

Abstract
Hyperpolarization-activated cyclic nucleotide-regulated (HCN) channels in the brain associate with their auxiliary subunit TRIP8b (also known as PEX5R), a cytoplasmic protein expressed as a family of alternatively spliced isoforms. Recent in vitro and in vivo studies have shown that association of TRIP8b with HCN subunits both inhibits channel opening and alters channel membrane trafficking, with some splice variants increasing and others decreasing channel surface expression. Here, we address the structural bases of the regulatory interactions between mouse TRIP8b and HCN1. We find that HCN1 and TRIP8b interact at two distinct sites: an upstream site where the C-linker/cyclic nucleotide-binding domain of HCN1 interacts with an 80 aa domain in the conserved central core of TRIP8b; and a downstream site where the C-terminal SNL (Ser-Asn-Leu) tripeptide of the channel interacts with the tetratricopeptide repeat domain of TRIP8b. These two interaction sites play distinct functional roles in the effects of TRIP8b on HCN1 trafficking and gating. Binding at the upstream site is both necessary and sufficient for TRIP8b to inhibit channel opening. It is also sufficient to mediate the trafficking effects of those TRIP8b isoforms that downregulate channel surface expression, in combination with the trafficking motifs present in the N-terminal region of TRIP8b. In contrast, binding at the downstream interaction site serves to stabilize the C-terminal domain of TRIP8b, allowing for optimal interaction between HCN1 and TRIP8b as well as for proper assembly of the molecular complexes that mediate the effects of TRIP8b on HCN1 channel trafficking.
Introduction
The function and expression of many ion channels are regulated by subunits that do not contribute to the channel pore, but stably associate with pore-forming subunits. Recent studies provide strong evidence that the cytoplasmic tetratricopeptide repeat-containing Rab8b interacting protein TRIP8b (also known as PEX5R) is a regulatory subunit of the hyperpolarization-activated cyclic nucleotide-regulated (HCN) channels in the brain (Santoro et al., 2004, 2009: Zolles et al., 2009; Lewis et al., 2009). TRIP8b undergoes extensive alternative splicing at its N terminus. All splice forms inhibit HCN channel activation by antagonizing the ability of cyclic nucleotides to facilitate channel opening (Santoro et al., 2009; Zolles et al., 2009). In contrast, different splice variants produce diverse effects on HCN channel membrane trafficking (Lewis et al., 2009; Santoro et al., 2009).
TRIP8b isoforms contain a large constant domain (exons 5–16) preceded by a variable region at which exons 1a or 1b combine with subsets of exons 2–4. The C-terminal half of TRIP8b (exons 9–16) comprises an ∼350 aa tetratricopeptide repeat (TPR) protein-binding domain with 57% identity to the PEX5 peroxisome receptor TPR domain. In contrast, TRIP8b and PEX5 have no sequence similarity in their initial N-terminal ∼250 aa, consistent with their divergent functions. TRIP8b(1a-4) and TRIP8b(1a-2–4) (the naming convention lists the alternatively spliced exons) cause a fourfold to sixfold increase in surface expression of HCN1 channels when coexpressed in Xenopus oocytes. In contrast, TRIP8b(1a) decreases HCN1 surface expression by 10-fold, and both TRIP8b(1b-2) and TRIP8b(1b-2–4) virtually eliminate channel surface expression (>50-fold decrease) (Santoro et al., 2009). The decrease in surface expression requires N-terminal consensus sites for adaptor protein (AP) Complex binding in exons 2 and 5 that likely promote clathrin-dependent channel endocytosis (Santoro et al., 2004, 2009; Popova et al., 2008; Petrenko et al., 2010).
Our laboratory initially found that the TRIP8b TPR domain interacts with the SNL (Ser-Asn-Leu) tripeptide at the C terminus of HCN1, −2, and −4 (Santoro et al., 2004), which we refer to as the “downstream” interaction site. More recently, Lewis et al. (2009) identified a second “upstream” interaction site at which the C-linker region and contiguous cyclic nucleotide-binding domain (CNBD) of HCN1 binds to an internal sequence in the conserved middle region of TRIP8b. Here we map in finer detail the boundaries of the two physical interaction sites between HCN1 and TRIP8b, and define the roles of these interactions in protein binding, channel trafficking, and channel gating. We find that the upstream interaction site is necessary and sufficient to enable TRIP8b to downregulate channel surface expression and inhibit channel activation. In contrast, the downstream interaction site stabilizes the C-terminal domain of TRIP8b, allowing for optimal interaction between HCN1 and TRIP8b. In addition, an intact TPR domain is required for the effect of certain TRIP8b splice forms to enhance HCN1 channel surface expression. Finally, we identify an 80 aa fragment within the core of TRIP8b that is sufficient to inhibit the action of cyclic nucleotides to facilitate HCN1 channel opening.
Materials and Methods
Plasmids and heterologous expression in Xenopus oocytes.
