variation in CTNNBL1 affects the susceptibility to CRC and searched for signals of selective pressure on this particular gene.
Material and methods
Study Populations
In this project, two case-control studies were carried out, one containing newly diagnosed, incident cases from the Czech Republic, the other familial cases from Germany. The first population was a hospital-based sample set from the Czech Republic. DNA extracts from peripheral leukocytes from the Czech study participants were collected between December 2004 and December 2007 [18]. The CRC case group contained 751 patients recruited in nine oncological departments in the Czech Republic (two in Prague, one each in Benesov, Brno, Liberec, Pies, Pribram, Usti nad Labem and Zlin). The patients showed positive colonoscopic results for malignancy, histologically confirmed as colon or rectal carcinomas. Patients who met the Amsterdam criteria I and II for hereditary nonpolyposis colorectal cancer (HNPCC) [19] were not included in the study population. The control group contained 755 individuals that underwent colonoscopy for various gastrointestinal complaints, such as macroscopic bleeding, positive fecal occult blood test (FOBT) or abdominal pain of unknown origin, in five gastro-enterological departments of the Czech Republic (Prague, Brno, Jihlava, Liberec and Pribram).
Both the case and the control populations represent the entire Czech Republic. Only individuals with negative coionoscopic results for malignancies, colorectal adenomas, benign polyps or inflammatory bowel disease (IBD) were chosen for the control group. Beside general information about gender and age, information about BMI was available for the majority of individuals (61.7% of the case population and 69.1% of the control population). Additionally, similar proportion of the study participants provided information about their diabetes status (62% of the case population and 70% of the control population). All data about the individuals were collected at the time of diagnosis for cases and at the time of sampling for controls. Table 1 outlines the available characteristics of the Czech study population at the time of recruitment.
Table 1.
Characteristics of the study populations at the time of diagnosis for cases and at the time of sampling for controls
| Czech population | Cases | Controls | p value |
|---|---|---|---|
| total | 751 | 755 | |
| male [n (%)] | 418 (56%) | 446 (59%) | 0.50 |
| female [n (%)] | 311 (41%) | 309 (41%) | |
| missing gender [n (%)] | 22 (3%) | 0 | |
| median age [range] | 60.8 [27-85] | 54.4 [28-91] | <0.001 |
| missing age [n (%)] | 23 (3%) | 0 | |
| median BMI [range] | 27 [13-53] | 27 [17-44] | 0.49 |
| missing BMI [n (%)] | 288 (38%) | 233 (31%) | |
| diabetes [n (%)] | 78 (10%) | 58 (8%) | 0.01 |
| no diabetes [n (%)] | 391 (52%) | 468 (62%) | |
| missing diabetes information [n (%)] | 282 (38%) | 229 (30%) | 0.003* |
| German population | Cases | Controls | |
| total | 697 | 672 | |
| male [n (%)] | 348 (50%) | 288 (43%) | 0.01 |
| female [n (%)] | 349 (50%) | 384 (57%) | |
| missing gender [n (%)] | 3 (0%) | 0 | |
| median age [range] | 44 [9-82] | 44 [26-68] | <0.001 |
| missing age [n (%)] | 30 (4%) | 0 | |
BMI, body mass index;
“with” compared to “without” self-reported diabetes information.
The SNPs which showed significant results in the Czech population were additionally analyzed in a German sample set based on familial CRC cases. Blood samples from the 697 German cases were collected as part of a large study on susceptibility to HNPCC [20] and they were recruited by six German university hospitals (Bochum, Bonn, Dresden, Düsseldorf, Heidelberg and Munich/Regensburg). All case patients fulfilled Bethesda Guidelines to be screened for HNPCC [21]. The patients selected for this case-control study were all found to be microsatellite stable and therefore negative for germ line mutations in MSH2 and MLH1 through systematic screening [20]. Additionally, only patients with at least one first degree relative affected by CRC (62.4% of the cases) or patients diagnosed below the age 50 years (33.6%) were included to the study. For 4% of the cases, the inclusion criterion was missing. The cases eligible for the study were unrelated. Data regarding the BMI and diabetes status were not available. The control population was composed of 672 healthy, unrelated and ethnicity-, gender- and age-matched German blood donors who were recruited between 2004 and 2006 by the Institute of Transfusion Medicine and Immunology, University of Mannheim, Germany [22]. Table 1 outlines the available characteristics of the German study population at the time of recruitment.
Gene characterization
Several databases were used to investigate distinct characteristics of CTNNBL1. The degree of overall conservation was measured by Pupasuite 2.0.0 using BLASTZ [23], information about the LD/r2 of all tagSNPs and captured SNPs were obtained from HapMap Genome Browser (HapMap3; release #2, Phase 3; http://hapmap.ncbi.nlm.nih.gov;) [24] and the recombination rate and the regulatory potential were analyzed using UCSC Genome Browser on Human (Mar. 2006 Assembly; hg18) [25-27].
Phylogenetic trees of the coding DNA (cDNA) sequences and intronic DNA sequences of different species were created with the program SplitsTree V4.10 [28]. The sequences of Homo sapiens sapiens, Pan troglodytes, Pongo pygmaeus, Macaca mulatta, Mus musculus, Rattus norvegicus, Bos Taurus, Equus freus caballus, Canis lupus familiaris and Monodelphis domestica gained from the Ensembl Genome Browser [29] were aligned using MEGA4 software [30]. For the cDNA, the sequence of Gallus gallus domesticus was additionally used for alignment. The sequence alignments were utilised to calculate phylogenetic consensus trees using the neighbour joining method and to calculate bootstrap values (SplitsTree V4.10). In the resulting trees, taxa are represented by nodes and their evolutionary relationships are represented by branches. A group of species that includes all descendants of one common ancestor is a denominated clade. Bootstrap values indicate how reliable a clade is [31].
Selection of SNPs
This study focused on SNPs within the CTNNBL1 gene region. Eight unlinked tagging SNPs (tagSNPs) with a minor allele frequency (MAF) > 5% were selected using the genotyping data of the CEU population in HapMap (NCBI dbSNP35) (rs6067377, rs2344481, rs238302, rs2281148, rs6067889, rs4811233, rs6067923) [24]. These tagSNPs represent a total of 123 SNPs annotated in the NCBI dbSNP (http://www.ncbi.nlm.nih.gov) encompassing the whole gene including 1.9kb up- and 1.1kb downstream of the first and the last exonic base, respectively (r2 value of LD >0.8). Including the captured SNPs, all polymorphisms were located in the non-coding regions of the CTNNBL1 gene. The tagSNP selection was performed using the HapMap Browser (NCBI dbSNP35)[24].
tagSNP characterization
Multiple statistical methods and databases were used to investigate the characteristics of the SNPs selected for the tagSNP approach. The worldwide allele distribution of the eight tagSNPs and all the 123 captured SNPs was analyzed for differences among the Caucasian population (CEU, Utah residents with Northern and Western European ancestry from the CEPH collection), the Sub-Saharan African population (YRI, Yoruba in Ibadan, Nigeria) and the East Asian population (HCB, Han Chinese in Beijing, China, and JPT, Japanese in Tokyo, Japan). The frequency data were derived from the NCBI database (http://www.ncbi.nlm.nih.gov) using the data available for the Submitter Population IDs: HapMap -CEU, -YRI, -HCB and -JPT.
Based on this data the Fixation indexes (FST values) and the corresponding probability values were estimated for the CRC associated SNPs (rs2344481, rs2281148 and rs2235460) using the Arlequin 3.1 Software [32]. FST values > 0.25 indicate strong genetic differentiation (i.e. the SNPs might have been targets of selection); FST values in the range of 0.05 to 0.1 indicate moderate genetic differentiation [33]. FST p values < 0,05 are generally considered to be evolu-tionarily significant [34].
Further, the Haplotter database was used to investigate additional parameters that reflect selective processes in CTNNBL1. In lack of directly available information about the SNP that showed the strongest association with CRC risk (rs2344481) we used data of six SNPs captured by rs2344481 (r2>0.8) (rs6020395, rs6021428, rs6020712, rs6125962, rs6020846, rs6512695). Strong negative Fay-Wu's H were considered as signatures for a selective sweep [35, 36]; iHS < -1.5 and > 1.5 give conclusive evidence for natural selection and iHS < -2 or > 2 give evidence for a powerful selection signal [17, 36]. Additionally, the derived allele frequencies of the six SNPs captured by rs2344481 were used as markers for the worldwide allele distribution, providing further evidence for a selective sweep.
Genotyping
Genotype analyses were performed using allele specific PCR based TaqMan assays, designed by Applied Biosystems (Applied Biosystems, Weiterstadt, Germany). PCR reactions were carried out using 5ng purified DNA per reaction. Thermo cycling was performed according to the Applied Biosystems PCR conditions with 35 to 50 cycles (depending on optimal performance). The genotyping was performed simultaneously for case- and control- samples. The genotype detection was performed using an ABI PRISM 7900 HT Sequence Detection System with SDS 2.2 software (Applied Biosystems, Weiterstadt, Germany). Direct sequencing was used to randomly control genotypes determined by TaqMan assays and to reveal the actual genotype of samples with an unclear classification by TaqMan assays. PCR amplification and sequencing reaction were performed as described in [37].
Statistical analysis of the genotyping data
The observed genotype frequencies in the controls were tested for Hardy-Weinberg equilibrium (HWE) and differences between the observed and the expected frequencies were tested for significance using X2 tests.
Statistical significance for different genotype distributions in cases and controls were determined by global X2 tests. The overall genotype effect of a SNP on the risk of CRC was considered statistically significant at the level p < 0.05. Odds ratios (ORs) and 95% confidence intervals (95% CIs) for association between genotypes and CRC risk were estimated based on logistic regression (PROC LOGISTIC, SAS Version 9.1; SAS Institute, Cary, NC). The calculated effects referred to the ancestral allele. Besides raw genotype effects, ORs were also adjusted for age, gender, nationality and BMI (age: conti-nous variable, BMI: grouped according to BMI <20; 20-24; 25-30; 30-35; 35-40; >40) since age, gender and BMI are the most important factors contributing to the risk of CRC.
Genotypes of SNPs in the ±500kb region around the genotyped polymorphisms were imputed based on the HapMap data of the CEU population, in order to detect possible associations with non-genotyped SNPs. Multiple imputation relied on inference of haplotypes by means of the expectation-maximization (EM) algorithm in the presence of partially missing genotypes. In brief, missing alleles were excluded from the calculation of allele frequencies. In the E-step, frequencies of partially missing genotypes were updated looping through all possible genotypes. In the M-step, all existing haplotypes that have alleles identical to the non -missing alleles of this haplotype were updated. The accuracy of imputation based on HapMap was evaluated by cross-validation, and it was represented by minus the logarithm of the probability value for Cohen's Kappa between true and imputed genotypes. Uncertainty in the imputed genotypes was taken into account in the subsequent logistic regression by bootstrapping from the multinomial distribution of the expected genotypes given the observed, directly genotyped variants (1,000 replicates). Probability values referred to a model-free, three-genotype model.
Results
Signatures of positive selection in CTNNBL1
The analyses of the linkage disequilibrium (LD) [24] and the recombination rate [27] indicated a high conservation of CTNNBL1. By the use of only eight tagSNPs, it was possible to analyze 123 SNPs (annotated in the NCBI dbSNP35; MAF>0.05, mean r2=0.95), encompassing the whole gene and the neighbouring regions 1,9 kb upstream and 1,1 kb downstream of the first and last exonic base, respectively (35,754kb-35,936kb) [24]. The average recombination rate in this region was very low, especially in males (0.0-0.7 in males, 1.5-2.7 in females). Furthermore, the regulatory potential of the CTNNBL1 sequence was estimated as high, encompassing the whole gene. The mean value of ESPERR Regulatory Potential within a sequence range from 35,755,848 to 35,933,934 was estimated to be 0.06 [25, 26]. In general, the range of the regulatory potential scores from 0.0 (low) to 0.1 (high).
Next to these characteristics, the database analysis showed a high degree of interspecific conservation (60.3%) within the gene region. This estimation refers to the comparison of the human-mouse alignment (Pupasuite 2.0.0 - Bioinfo 2008. CIPF; using BLASTZ) [23]. Maximal extent of the region analyzed was 35,754kb-35,939kb and contained both coding and in-tronic gene regions. Within this region 553 out of 917 SNPs were found to be unique. Ensemble reports this gene in 36 different species including zebrafish and chicken next to various mammalian species (http://www.ensembl.org). Besides the similarities within the analysed species the differences were species specific and were used to construct phylogenetic trees.
The phylogenetic analysis of the cDNA sequences of CTNNBL1 provided further about the location of the gene in a highly conserved gene region. The algorithm resulted in a highly resolved phylogenetic tree that reflected one of the today's valid phylogenetic trees of animals [38] (Figure 1). The analysis of intronic sequences of CTNNBL1 showed a phylogenetic tree that resembled the cDNA tree on many points. In Figure 2 the phylogentic tree of intron 3 is shown as an example. The order- and spe-cies-clades were equally well dissolved in the intronic tree as in the cDNA tree. In the case of the primates-clade it fitted the today's valid phylogenetic tree even better than the cDNA tree. Both trees failed in resolving the Laurasiatheria and Euarchontoglires clade [38].