All constructs were cloned in pGHE or pGH19 vectors, linearized, and transcribed into cRNA using T7 polymerase (MessageMachine, Ambion) as described previously (Santoro et al., 2004, 2009). cDNA clones encoding HCN1 and TRIP8b both corresponded to the Mus musculus sequence. Site-directed mutagenesis was performed using either the QuickChange Mutagenesis kit (Stratagene) or PCR cloning. The enhanced green fluorescent protein (GFP) tag (derived from vector pEGFP-C1, Clontech) was always inserted at the N-terminal end of the target protein (HCN1 or TRIP8b, wild-type or mutant) (Santoro et al., 2004), with the exception of the CD8 fusion construct, where the GFP sequence was inserted between the CD8 transmembrane domain and the HCN1776–910 domain (CD8-GFP-HCN1776–910). Mutant HCN1 constructs are as described previously (Wainger et al., 2001; Santoro et al., 2004), with HCN1ΔSNL comprising residues HCN11–907, HCN1ΔCX comprising residues HCN11–610, and HCN1ΔCNBD comprising residues HCN11–472. Specific residue numbers for corresponding mutations introduced in the background of the three TRIP8b splice variants are as follows: TRIP8bΔNX, which lacks most of the initial N-terminal region that is divergent from PEX5, corresponds to TRIP8b(1b-2)204–615 [identical to TRIP8b(1a-4)191–602 or TRIP8b(1a)156–567]; TRIP8bΔNter, a larger N-terminal deletion extending into the PEX5 homology domain, corresponds to TRIP8b(1b-2)272–615 [identical to TRIP8b(1a-4)259–602 or TRIP8b(1a)224–567]; TRIP8bΔCter, the complementary fragment to TRIP8bΔNter (see also supplemental Fig. 1, available at www.jneurosci.org as supplemental material), corresponds to TRIP8b(1b-2)1–271, TRIP8b(1a-4)1–258, and TRIP8b(1a)1–223; TRIP8bΔTPR, which lacks all but the first two helices of the C-terminal TPR domain, corresponds to TRIP8b(1b-2)1–346, TRIP8b(1a-4)1–333, and TRIP8b(1a)1–298; the minimal TRIP8b fragment (TRIP8bmini), which consists of an 81 aa stretch immediately upstream of the TPR domain, corresponds to TRIP8b(1b-2)236–316 [identical to TRIP8b(1a-4)223–303 or TRIP8b(1a)188–268]; TRIP8bΔint, in which a 22 aa region in the conserved core of TRIP8b is internally deleted, corresponds to TRIP8b(1b-2)Δ250–271, TRIP8b(1a-4)Δ237–258, and TRIP8b(1a)Δ202–223; and TRIP8bNK, in which a conserved asparagine in the TPR domain is mutated to lysine, corresponds to TRIP8b(1b-2)N501K, TRIP8b(1a-4)N488K, and TRIP8b(1a)N453K. Oocytes were injected with 50 nl of cRNA solution each, at a concentration of 0.1–1.0 μg/μl for HCN1 channel constructs (as specified in figure legends) and 0.2 μg/μl for TRIP8b constructs and GFP (or as specified in figure legends). In each given set of experiments, we always used a constant ratio of HCN1 cRNA to TRIP8b or GFP cRNA.
Coimmunoprecipitation and Western blots.
Oocytes were screened for GFP expression by live confocal microscopy 3 d after cRNA injection. Positive oocytes were collected, briefly rinsed in Barth's Saline (Millipore Bioscience Research Reagents), and homogenized in ice-cold lysis buffer (150 mm NaCl, 10 mm Tris-Cl pH 7.4, 1 mm EGTA, 2 mm EDTA, 1% Triton) with protease inhibitors (Complete Mini, Roche) using a glass Teflon homogenizer, at a constant ratio of 12 oocytes per 300 μl of buffer. The homogenate was spun twice for 5 min at 12,000 × g (4°C), and the second supernatant was used for Western blot analysis of protein expression levels (input) or coimmunoprecipitation. For coimmunoprecipitation, 30 μl of extract was mixed with 120 μl of coimmunoprecipitation buffer (150 mm NaCl, 10 mm Tris), precleared for 1 h at 4°C, and then incubated overnight at 4°C with 15 μl of Protein-A-Sepharose beads (GE Healthcare) bound to anti-GFP antibody (Abcam 290; 1.5 μl of for each sample) or anti-TRIP8b antibody (rabbit polyclonal 794, 2 μl of serum for each sample) (Santoro et al., 2009). Beads were washed three times with TBST (150 mm NaCl, 10 mm Tris-Cl pH 7.4, 0.1% Triton) and proteins eluted in Laemmli sample buffer. Proteins were resolved by 8% SDS-PAGE, transferred to a polyvinylidene difluoride membrane (PolyScreen, PerkinElmer) in CAPS buffer (10 mm CAPS, pH 11, 10% methanol) and Western blot analysis performed as described previously (Santoro et al., 2009). Primary antibody dilutions were as follows: anti-HCN1 (rat monoclonal 7C3, generous gift from Frank Müller, Forschungszentrum Jülich, Jülich, Germany) 1:500; anti-TRIP8b (rabbit polyclonal 794) (Santoro et al., 2009) 1:5000; and anti-GFP (Abcam 290) 1:5000. HRP-anti-rat conjugate (Jackson ImmunoResearch) or HRP-anti-rabbit conjugate (Cell Signaling Technology) were used as secondary antibodies. The protein bands were visualized by chemiluminescence using SuperSignal reagent (Pierce).
Yeast two-hybrid assay.
Assays were performed using the Grow'N′Glow Two-Hybrid kit (Bio 101) (Santoro et al., 2004) and yeast strain EGY48. Bait constructs representing the indicated HCN1 channel domains were cloned into vector pEG202, and prey constructs representing the indicated TRIP8b domains (or mutants thereof) were cloned in vector pJG4–5. Channel domains representing successive deletions of the six (A′–F′) helixes that comprise the HCN1 C-linker region (a highly conserved 84 aa region, residues 387–471, connecting the channel transmembrane domain to the 120 aa CNBD) are as follows: CNBD alone, HCN1472–592; E′–F′ C-linker helixes plus the CNBD, HCN1453–592; C′–F′ C-linker helixes plus the CNBD, HCN1429–592; and A′–F′ C-linker helixes plus the CNBD, HCN1387–592. The construct representing the C-terminal domain of HCN1 is as described previously (Santoro et al., 2004) and comprises residues HCN1776–910. The C-linker/CNBD region construct (see Fig. 4B,C) comprises residues HCN1390–610 (same C-terminal ending as HCN1ΔCX). Note that all TRIP8b constructs used for the yeast two-hybrid assay comprise the constant domain of TRIP8b only (exons 5–16); that is, they exclude all N-terminal alternatively spliced exons. To facilitate referencing, however, numbering in Figure 4 is based on full-length TRIP8b(1b-2). Bait and prey plasmids were cotransformed with reporter plasmid pGNG1, and cells were plated onto glucose-containing medium. Transformants were restreaked (in triplicate) on galactose+/Leu− selective medium and screened for positive GFP expression under a UV light following 3–5 d of growth.
Figure 4.

Minimal HCN1 and TRIP8b domains required for binding at the upstream and downstream interaction sites. The binding activity of a set of HCN1 and TRIP8b fragments was assessed using the yeast two-hybrid assay. Activity was detected by transactivation of a GFP reporter gene. ++, Strong fluorescence; +++, very strong fluorescence; −, no detectable fluorescence. A, Binding of TRIP8b to HCN1 C-linker and CNBD region. The TRIP8b construct (see Materials and Methods) failed to interact with the CNBD alone, but showed strong binding to the CNBD plus two (E and F), four (C–F) or all six C-linker α helixes (A–F). B, Binding of different TRIP8b wild-type and mutant constructs to the HCN1 C-linker/CNBD region (HCN1390–611) or extreme C-terminal 134 aa region, including the SNL tripeptide (HCN1776–910). C, Binding of TRIP8b bearing different TPR domain point mutations to HCN1 C-linker/CNBD or extreme C-terminus regions (as in B). All TPR mutants prevented interaction with the extreme C terminus of HCN1 but did not alter binding to the C-linker/CNBD fragment. TRIP8b numbering is based on isoform TRIP8b(1b-2), as used in Figure 3 (see Materials and Methods).
Affinity purification of protein complexes from bacterial lysates.
A cDNA fragment encoding residues 470–672 of hHCN2 (Ludwig et al., 1999) was cloned into a modified pET-24b vector (generous gift from Dan Minor, University of California, San Francisco, San Francisco, CA) downstream of a double His6-maltose binding protein (MBP) tag, while cDNA fragments corresponding to residues 236–316 (TRIP8bmini) and 272–615 (TRIP8bΔNter) of TRIP8b(1b-2) were cloned into vector pET-52b (Novagen) downstream of a Strep(II) tag sequence. Plasmids were cotransformed into Escherichia coli BL21 Rosetta strain (Novagen), under double ampicillin and kanamycin selection. Cells were grown at 37°C in Luria broth to 0.6 OD600 and induced with 0.4 mm isopropyl-1-thioβ-d-galactopyranoside. After 3 h, cells were collected by centrifugation, resuspended in ice-cold lysis buffer (150 mm NaCl, 100 mm Tris-Cl pH 8, 1 mm EDTA, 1 mm β-mercaptoethanol, 5 μg/ml leupeptin, 1 μg/ml pepstatin, 100 μm phenylmethylsulphonyl chloride) with the addition of 10 μg/ml DNase and 0.25 mg/ml lysozyme, sonicated on ice 12 × 20 s, and the lysate was cleared by centrifugation for 30 min at 20,000 × g. Proteins were purified by affinity chromatography using StrepTrap HP columns (GE Healthcare), according to the manufacturer's instructions, and eluted in 500 mm NaCl, 30 mm HEPES pH 7.5, 10% glycerol, plus 2.5 mm desthiobiotin. All purification steps were performed at 4°C and monitored using the ÄKTApurifier UPC 10 fast protein liquid chromatography system (GE Healthcare). Samples were analyzed by SDS-PAGE using a NuPAGE (Novex) Bis-Tris 4–12% gel, in MES buffer (Invitrogen).
Electrophysiology.
Two-microelectrode voltage-clamp recordings were obtained 3 d after cRNA injection, using a Warner Instruments OC-725C amplifier. Data were digitized and acquired with an ITC-16 interface (Instrutech), filtered at 1 kHz and sampled at 2 kHz, and analyzed using Pulse software (HEKA). Microelectrodes filled with 3 m KCl had resistances of 0.5–2.0 MΩ. Oocytes were bathed in extracellular solution containing the following (in mm): 94 NaCl, 4 KCl, 10 HEPES, and 2 MgCl2, pH 7.5. Three-second voltage steps were applied from a holding potential of −30 mV to a range of test potentials between −15 and −125 mV in 10 mV decrements, followed by a depolarizing step to 0 mV. Peak tail-current amplitudes were measured at 0 mV after the decay of the capacitive transient, and tail-current current–voltage curves fitted using the Boltzmann equation, I(V) = A1 + A2/{1 + exp[(V − V1/2)/s]}, in which A1 is the offset caused by holding current, A2 is the maximal tail current amplitude, V is the test pulse voltage, V1/2 is the midpoint voltage of activation, and s is the slope factor (in mV). All recordings were obtained at room temperature (22–24°C).
Results
Structural mapping
Previous studies have identified upstream and downstream HCN1/TRIP8b interaction sites largely based on the binding activity of isolated protein fragments (Santoro et al., 2004; Lewis et al., 2009). Here we have addressed the relative contribution of the two putative interaction domains to binding under more physiological conditions by performing a series of coimmunoprecipitation experiments with wild-type and mutant TRIP8b and HCN1 proteins coexpressed in Xenopus oocytes from injected cRNAs. In addition to expressing the channel in its native membrane context, this approach allowed us to titrate the amount of HCN1 and TRIP8b protein in each cell to ensure that wild-type and mutant proteins were expressed at similar levels, thus allowing their binding activity to be compared in a semiquantitative manner. Further, we tagged the N terminus of wild-type and mutant constructs with GFP so that, in addition to monitoring expression levels, all experiments could be performed using the same antibody, independent of the position of the deletion being studied.