Figure 1.
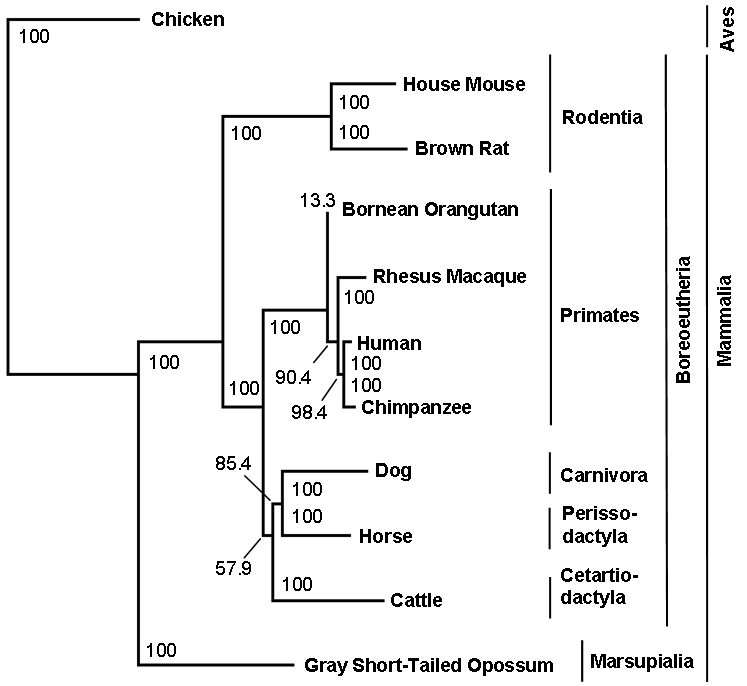
Phylogenetic tree of the cDNA sequence of CTNNBL1. Different taxa are represented by nodes and their evolutionary relationships are represented by branches. Bootstrap values indicate how reliable a clade is.
Figure 2.
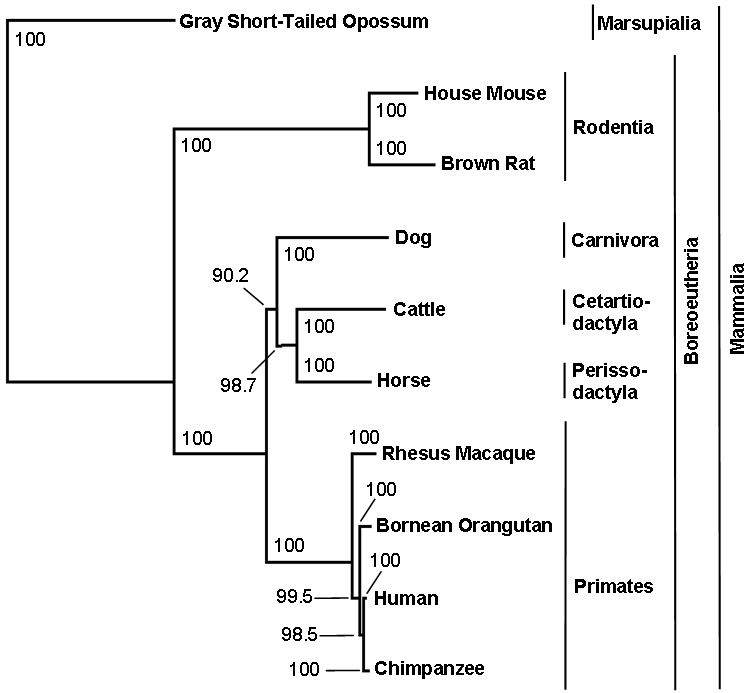
Phylogenetic tree of the intronic DNA (intron3) sequence of CTNNBL1. Different taxa are represented by nodes and their evolutionary relationships are represented by branches. Bootstrap values indicate how reliable a clade is. All tested introns gave similar phylogenetic trees as intron 3, exem-plarily shown here.
The analysis of the allele frequencies of the tagSNPs selected for the study showed distinct differences among the three populations analyzed by the International HapMap Project (YRI, CEU, HCB and JPT) [24]. The frequencies of the ancestral alleles declined from the YRI to the CEU and the HCB/JPT populations in about half of the SNPs captured in this study (60/123). The strongest decline in the ancestral allele frequency was found in tagSNP rs2344481 and in SNPs captured by it. The ancestral allele of rs2344481 was the major allele in YRI (52%) and the minor allele in CEU (5%), JPT (4.7%) and HCB (0%).
To investigate whether SNPs in CTNNBL1 might have been targets of selection, we further investigated three parameters: FST, Fay-Wu's H and iHS The FST statistics showed values above 0.25 for rs2344481 and rs2281148 in the comparison of the African population with the three non-African populations indicating strong genetic differentiation (Table 2). For rs2235460, the comparison of the African and the Japanese population showed moderate genetic differentiation (FST value 0.05). A moderate genetic differentiation was also found for rs2344481 (European vs. Chinese) and rs2281148 (European vs. Chinese). The FST p values were statistically significant for rs2344481 and rs2281148 in the comparison of the African population with the three non-African populations and for rs2239460 in the comparison of the African population to the Japanese populations, indicating evolutionary significance. To investigate the two other parameters, Fay-Wu's H and iHS, we used data of six SNPs (rs6020395, rs6021428, rs6020712, rs6125962, rs6020846, rs6512695) captured by rs2344481, the SNP with strongest association with CRC, because no direct information about rs2344481 was available (Table 3). All six SNPs showed strong negative Fay-Wu's H with a sizeable decrease from the YRI via the CEU to the ANS (East Asians) population indicating a selective sweep. Two SNPs (rs6020395 and rs6125962) showed an iHS >1.5 giving suggestive evidence for natural selection. Additionally, four SNPs (rs6020395, rs6020712, rs6020846, rs6125962) showed a distinct rise of the derived allele frequency up to 52% from the YRI via the CEU to the ANS population, providing further evidence for a selective sweep (Table 3).
Table 2.
Fixation index (FST estimates with probability values in brackets) for CRC associated SNPs among African, European, Chinese and Japanese population
| Population | African | European | Chinese | Japanese | |
|---|---|---|---|---|---|
| African | * | ||||
| rs2344481 | European | 0.50 (<0.001) | * | ||
| Chinese | 0.61 (<0.001) | 0.07 (<0.001) | * | ||
| Japanese | 0.49 (<0.001) | -0.01 (0.80) | -0.08 (<0.001) | * | |
| African | * | ||||
| rs2281148 | European | 0.36 (<0.001) | * | ||
| Chinese | 0.55 (<0.001) | 0.10 (<0.001) | * | ||
| Japanese | 0.54 (<0.001) | 0.04 (<0.001) | 0.02 (0.06) | * | |
| African | * | ||||
| rs2235460 | European | 0.04 (0.02) | * | ||
| Chinese | -0.01 (0.59) | 0.03 (0.02) | * | ||
| Japanese | 0.05 (<0.001) | -0.01 (0.95) | 0.05 (0.02) | * | |
Bold numbers indicate strong/moderate genetic differentiation at 5% statistical significance.