In a first set of experiments, we compared the binding of different HCN1 deletion mutants with full-length, wild-type TRIP8b (Fig. 1). Consistent with previous results, truncation of the C-terminal SNL tripeptide of HCN1 strongly reduced, but did not abolish, channel binding to TRIP8b. Binding activity was still retained in a larger HCN1 C-terminal truncation in which the CNBD was intact but all downstream 300 aa were deleted (HCN1ΔCX, containing residues 1–610) (Wainger et al., 2001). However, an even larger truncation in which both the CNBD and downstream C-terminal amino acids were deleted (HCN1ΔCNBD, containing residues 1–472) (Wainger et al., 2001) essentially abolished TRIP8b binding. These findings suggest that the CNBD is an essential component of the upstream interaction site, and that the N terminus, transmembrane domain, and C-linker region of the channel are not, by themselves, sufficient to support significant HCN1/TRIP8b interaction.
Figure 1.

Contribution of HCN1 upstream and downstream binding sites to the interaction with TRIP8b. Western blots show binding of HCN1, its deletion mutants, and other membrane proteins to wild-type TRIP8b(1b-2) assessed by coimmunoprecipitation from Xenopus oocyte extracts. Oocytes were coinjected with TRIP8b cRNA (0.2 μg/μl) and cRNAs (at indicated concentration) of various constructs, all tagged with GFP: GFP-HCN1 (1.0 μg/μl), GFP-HCN1ΔSNL (1.0 μg/μl), GFP-HCN1ΔCX (0.1 μg/μl), GFP-HCN1ΔCNBD (0.1 μg/μl), GFP-CNGA1 (0.5 μg/μl), and CD8-GFP-HCN1776–910 (0.5 μg/μl). The left column shows Western blot using GFP antibody as measure of input signal. The middle column shows the TRIP8b input signal using an anti-TRIP8b antibody. The right column shows the amount of target protein coimmunoprecipitated with TRIP8b antibody (Western blot using GFP antibody). Note that exposure times are directly comparable down each column, but not along each row. The two bands in the CD8-GFP-HCN1776–910 samples likely represent the unglycosylated (lower) and glycosylated (upper) forms of the CD8 receptor moiety (Pascale et al., 1992). Individual bands have been cut from intact gel pictures and aligned to allow direct comparison of intensities for wild-type and mutant constructs with differing molecular weights. Relevant HCN1 channel domains and amino acid positions are indicated in the icons on the left (black, transmembrane domain; orange, C-linker region; purple, cyclic nucleotide binding domain; light green, C-terminal SNL tripeptide).
Given the high degree of conservation between the CNBD of HCN channels and that of cyclic nucleotide-gated (CNG) channels, we asked whether these close relatives might also display TRIP8b binding activity. However, when assayed in the same conditions, no binding was observed between the CNGA1 channel and TRIP8b, demonstrating the specificity of the HCN/TRIP8b interaction (Fig. 1). Finally, we asked whether interaction at the downstream SNL/TPR site was sufficient for protein binding in native conditions. To address this question, we attached the extreme C-terminal 134 aa of HCN1 (residues 776–910) to the C terminus of a fusion protein in which the single-pass membrane protein CD8 was fused at its C terminus to GFP (CD8-GFP-HCN1776–910). Coimmunoprecipitation experiments showed that TRIP8b was able to bind efficiently to the HCN1 C terminus in the context of this minimal membrane protein (Fig. 1). When TRIP8b(1b-2) is coexpressed with HCN channels, in either Xenopus oocytes or HEK293 cells, the channel protein relocalizes to intracellular puncta (Fig. 2A,C) (Santoro et al., 2004, 2009). Interestingly, coexpression in oocytes of TRIP8b(1b-2) with CD8-GFP-HCN1776–910 similarly resulted in the formation of distinct puncta (Fig. 2B,D). The redistribution of CD8-GFP-HCN1776–910 into puncta confirms that TRIP8b(1b-2) is able to interact functionally with the HCN1 extreme C terminus, as the formation of the puncta was clearly dependent on the presence of the HCN1776–910 fragment (Fig. 2E).
Figure 2.

Functional assay of the interaction between TRIP8b(1b-2) and the HCN1 extreme C terminus in live cells. A–D, Live confocal imaging of Xenopus oocytes injected with cRNA encoding either a GFP-HCN1 (A) or a CD8-GFP-HCN1776–910 fusion protein alone (B), or in combination with TRIP8b(1b-2) (C, D). A redistribution of the GFP fluorescence into distinct puncta is visible in both cases, indicating the ability of TRIP8b(1b-2) to alter the localization of both fusion proteins. E, Coexpression of TRIP8b(1b-2) with a CD8-GFP fusion protein lacking the C-terminal domain of HCN1 does not show such redistribution, indicating that the action of TRIP8b(1b-2) is specifically dependent on its binding to the target HCN1776–910 sequence.
In a complementary set of experiments, we compared the binding of different TRIP8b mutants, in the background of isoform TRIP8b(1b-2), to full-length wild-type HCN1 (Fig. 3). We first sought to selectively disrupt the interaction between HCN1 and TRIP8b(1b-2) by introducing a point mutation in a conserved TPR residue, N501K, which greatly diminishes the binding of the PEX5 TPR domain to the C-terminal SKL (Ser-Lys-Leu) tripeptide in PEX5 target proteins (Gatto et al., 2000). In the crystal structure of the PEX5/target peptide complex, the side-chain amide group of this asparagine forms a direct hydrogen bond with the C-terminal carboxylate of the SKL peptide. As expected, TRIP8b(1b-2)N501K binding to HCN1 was strongly reduced, although not abolished, similar to what we observed upon deletion of the HCN1 SNL tripeptide (compare Figs. 1, 3). This suggests that the interaction between the TRIP8b TPR domain and the HCN channel SNL sequence is similar to the interaction of PEX5 with its SKL-containing targets. Furthermore, this result indicates that an intact TPR domain is not required for an interaction between HCN1 and TRIP8b at the upstream CNBD/core binding site in native conditions.
Figure 3.
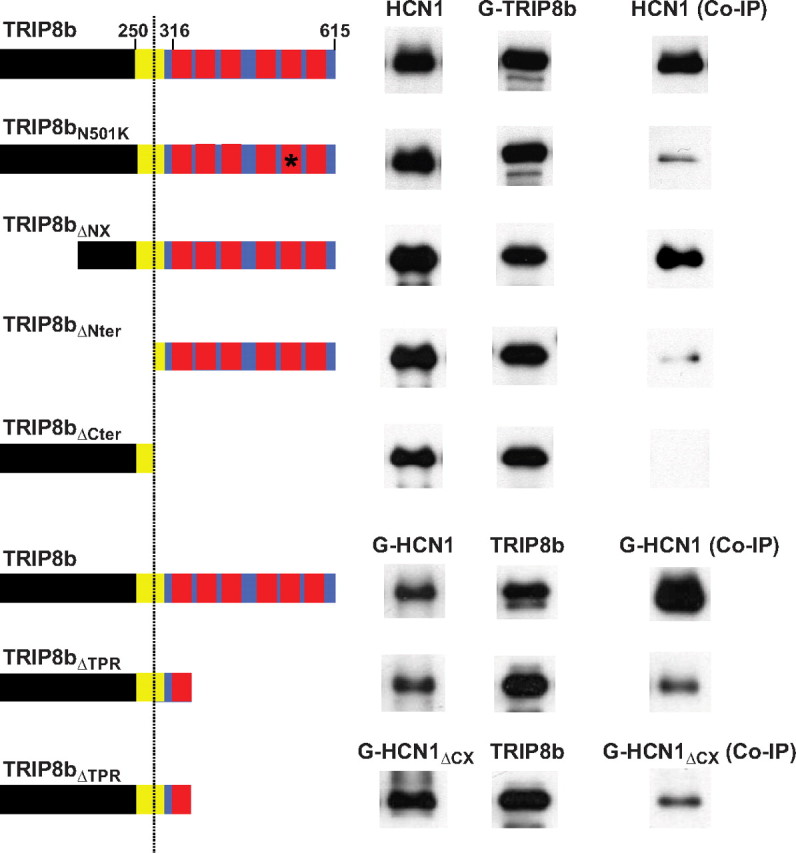
Contribution of TRIP8b upstream and downstream binding sites to the interaction with HCN1. Western blots show binding of a set of TRIP8b(1b-2) mutants to wild-type HCN1 assessed by coimmunoprecipitation from Xenopus oocyte extracts, as in Figure 1. Oocytes were coinjected with cRNA encoding HCN1 (0.5 μg/μl), GFP-HCN1 (1.0 μg/μl), or GFP-HCN1ΔCX (0.1 μg/μl), as indicated, and cRNA for TRIP8b N-terminally tagged with GFP (GFP-TRIP8b; 0.2 μg/μl, first five rows) or untagged TRIP8b (0.2 μg/μl, last three rows). Left column, HCN1 input signal (Western blot using HCN1 antibody for first five rows or GFP antibody for last three rows). Middle column, TRIP8b input signal (Western blot using GFP antibody for first five rows or TRIP8b antibody for last three rows). Right column, amount of HCN1 coimmunoprecipitated with TRIP8b (immunoprecipitation performed using anti-GFP for first five rows or anti-TRIP8b antibody for last three rows). As in Figure 1, exposure times are directly comparable down each column, but not along each row. Individual bands have been cut from intact gel pictures and aligned to allow direct comparison of intensities for wild-type and mutant constructs with differing molecular weights. Relevant protein domains and amino acid positions are indicated in the icons on the left. The black color denotes the N-terminal region that is unique to TRIP8b. The blue color denotes the region of TRIP8b that is homologous to PEX5, with the red squares representing the tetratricopeptide repeats. The yellow square represents a highly conserved core region, homologous to the corresponding region of PEX5, immediately preceding the TPR repeats (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The vertical dotted line marks the position of residue 272 in isoform TRIP8b(1b-2), corresponding to residue 259 in isoform TRIP8b(1a-4).
We next examined whether the N-terminal region of TRIP8b, which is not homologous with PEX5, plays any role in HCN1 binding. We first deleted the initial 203 aa of TRIP8b(1b-2), which eliminates all alternatively spliced exons as well as the consensus motifs required for the putative binding of TRIP8b to intracellular trafficking factors (Santoro et al., 2009; Petrenko et al., 2010). This TRIP8b(1b-2) N-terminal deletion mutant (TRIP8bΔNX) still strongly binds to HCN1, indicating that the extreme N terminus of TRIP8b is not necessary for channel binding. However, a larger N-terminal deletion mutant lacking the first 271 residues (TRIP8bΔNter), a deletion that extends into the initial segment of the homology region between TRIP8b and PEX5 but leaves the TPR domain intact, exhibited a significant reduction in binding activity (Fig. 3). This finding is consistent with previous observations suggesting that the TRIP8bΔNter fragment binds to the HCN channel extreme C terminus, but not to the C-linker/CNBD region (Lewis et al., 2009). It further suggests that the contribution of the upstream interaction site to TRIP8b/HCN channel binding in native conditions is substantial and comparable to that of the downstream TPR/SNL interaction site. The portion of the homology region removed in TRIP8bΔNter is not sufficient to confer HCN binding activity, as the complementary N-terminal fragment (TRIP8bΔCter) failed to coimmunoprecipitate HCN1 (Fig. 3). However, a larger N-terminal fragment that includes the first tetratricopeptide repeat of TRIP8b (TRIP8bΔTPR) did efficiently bind both wild-type HCN1 as well as the HCN1 extreme C-terminal truncation mutant (HCN1ΔCX), presumably due to its efficient interaction with the CNBD of the channel at the upstream binding site (Fig. 3).