Table 3.
Haplotter Data for SNPs captured by rs2344481 (r2>0.8)
| rs6020395 | YRI | CEU | ANS | rs6021428 | YRI | CEU | ANS |
|---|---|---|---|---|---|---|---|
| Derived allele frequency | 0.467 | 0.933 | 0.978 | Derived allele frequency | 0.083 | 0.058 | 0.011 |
| Standardized iHS | 1.626 | 0.984 | -0.232 | Standardized iHS | -0.117 | -0.957 | - |
| Fay-Wu's H | -1.417 | -7.631 | -25.785 | Fay-Wu's H | -11.649 | -13.473 | -29.167 |
| rs6020712 | YRI | CEU | ANS | rs6125962 | YRI | CEU | ANS |
| Derived allele frequency | 0.717 | 0.950 | 0.989 | Derived allele frequency | 0.458 | 0.933 | 0.978 |
| Standardized iHS | 0.345 | 1.051 | - | Standardized iHS | 1.860 | 0.730 | - |
| Fay-Wu's H | -21.846 | -36.428 | -72.758 | Fay-Wu's H | -19.302 | -37.773 | -83.117 |
| rs6020846 | YRI | CEU | ANS | rs6512695 | YRI | CEU | ANS |
| Derived allele frequency | 0.717 | 0.933 | 0.989 | Derived allele frequency | 0.242 | 0.067 | 0.011 |
| Standardized iHS | 0.420 | 0.391 | - | Standardized iHS | 0.159 | 0.138 | - |
| Fay-Wu's H | -11.749 | -36.683 | -69.796 | Fay-Wu's H | -11.258 | -35.959 | -65.078 |
iHS integrated Haplotype Score; YRI Yoruba in Ibadan, Nigeria; CEU, Utah residents with Northern and Western European ancestry from the CEPH collection; ANS combined Asian Population.
Case-control study
Czech population - sporadic CRC
The genotype distribution of all eight tagSNPs measured in the Czech control population was according to Hardy-Weinberg equilibrium (HWE) and the allele frequencies did not differ significantly from the allele frequencies for the Caucasian population given in the NCBI database (http://www.ncbi.nlm.nih.gov/). Therefore, it was feasible to use the NCBI data for the phylogenetic investigation.
Three tagSNPs were found to be statistically significantly associated with CRC: rs2344481 (OR 1.44, 95% Cl 1.06-1.95, dominant model), rs2281148 (OR 0.59, 95% Cl 0.36-0.96, dominant model) and rs2235460 (OR 1.38, 95% Cl 1.01-1.89, derived allele homozygote vs. ancestral allele homozygote) (Table 4). The analysis of the data using a recessive model was also conducted and yielded no significant results (data not shown). After the adjustment of the data for age and for age, gender and BMI in the subgroup of individuals with data of BMI, only rs2344481 showed a statistically significant association with CRC (OR 1.38, 95% Cl 1.00-1.91; dominant model and OR 1.55, 95% Cl 1.01-2.36; dominant model, respectively).
Table 4.
Genotype distributions and estimated risk of the ancestral allele of the polymorphisms in CTNNBL1 in the hospital-based Czech sample population: unadjusted and age adjusted results
| SNP | Allelea | Sample | Genotype distribution | Codominat Model | Dominant Model | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DD-DA-AA | DD vs DA | DD vs AA | DD vs DA+AA | ||||||||||
| Cases | Controls | OR | 95% CI | P | OR | 95% CI | P | OR | 95% CI | P | |||
| rs6067377 | T/C | all samples | 252-368-127 | 266-360-115 | 1.08 | 0.86-1.35 | 0.51 | 1.17 | 0.86-1.58 | 0.33 | 1.10 | 0.89-1.36 | 0.38 |
| adjusted for age | 240-359-125 | 266-360-115 | 1.11 | 0.87-1.41 | 0.42 | 1.12 | 0.81-1.54 | 0.50 | 1.11 | 0.88-1.39 | 0.39 | ||
| rs2344481 | A/G | all samples | 628-112-2 | 658-80-3 | 1.47 | 1.08-1.99 | 0.01 | 0.70 | 0.12-4.19 | 0.69 | 1.44 | 1.06-1.95 | 0.02 |
| adjusted for age | 606-111-2 | 658-80-3 | 1.40 | 1.01-1.94 | 0.04 | 0.77 | 0.12-5.09 | 0.79 | 1.38 | 1.00-1.91 | 0.05 | ||
| rs238302 | A/G | all samples | 82-335-327 | 85-330-324 | 1.05 | 0.75-1.48 | 0.77 | 1.05 | 0.75-1.47 | 0.79 | 1.05 | 0.76-1.45 | 0.77 |
| adjusted for age | 82-335-327 | 85-330-324 | 1.12 | 0.78-1.61 | 0.54 | 1.09 | 0.76-1.57 | 0.63 | 1.11 | 0.79-1.56 | 0.56 | ||
| rs2281148 | C/T | all samples | 45-242-457 | 27-237-478 | 0.61 | 0.37-1.02 | 0.06 | 0.57 | 0.35-0.94 | 0.03 | 0.59 | 0.36-0.96 | 0.03 |
| adjusted for age | 42-234-445 | 27-237-478 | 0.64 | 0.37-1.10 | 0.11 | 0.62 | 0.37-1.05 | 0.08 | 0.63 | 0.37-1.05 | 0.08 | ||
| rs2235460 | A/G | all samples | 100-349-270 | 124-368-243 | 1.18 | 0.87-1.59 | 0.29 | 1.38 | 1.01-1.89 | 0.05 | 1.26 | 0.94-1.67 | 0.12 |
| adjusted for age | 96-342-262 | 124-368-243 | 1.16 | 0.83-1.61 | 0.38 | 1.29 | 0.92-1.82 | 0.14 | 1.21 | 0.89-1.66 | 0.22 | ||
| rs6067889 | A/G | all samples | 448-262-32 | 448-255-30 | 1.03 | 0.83-1.28 | 0.81 | 1.07 | 0.64-1.79 | 0.81 | 1.03 | 0.84-1.27 | 0.77 |
| adjusted for age | 432-257-30 | 448-255-30 | 1.07 | 0.85-1.36 | 0.55 | 1.10 | 0.63-1.92 | 0.75 | 1.08 | 0.86-1.35 | 0.52 | ||
| rs4811233 | C/G | all samples | 358-295-80 | 378-265-81 | 1.18 | 0.94-1.47 | 0.15 | 1.04 | 0.74-1.47 | 0.81 | 1.14 | 0.93-1.41 | 0.20 |
| adjusted for age | 343-289-78 | 378-265-81 | 1.20 | 0.96-1.50 | 0.10 | 1.06 | 0.75-1.50 | 0.74 | 1.17 | 0.95-1.44 | 0.14 | ||
| rs6067923 | G/Ab | all samples | 339-322-84 | 356-291-81 | 1.16 | 0.94-1.44 | 0.18 | 1.09 | 0.78-1.53 | 0.62 | 1.15 | 0.93-1.41 | 0.19 |
| adjusted for age | 330-331-79 | 356-291-81 | 1.23 | 0.99-1.53 | 0.07 | 1.05 | 0.75-1.48 | 0.77 | 1.19 | 0.97-1.46 | 0.10 | ||
OR odds ratio; CI confidence interval; A ancestral allele, D derived allele.