To localize further the sequences required for interaction at the upstream and downstream binding sites, we used the yeast two-hybrid system to assay the binding activities of a series of HCN1 and TRIP8b constructs (Fig. 4). First, we explored the relative importance of the HCN1 120 aa CNBD and 80 aa C-linker region that connects the transmembrane domain of the channel to the CNBD (Fig. 4A). Although a construct expressing the CNBD alone produced large amounts of protein in yeast, as assessed by Western blot analysis (data not shown), it failed to bind to TRIP8b. We therefore explored CNBD constructs that included variable lengths of the C-linker region, which consists of six α helixes (A′–F′) (Zagotta et al., 2003). We found that the minimal construct that bound to TRIP8b comprised the CNBD plus the two immediate upstream E′–F′ helixes. Given that a channel truncation mutant that contains an intact C-linker but lacks the CNBD was not able to bind to TRIP8b (Fig. 1, HCN1ΔCNBD), the E′-F′ helix pair probably does not directly interact with TRIP8b but rather enables the proper folding of the CNBD. Indeed, in the HCN2 crystal structure the E′-F′ helix pair directly contacts the CNBD, whereas the A′-D′ helixes do not (Zagotta et al., 2003).
Next, we dissected the regions of TRIP8b involved in the upstream interaction with the HCN1 C-linker/CNBD domain and the downstream interaction with the C-terminal SNL tripeptide (HCN1776–910) (Fig. 4B). We found that a highly conserved ∼80 aa TRIP8b fragment extending from position 236 to position 316 in TRIP8b(1b-2), located just upstream of the first tetratricopeptide repeat (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material), was sufficient to bind to the C-linker/CNBD of HCN1. In contrast, this minimal TRIP8b fragment (TRIP8bmini) failed to interact with the HCN1 extreme C terminus, confirming that the 80 aa core region of TRIP8b interacts with HCN1 only at the upstream site. Conversely, a TRIP8b(1b-2) internal deletion mutant lacking 22 aa within the middle of the core region (TRIP8bΔint, which is missing amino acids 250–271) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) lost the ability to bind to the upstream C-linker/CNBD site of HCN1 but retained the capacity to interact with the downstream SNL site of the channel. Thus, sequences within TRIP8bmini are both necessary and sufficient to interact with the HCN1 C-linker/CNBD site, as assayed through the yeast two-hybrid screen.
To rule out potential artifacts of the yeast two-hybrid assay, we sought to confirm the interaction between the TRIP8bmini fragment and the HCN C-linker/CNBD region by direct biochemical analysis. To this end, we coexpressed in E. coli the HCN2 C-linker/CNBD region (Zagotta et al., 2003) tagged with a double His6-MBP sequence, together with Strep-tagged TRIP8b fragments corresponding to either TRIP8bmini or TRIP8bΔNter. As shown in Figure 5, the HCN2 C-linker/CNBD fragment was efficiently copurified following passage over a Strep-tactin affinity column of the bacterial lysate containing TRIP8bmini but not TRIP8bΔNter. These results confirm that the binding of the HCN C-linker/CNBD region to TRIP8bmini is direct and specific, and that removal of the initial segment of the TRIP8b/PEX5 homology domain (as in TRIP8bΔNter) completely abolishes interaction at the upstream site (Lewis et al., 2009).
Figure 5.
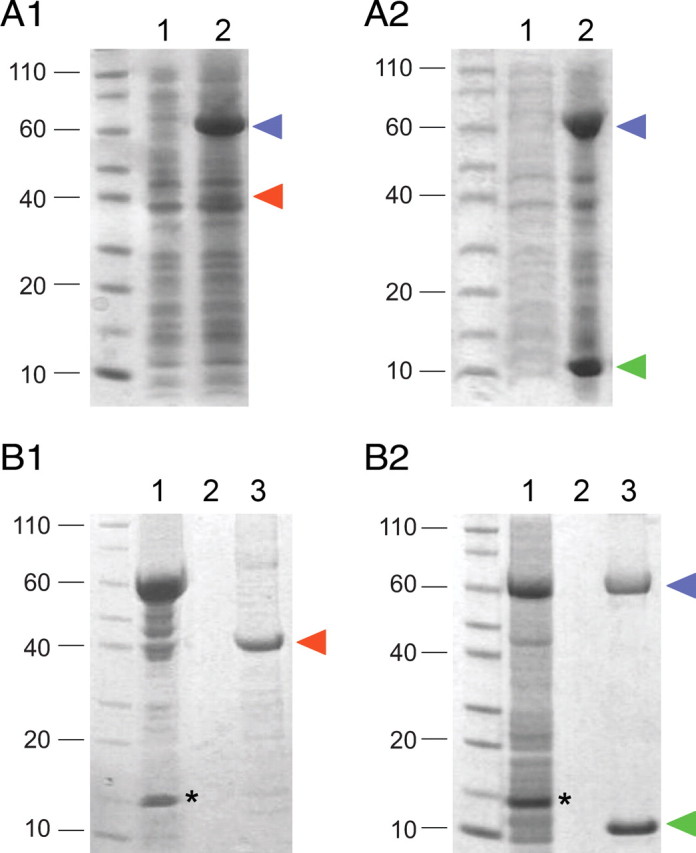
Association of TRIP8bmini and HCN2470–672 fragments in bacterial lysates. A1, A2, Coexpression of the MBP-tagged HCN2 C-linker/CNBD region (HCN2470–672) with either Strep-tagged TRIP8bΔNter (A1) or TRIP8bmini (A2) in E. coli cells. Coomassie staining following SDS/PAGE shows uninduced bacterial lysate (1) and IPTG-induced bacterial lysate (2). In each case, an amount corresponding to 100 μl of bacterial culture was loaded per lane. Blue arrowhead marks position of MBP-HCN2 band, red arrowhead Strep-TRIP8bΔNter and green arrowhead Strep-TRIP8bmini. B1, B2, Bacterial lysates from cells coexpressing MBP-HCN2 and Strep-TRIP8bΔNter (B1) or MBP-HCN2 and Strep-TRIP8bmini (B2) were loaded onto a Strep-Tactin column for Strep-tag purification. Equivalent samples were analyzed by Coomassie staining following SDS-PAGE separation, at the following stages: flow-through (1), last wash (2), eluate (3). The MBP-HCN2 fragment is found in the eluate along with the purified Strep-TRIP8bmini fragment, indicating the formation of a stable complex between the two proteins, but not with the purified Strep-TRIP8bΔNter fragment which lacks the conserved core region required for upstream site interaction. Labeling as in A. Asterisks mark the band corresponding to the lysozyme protein, which is added during the lysis stage. Molecular weight marker positions are indicated to the left of each panel.