Second allele represents the ancestral allele for each locus.
ancestral allele unknown, the major allele was used as reference.
Total numbers of cases and controls may slightly vary among SNPs due to missing genotypes. Bold numbers indicate statistical significance at 5% level. The results were adjusted for age due to significant difference in the median age distribution among the case and the control group.
The imputation of genotypes based on HapMap data revealed three SNPs with a lower p value than rs2344481: rs6012770 (p 0.02; MAF 0.08; ∼18kb upstream of CTNNBL1), rs928199 (p 0.02; MAF 0.05; ∼16kb upstream of CTNNBL1) and rs6096781 (p 0.02; MAF 0.08; ∼30kb downstream CTNNBL1). These three SNPs are in high LD with rs2344481 (rs6012770: r2 0.9, rs928199: r2 0.95, rs6096781: r2 0.7).
German population - familial CRC
In order to analyze whether the SNPs affecting sporadic CRC risk would also affect the risk of familial/early onset CRC, we genotyped three SNPs (rs2344481, rs2281148, rs2235460) in the German sample set, with familial/early onset CRC cases. The allele frequencies of the tested SNPs did not differ significantly from the allele frequencies of the Caucasian population given in the NCBI database (http://www.ncbi.nlm.nih.gov/) and the genotype distribution in the control population was in Hardy-Weinberg equilibrium (HWE). None of the three tagSNPs showed a statistically significant association with CRC (Table 5). The results were adjusted for age and gender due to significant difference in the median age and gender distribution among the case and the control group (Table 1). The imputation of genotypes based on HapMap data did not reveal any SNP to be associated with the risk of CRC.
Table 5.
Genotype distributions of the polymorphisms in CTNNBL1 in the hospital-based Czech and the family/early onset-based German sample population showing combined results adjusted for age, gender and nationality
| SNP | Allelea | Sample | Genotype distributionb | Codominat Model | Dominant Model | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DD-DA-AA | DD vs DA | DD vs AA | DD vs DA+AA | ||||||||||
| Cases | Controls | OR | 95% CI | P | OR | 95% | CI | P | OR | 95% | |||
| Czech | 606-111-2 | 658-80-3 | 1.47 | 1.07-2.02 | 0.02 | 0.82 | 0.13-5.29 | 0.84 | 1.45 | 1.06-1.98 | 0.02 | ||
| rs2344481 | A/G | German | 561-81-4 | 554-69-4 | 1.16 | 0.82-1.63 | 0.40 | 0.95 | 0.23-3.83 | 0.94 | 1.15 | 0.82-1.61 | 0.42 |
| Czech & German | 1167-192-6 | 1212-149-7 | 1.34 | 1.06-1.68 | 0.01 | 0.99 | 0.33-2.98 | 0.98 | 1.32 | 1.05-1.66 | 0.02 | ||
| Czech | 42-234-445 | 27-237-478 | 0.60 | 0.35-1.03 | 0.06 | 0.57 | 0.34-0.95 | 0.03 | 0.58 | 0.35-0.97 | 0.04 | ||
| rs2281148 | C/T | German | 37-199-418 | 41-212-408 | 1.08 | 0.66-1.76 | 0.76 | 1.15 | 0.72-1.83 | 0.57 | 1.12 | 0.71-1.77 | 0.64 |
| Czech & German | 79-433-863 | 68-449-886 | 0.81 | 0.56-1.15 | 0.23 | 0.83 | 0.59-1.16 | 0.27 | 0.82 | 0.59-1.15 | 0.25 | ||
| Czech | 96-342-262 | 124-368-243 | 1.20 | 0.89-1.65 | 0.26 | 1.39 | 1.00-1.93 | 0.05 | 1.28 | 0.95-1.72 | 0.11 | ||
| rs2235460 | A/G | German | 97-302-207 | 120-311-214 | 1.21 | 0.88-1.65 | 0.24 | 1.19 | 0.86-1.66 | 0.30 | 1.20 | 0.89-1.61 | 0.22 |
| Czech & German | 193-644-469 | 244-679-457 | 1.21 | 0.97-1.51 | 0.09 | 1.31 | 1.04-1.65 | 0.02 | 1.25 | 1.01-1.54 | 0.04 | ||
OR odds ratio; CI confidence interval; A ancestral allele, D derived allele.
Second allele represents the ancestral allele for each locus.
Total numbers of cases and controls may slightly vary among SNPs due to missing genotypes. Bold numbers indicate statistical significance at 5% level. The results were adjusted for age due to significant difference in the median age distribution among the case and the control group.
Joint analysis of the two sample sets
The allele frequencies of the three tagSNPs genotyped in the two populations did not differ significantly between the two control groups. For two tagSNPs, the ORs were increased for the same genotypes in both the Czech and the German population (Table 5). The joint anlysis indicated a statistically significant association of rs2344481 with an OR of 1.32 (95% Cl 1.05-1.66, dominant model) and of rs2235460 with an OR of 1.31 (95% Cl 1.04-1.66, AA vs GG) after adjustment of the data for age, gender and nationality (Table 5).
Discussion
In this study, we found that polymorphisms in CTNNBL1 may be associated with CRC risk. In particular, tagSNP rs2344481 showed a statistically significant association with CRC in the Czech population before and after we adjusted the data for age, gender and BMI. The increased risk was conferred by the ancestral G allele of the polymorphism. This result was supported by imputation based on the HapMap data [24] which additionally revealed three SNPs up- and downstream CTNNBL1 to be associated with CRC. All three SNPs are in high LD with rs2344481. In contrast, no statistically significant association was found for familial/early onset CRC of German origin, although in the joint analysis of the two populations the association remained statistically significant. The results may indicate that the SNPs in CTNNBL1 are associated with the risk of non-familial, sporadic CRC or they may reflect the different geographical origins of the study populations. However, the different geographical origin of the study populations may play only a subordinate role for interpreting the results. Recent studies have shown that, regarding SNPs, the auto-somal gene pool in Europe is relatively homogeneous with a slightly distinct gradient in the North-South direction [39, 40]. In addition, mi-crosatellite data have shown that genetic makeup of the Czech and German population does not differ significantly [41]. Accordingly, the two sample populations may be attributed as central European without a significant stratification according to their geographic origin. Thus, the partly discordant association observed in the German and the Czech sample set might more probably be attributed to the different etiology of familial/early onset versus non-familial CRC, respectively.
The results may also indicate a chance finding, because the detected effect would have been lost in multiple comparison correction. However, considering the previously reported association of several SNPs in the CTNNBL1 with obesity [9,12,13], the ancestral nature of the risk alleles, and the fact that the gene itself may have been a target of positive selection, a true nature of the modest effect cannot be excluded.
The previously reported associations of SNPs in CTNNBL1 with an increased risk of obesity, increased BMI and fat mass [9, 12, 13], is also conferred by the ancestral alleles as the risk alleles (OR -1.3). The same SNPs are in high LD (r2>0.85) with the SNP rs2344481 which showed the most significant association with CRC in the present study. The non-existing differences in the median BMI of the Czech case and control population may be due to the fact that the blood samples and the anthropometric data were collected from the CRC patients at the time of diagnosis and the controls were individuals with gastrointestinal complaints, which might have influenced the patient's weight even before it was measured for this study.
Combining the results, it is reasonable that CTNNBL1, particularly rs2344481 and its linked SNPs, might play a role in at least two nutrition-related complex diseases. There is strong evidence that nutrition-related complex diseases can be associated with an ancestral risk allele [16]. In this study, we used data derived from several databases, statistical tests, and phylogenetic analyses, which consistently pointed to a likely shaping of CTNNBL1 by positive selection. First, the distinct change in the frequency of the ancestral alleles (up to more than 50% differences from YRI to CEU and to CHB/JPT), found in several SNPs captured by rs2344481, indicated that a selective process could have driven the development of the gene [42, 43]. Second, statistically significant fixation indices (FST) supported the assumption that positive selection has shaped CTNNBL1 [33, 34, 44]. The same applies for the observed combination of extreme standardized integrated haplotype scores (|iHS|) and strongly negative values of Fay and Wu's H for several SNPs captured in our study [36]. A comparison of approximately 800.000 SNPs has shown that a high |iHS| >2.5 corresponds to the most extreme 1% of the iHS values. The proportion of |iHS| >2 is higher in genie than in non-genic SNPs, showing varying values in different population (1.23 in YRI, 1.16 in CEU, 1.13 in CHB/JPT)[36]. Furthermore, the strong linkage disequilibrium and the low recombination rate implied a high intras-pecific conservation of CTNNBL1. At the same time, the human-mouse alignment indicated a high interspecific conservation within the whole gene region, including coding and non-coding sequences. In the human-mouse alignments, the coding regions of genes are well conserved in general, while intronic regions tend to be the least conserved feature type of non-coding but intergenic sequences, showing an average of 23% identity [45]. Interestingly, CTNNBL1 shows both features of interspecific and intras-pecific conservation encompassing the whole gene region. The findings were supported by the results of the phylogenetic analyses to the coding and non-coding sequences of CTNNBL1. The sequence differences of both the cDNA and intronic DNA of CTNNBL1 were species specific and allowed construction of phylogenetic trees that reflected one of the today's valid phylogenetic trees of animals quite well. Studies have shown that conserved non-genic sequences are an unexpected feature of mammalian genomes [46] and that the phylogenetic performance of single genes is highly variable [47, 48].
The characteristics of CTNNBL1 and the association of the ancestral alleles of several SNPs in this gene with CRC and obesity indicate that the ancestral susceptibility model may apply to this gene. As the actual function of CTNNBL1 remains unknown, one may speculate that both an adaptation to a new dietary composition and an adaptation of the immune system for the new requirements of the environment are possible triggers of the positive selection in CTNNBL1 [9, 48, 49]. The fact that the associated SNP was non-coding or that CRC is a late-onset disease does not negate the hypothesis. Several functional non-protein-coding DNA sequences, such as cis-regulatory sequences [50] and miRNAs [51], are known to be targets of selection. Polymorphisms associated with multifactorial traits are often located in non-coding sequences, such as in regulatory sequences, 3’ untranslated regions, introns or intergenic sequences of unknown transcriptional status [52]. Especially the new discoveries in the field of epigenetics highlight the role of non-coding DNA sequences in regulatory processes and associations of polymorphisms in these regions with complex diseases [52]. It should also be considered that the detected SNP may be linked to a still unknown functional variant as the actual target of selection, or that the SNP itself may have a so far unknown regulatory function. Furthermore, it has to be taken into account that CRC itself may not have been the trigger of the selection but a so far unknown trait. Accordingly, a mutation leading to a primary effect late in life may have a weak deleterious effect early in life [53].
Conclusion
In conclusion, our study suggests that polymorphisms in CTNNBL1 may be associated with CRC, possibly through a gene-environment interaction. Carriers of the ancestral G allele in rs2344481 had an increased risk of CRC. The effect was more prominent in the hospital-based Czech sample population than in the family/early onset-based German sample population. Additionally, we showed that CTNNBL1 was shaped by positive selection. Further studies are needed to identify the actual function of CTNNBL1, to define the selective trait and to validate our results in other sample populations. The analysis of the relationship between CTNNBL1, CRC and other nutrition related diseases may provide further insights into the evolution of common complex diseases.
Ethical standards
The study protocols were approved by the ethical committees of all participating clinical centers in Germany (Bochum, Bonn, Dresden, Düsseldorf, Heidelberg, Mannheim and Munich/Regensburg) and in Prague, Czech Republic. Written informed consent was obtained from all study participants. The ethical committees were the following: Ethik Kommission der Medizinischen Fakultät der Ruhr Universität Bochum [Reg.-Nr.:1514]; Ethik Kommission – Medizinische Fakultät Bonn [Lfd. Nr. 115/09]; Ethik Kommission der Medizinische Fakultät der Technischen Universität Dresden [Bearbeitungs- Nr. EK170102000]; Ethikkommission der Medizinische Fakultät der Heinrich Heine Universität Düsseldorf [Studiennummer: 1172]; Ethikkommission I der Universität -Medizinische Fakultät Heidelberg [Antrags- Nr.: 220/2002]; Ethikkommission II an der Fakultät für Klinische Medizin der Ruprechts-Karl-Universität Heidelberg (concerning samples from Mannheim) [Antrags- Nr.: 87/04]; Ethikkommission der Medizinische Fakultät Universität München [Projekt Nr. 255/98]; Etická komise Ústravu experimentální medicí AV ĈR; Ethics Committee of the Institute for clinical and Experimental Medicine and Faculty Thomayer Hospital [Ĉ.j. 786/09 (09-0409)].