Finally, we extended our analysis of the interaction between the TRIP8b TPR domain and HCN1 by using the yeast two-hybrid system to screen the ability of a range of TPR point mutations, which have been found to disrupt the interaction between PEX5 and its SKL-containing targets (Gatto et al., 2000; Klein et al., 2001), to alter the binding of TRIP8b to the HCN1 C-linker/CNBD or extreme C terminus (Fig. 4C). In addition to N501K, three mutations designed to disrupt specific contact sites between the TPR domain and the SNL tripeptide (V391D, N395D, R532A) abolished binding to HCN1776–910. Furthermore, two mutations designed to disrupt the formation of specific α-helixes within the tetratricopeptide repeats (G356C, G504E) also abolished binding to HCN1776–910, strongly suggesting that the interaction between the TRIP8b TPR domain and the HCN channel SNL tripeptide is very similar to the PEX5/SKL interaction. Strikingly, none of the point mutations in the TPR domain altered the binding of TRIP8b to the HCN1 C-linker/CNBD region (Fig. 4C), providing strong evidence that the upstream and downstream interaction domains are independent sites of binding.
Role of upstream and downstream interaction sites in the regulation of HCN1 gating and trafficking by TRIP8b
How do the upstream and downstream interaction sites contribute to the actions of TRIP8b to regulate HCN1 trafficking and gating? To address this question, we examined the functional effects of the mutations characterized in the above interaction studies. HCN1 hyperpolarization-activated currents were measured by two-microelectrode voltage-clamp from Xenopus oocytes coexpressing wild-type or mutant TRIP8b with wild-type or mutant HCN1 cRNAs, under the same conditions used for biochemical analysis. Normally, coexpression of wild-type TRIP8b with wild-type HCN1 in Xenopus oocytes shifts the voltage-dependent activation of HCN1 channels to more negative potentials by 10–15 mV (Santoro et al., 2009). This negative shift is caused by TRIP8b antagonizing the facilitatory effect on HCN1 opening that is exerted by the basal levels of cAMP present in the oocyte (Chen et al., 2001b). We therefore assayed the activity of wild-type and mutant TRIP8b (following coexpression with wild-type or mutant HCN1 channels) by testing their effects on the HCN1 midpoint voltage of activation (V1/2), a measure of the voltage dependence of channel gating, and on the HCN1 maximal tail-current amplitude (Imax) following strong hyperpolarizations, a measure of channel surface expression (Santoro et al., 2004, 2009).
Regulation of HCN1 channel gating by TRIP8b depends selectively on the upstream CNBD/core interaction site
To assess the gating phenotypes of HCN1 or TRIP8b mutations, we focused on TRIP8b(1a-4) as this is the most abundant isoform expressed in the brain and strongly enhances HCN1 surface expression (Santoro et al., 2009), ensuring the presence of large, measurable hyperpolarization-activated currents. We first analyzed the effects of coexpressing wild-type TRIP8b(1a-4) with two HCN1 C-terminal deletions that retain some TRIP8b binding activity (Fig. 1), HCN1ΔSNL and HCN1ΔCX. As illustrated in Figure 6, neither of these deletions affected the ability of TRIP8b(1a-4) to shift the channel V1/2 to more negative potentials. Thus, TRIP8b binding to HCN1 at the upstream CNBD interaction site must be sufficient to inhibit gating.
Figure 6.
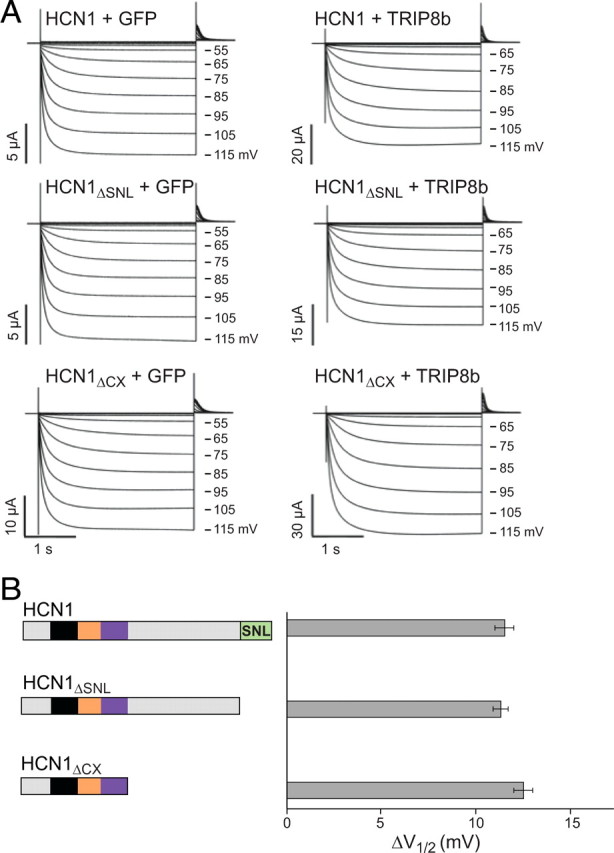
TRIP8b inhibition of HCN1 opening is not affected by deletion of HCN1 extreme C terminus. TRIP8b(1a-4) (0.2 μg/μl) or GFP as a baseline control (0.2 μg/μl) were coinjected in Xenopus oocytes with wild-type HCN1 (0.5 μg/μl), HCN1ΔSNL (0.5 μg/μl), or HCN1ΔCX (0.1 μg/μl). A, Two-microelectrode voltage-clamp current traces elicited from a holding potential of −30 mV to a range of test potentials between −15 and −115 mV in 10 mV decrements (as indicated), followed by a depolarizing step to 0 mV. Note that TRIP8b coexpression slows the rate of current activation and shifts opening to more negative potentials. B, Population data showing effect of TRIP8b to shift voltage at which channels are half activated (V1/2). ΔV1/2 was calculated by subtracting V1/2 values obtained when each HCN1 construct was coexpressed with GFP from corresponding V1/2 values when same HCN1 constructs were coexpressed with TRIP8b(1a-4). Note that V1/2 values used for subtraction were always obtained from the same batch of oocytes to minimize variability. Error bars show SEM. Mean ΔV1/2 values ± SEM (n = number of observations) are as follows: HCN1, 11.5 ± 0.5 mV (n = 26, GFP; n = 26, TRIP8b); HCN1ΔSNL, 11.3 ± 0.4 mV (n = 21, GFP; n = 21, TRIP8b); HCN1ΔCX, 12.5 ± 0.5 mV (n = 28, GFP; n = 28, TRIP8b).