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.JemalA, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Huxley RR, Ansary-Moghaddam A, Clifton P, Czernichow S, Parr CL, Woodward M. The impact of dietary and lifestyle risk factors on risk of colorectal cancer: a quantitative overview of the epidemiological evidence. Int J Cancer. 2009;125:171–180. doi: 10.1002/ijc.24343. [DOI] [PubMed] [Google Scholar]
- 4.de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer. 2004;4:769–780. doi: 10.1038/nrc1453. [DOI] [PubMed] [Google Scholar]
- 5.Hemminki K, Forsti A, Lorenzo Bermejo J. Surveying the genomic landscape of colorectal cancer. Am J Gastroenterol. 2009;104:789–790. doi: 10.1038/ajg.2008.100. [DOI] [PubMed] [Google Scholar]
- 6.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 7.Webb EL, Rudd MF, Sellick GS, El Galta R, Bethke L, Wood W, Fletcher O, Penegar S, Withey L, Qureshi M, Johnson N, Tomlinson I, Gray R, Peto J, Houlston RS. Search for low penetrance alleles for colorectal cancer through a scan of 1467 non-synonymous SNPs in 2575 cases and 2707 controls with validation by kin-cohort analysis of 14 704 first-degree relatives. Hum Mol Genet. 2006;15:3263–3271. doi: 10.1093/hmg/ddl401. [DOI] [PubMed] [Google Scholar]
- 8.Tomlinson IP, Dunlop M, Campbell H, Zanke B, Gallinger S, Hudson T, Koessler T, Pharoah PD, Niittymaki I, Tuupanen S, Aaltonen LA, Hemminki K, Lindblom A, Forsti A, Sieber O, Upton L, van Wezel T, Morreau H, Wijnen JT, Devilee P, Matsuda K, Nakamura Y, Castellvi-Bel S, Ruiz-Ponte C, Castells A, Carracedo A, Ho JW, Sham P, Hofstra RM, Vodicka P, Brenner H, Hampe J, Schafmayer C, Tepel J, Schreiber S, Volzke H, Lerch MM, Schmidt CA, Buch S, Moreno V, Villanueva CM, Peterlongo P, Radice P, Echeverry MM, Velez A, Carvajal-Carmona L, Scott R, Penegar S, Broderick P, Tenesa A, Houlston RS. COGENT (COIorectal cancer GE-NeTics): an international consortium to study the role of polymorphic variation on the risk of colorectal cancer. Br J Cancer. 2010;102:447–454. doi: 10.1038/sj.bjc.6605338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu YJ, Liu XG, Wang L, Dina C, Yan H, Liu JF, Levy S, Papasian CJ, Drees BM, Hamilton JJ, Meyre D, Delplanque J, Pei YF, Zhang L, Recker RR, Froguel P, Deng HW. Genome-wide association scans identified CTNNBL1 as a novel gene for obesity. Hum Mol Genet. 2008;17:1803–1813. doi: 10.1093/hmg/ddn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- 11.Prestwich TC, Macdougald OA. Wnt/beta-catenin signaling in adipogenesis and metabolism. Curr Opin Cell Biol. 2007;19:612–617. doi: 10.1016/j.ceb.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban HJ, Yoon D, Lee MH, Kim DJ, Park M, Cha SH, Kim JW, Han BG, Min H, Ahn Y, Park MS, Han HR, Jang HY, Cho EY, Lee JE, Cho NH, Shin C, Park T, Park JW, Lee JK, Cardon L, Clarke G, McCarthy MI, Lee JY, Oh B, Kim HL. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet. 2009;41:527–534. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 13.Andreasen CH, Mogensen MS, Borch-Johnsen K, Sandbaek A, Lauritzen T, Almind K, Hansen L, Jorgensen T, Pedersen O, Hansen T. Studies of CTNNBL1 and FDFT1 variants and measures of obesity: analyses of quantitative traits and case-control studies in 18,014 Danes. BMC Med Genet. 2009;10:17. doi: 10.1186/1471-2350-10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellisari A. Evolutionary origins of obesity. Obes Rev. 2008;9:165–180. doi: 10.1111/j.1467-789X.2007.00392.x. [DOI] [PubMed] [Google Scholar]
- 15.Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, Drake JA, Rhodes M, Reich DE, Hirschhorn JN. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Rienzo A, Hudson RR. An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends Genet. 2005;21:596–601. doi: 10.1016/j.tig.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Southam L, Soranzo N, Montgomery SB, Frayling TM, McCarthy MI, Barroso I, Zeggini E. Is the thrifty genotype hypothesis supported by evidence based on confirmed type 2 diabetes-and obesity-susceptibility variants? Diabetologia. 2009;52:1846–1851. doi: 10.1007/s00125-009-1419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pechlivanis S, Bermejo JL, Pardini B, Naccarati A, Vodickova L, Novotny J, Hemminki K, Vodicka P, Forsti A. Genetic variation in adipokine genes and risk of colorectal cancer. Eur J Endocrinol. 2009;160:933–940. doi: 10.1530/EJE-09-0039. [DOI] [PubMed] [Google Scholar]
- 19.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 20.Mangold E, Pagenstecher C, Friedl W, Mathiak M, Buettner R, Engel C, Loeffler M, Holinski-Feder E, Muller-Koch Y, Keller G, Schackert HK, Kruger S, Goecke T, Moeslein G, Kloor M, Gebert J, Kunstmann E, Schulmann K, Ruschoff J, Propping P. Spectrum and frequencies of mutations in MSH2 and MLH1 identified in 1,721 German families suspected of hereditary nonpolyposis colorectal cancer. Int J Cancer. 2005;116:692–702. doi: 10.1002/ijc.20863. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin L, Srivastava S. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997;89:1758–1762. doi: 10.1093/jnci/89.23.1758. [DOI] [PubMed] [Google Scholar]
- 22.Frank B, Burwinkel B, Bermejo JL, Forsti A, Hemminki K, Houlston R, Mangold E, Rahner N, Friedl W, Friedrichs N, Buettner R, Engel C, Loeffler M, Holinski-Feder E, Morak M, Keller G, Schackert HK, Kruger S, Goecke T, Moeslein G, Kloor M, Gebert J, Kunstmann E, Schulmann K, Ruschoff J, Propping P. Ten recently identified associations between nsSNPs and colorectal cancer could not be replicated in German families. Cancer Lett. 2008;271:153–157. doi: 10.1016/j.canlet.2008.05.043. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The.International.HapMap.Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolbe D, Taylor J, Elnitski L, Eswara P, Li J, Miller W, Hardison R, Chiaromonte F. Regulatory potential scores from genome-wide three-way alignments of human, mouse, and rat. Genome Res. 2004;14:700–707. doi: 10.1101/gr.1976004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.King DC, Taylor J, Elnitski L, Chiaromonte F, Miller W, Hardison RC. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 2005;15:1051–1060. doi: 10.1101/gr.3642605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 28.Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998;14:68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- 29.Hubbard TJ, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Clarke L, Coates G, Fairley S, Fitzgerald S, Fernandez-Banet J, Gordon L, Graf S, Haider S, Hammond M, Holland R, Howe K, Jenkinson A, Johnson N, Kahari A, Keefe D, Keenan S, Kinsella R, Kokocinski F, Kulesha E, Lawson D, Longden I, Megy K, Meidl P, Overduin B, Parker A, Pritchard B, Rios D, Schuster M, Slater G, Smedley D, Spooner W, Spudich G, Trevanion S, Vilella A, Vogel J, White S, Wilder S, Zadissa A, Birney E, Cunningham F, Curwen V, Durbin R, Fernandez-Suarez XM, Herrero J, Kasprzyk A, Proctor G, Smith J, Searle S, Flicek P. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 31.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 32.Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bio-inform Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 33.Coop G, Pickrell JK, Novembre J, Kudaravalli S, Li J, Absher D, Myers RM, Cavalli-Sforza LL, Feldman MW, Pritchard JK. The role of geography in human adaptation. PLoS Genet. 2009;5:el000500. doi: 10.1371/journal.pgen.1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright S. Chicago: Univ. of Chicago Press; 1978. Evolution and the Genetics of Populations: Variability Within and Among Natural Populations. [Google Scholar]
- 35.Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pechlivanis S, Pardini B, Bermejo JL, Wagner K, Naccarati A, Vodickova L, Novotny J, Hemminki K, Vodicka P, Forsti A. Insulin pathway related genes and risk of colorectal cancer: INSR promoter polymorphism shows a protective effect. Endocr Relat Cancer. 2007;14:733–740. doi: 10.1677/ERC-07-0107. [DOI] [PubMed] [Google Scholar]
- 38.Murphy WJ, Eizirik E, O'Brien SJ, Madsen O, Scally M, Douady CJ, Teeling E, Ryder OA, Stanhope MJ, de Jong WW, Springer MS. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science. 2001;294:2348–2351. doi: 10.1126/science.1067179. [DOI] [PubMed] [Google Scholar]
- 39.Lao O, Lu TT, Nothnagel M, Junge O, Freitag-Wolf S, Caliebe A, Balascakova M, Bertranpetit J, Bindoff LA, Comas D, Holmlund G, Kouvatsi A, Macek M, Mollet I, Parson W, Palo J, Ploski R, Sajantila A, Tagliabracci A, Gether U, Werge T, Rivadeneira F, Hofman A, Uitterlinden AG, Gieger C, Wichmann HE, Ruther A, Schreiber S, Becker C, Nurnberg P, Nelson MR, Krawczak M, Kayser M. Correlation between genetic and geographic structure in Europe. Curr Biol. 2008;18:1241–1248. doi: 10.1016/j.cub.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 40.Nelis M, Esko T, Magi R, Zimprich F, Zimprich A, Toncheva D, Karachanak S, Piskackova T, Balascak I, Peltonen L, Jakkula E, Rehnstrom K, Lathrop M, Heath S, Galan P, Schreiber S, Meitinger T, Pfeufer A, Wichmann HE, Melegh B, Polgar N, Toniolo D, Gasparini P, D'Adamo P, Klovins J, Nikitina-Zake L, Kucinskas V, Kasnauskiene J, Lubinski J, Debniak T, Limborska S, Khrunin A, Estivill X, Rabionet R, Marsal S, Julia A, Antonarakis SE, Deutsch S, Borel C, Attar H, Gagnebin M, Macek M, Krawczak M, Remm M, Metspalu A. Genetic structure of Europeans: a view from the North-East. PLoS One. 2009;4:e5472. doi: 10.1371/journal.pone.0005472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zastera J, Roewer L, Willuweit S, Sekerka P, Benesova L, Minarik M. Assembly of a large Y-STR haplotype database for the Czech population and investigation of its substructure. Forensic Sci Int Genet. 2010;4:e75–78. doi: 10.1016/j.fsigen.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, Ackerman HC, Campbell SJ, Altshuler D, Cooper R, Kwiatkowski D, Ward R, Lander ES. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 43.Novembre J, Di Rienzo A. Spatial patterns of variation due to natural selection in humans. Nat Rev Genet. 2009;10:745–755. doi: 10.1038/nrg2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting F(ST) Nat Rev Genet. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jareborg N, Birney E, Durbin R. Comparative analysis of noncoding regions of 77 orthologous mouse and human gene pairs. Genome Res. 1999;9:815–824. doi: 10.1101/gr.9.9.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dermitzakis ET, Reymond A, Antonarakis SE. Conserved non-genic sequences - an unexpected feature of mammalian genomes. Nat Rev Genet. 2005;6:151–157. doi: 10.1038/nrg1527. [DOI] [PubMed] [Google Scholar]
- 47.Aguileta G, Marthey S, Chiapello H, Lebrun MH, Rodolphe F, Fournier E, Gendrault-Jacquemard A, Giraud T. Assessing the performance of single-copy genes for recovering robust phylogenies. Syst Biol. 2008;57:613–627. doi: 10.1080/10635150802306527. [DOI] [PubMed] [Google Scholar]
- 48.Graybeal A. Evaluating the Phylogenetic Utility of Genes: A Search for Genes Informative About Deep Divergences among Vertebrates. Systematic Biology. 1994;43:174–193. [Google Scholar]
- 49.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibody-diversification enzyme AID and spli-ceosome-associated factor CTNNBL1. Mol Cell. 2008;31:474–484. doi: 10.1016/j.molcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 50.Wray GA. The evolutionary significance of cisregulatory mutations. Nat Rev Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- 51.Quach H, Barreiro LB, Laval G, Zidane N, Patin E, Kidd KK, Kidd JR, Bouchier C, Veuille M, Antoniewski C, Quintana-Murci L. Signatures of purifying and local positive selection in human miRNAs. Am J Hum Genet. 2009;84:316–327. doi: 10.1016/j.ajhg.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mattick JS. The genetic signatures of noncoding RNAs. PLoS Genet. 2009;5:el000459. doi: 10.1371/journal.pgen.1000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]