Next, we analyzed the effects of a series of mutations in TRIP8b on its ability to alter the gating of wild-type HCN1 (Fig. 7) [see Materials and Methods for exact positions of mutations in TRIP8b(1a-4)]. Deletion of the extreme N-terminal domain of TRIP8b (TRIP8bΔNX), which contains sequences important for the regulation of channel trafficking but does not contribute to HCN1 binding (Fig. 3), had no effect on the ability of the protein to induce a negative shift in the V1/2 of HCN1. In contrast, the larger N-terminal deletion extending into the 80 aa TRIP8b core domain (TRIP8bΔNter) completely blocked the capacity of the mutant protein to alter HCN1 gating. This result is consistent with a previous report that the same TRIP8b fragment failed to alter the facilitatory effect of cAMP on gating of HCN2 in inside-out patches (Zolles et al., 2009).
Figure 7.

Effect of various TRIP8b(1a-4) mutants on the gating of wild-type HCN1. Xenopus oocytes were injected with cRNA encoding wild-type or mutant TRIP8b(1a-4) (0.2 μg/μl) or GFP as a baseline control (0.2 μg/μl), together with cRNA encoding wild-type HCN1 (0.5 μg/μl). Population data show the difference between the V1/2 observed when HCN1 was coexpressed with GFP and the V1/2 when HCN1 was coexpressed with indicated TRIP8b construct, with data points matched by batch of oocytes as described for Figure 6. Specific residue numbers for mutations introduced in the background of isoform TRIP8b(1a-4) are provided in Materials and Methods. Mean ΔV1/2 values ± SEM (n) are as follows: TRIP8b, 11.5 ± 0.5 mV (n = 26, GFP; n = 26, TRIP8b); TRIP8bΔNX, 10.9 ± 0.5 mV (n = 30, GFP; n = 30, TRIP8bΔNX); TRIP8bΔNter, 0.7 ± 0.7 mV (n = 11, GFP; n = 11, TRIP8bΔNter); TRIP8bΔint, 0.1 ± 0.6 mV (n = 16, GFP; n = 16, TRIP8bΔint); TRIP8bNK, 9.1 ± 0.8 mV (n = 12, GFP; n = 12, TRIP8bNK); TRIP8bΔTPR, 11.7 ± 0.5 mV (n = 18, GFP; n = 18, TRIP8bΔTPR).
As TRIP8bΔNterm can bind efficiently to HCN1 at the downstream SNL/TPR interaction site but cannot bind to HCN channels at the upstream C-linker/CNBD site (Figs. 3, 5) (Lewis et al., 2009), the upstream interaction must be necessary for the TRIP8b-dependent inhibition of HCN1 channel opening. We directly tested this idea by examining the ability of the 22 aa core deletion mutant, TRIP8bΔint [deleting residues 237–258 of TRIP8b(1a-4)], to inhibit HCN1 gating as this mutant protein binds to the downstream but not upstream HCN1 interaction site (Fig. 4B). Indeed, the internal deletion abolished the ability of TRIP8b(1a-4) to inhibit HCN1 gating (Fig. 7), confirming the importance of the upstream CNBD/core interaction in regulating channel opening. Conversely, selective disruption of the downstream interaction site, either by introducing the N to K mutation in the TRIP8b(1a-4) TPR domain or by ablating the TPR domain in the TRIP8b(1a-4)ΔTPR construct, had essentially no effect on the ability of TRIP8b to inhibit HCN1 activation (Fig. 7).
Next, we asked whether the interaction between HCN1 and the 80 aa core region of TRIP8b is sufficient to regulate channel opening. To obtain equimolar expression of this relatively small protein fragment and HCN1 subunits in Xenopus oocytes, we fused TRIP8bmini to the N terminus of HCN1 (TRIP8bmini-HCN1). We then compared the voltage dependence of the resulting fusion protein channel with that of two other fusion constructs we previously characterized: GFP-HCN1, which gates identically to wild-type HCN1, and TRIP8b(1a-4)-HCN1, which shows a negative shift in its V1/2 identical to the shift seen when TRIP8b(1a-4) is coexpressed with HCN1 as independent proteins (Santoro et al., 2009). As an additional control, we fused the TRIP8b(1a-4) internal deletion mutant lacking the central 22 aa of the core domain to the N terminus of HCN1 (TRIP8bΔint-HCN1).
When fused to HCN1, the 80 aa TRIP8bmini fragment was as effective as full-length TRIP8b(1a-4) in shifting the midpoint of channel activation to more negative potentials (Fig. 8). Conversely, the TRIP8bΔint internal deletion mutant caused only a minor shift in V1/2 when fused to the N terminus of HCN1. Moreover, TRIP8bmini also produced a full negative shift in V1/2 when fused to the N terminus of the HCN1ΔCX truncation mutant, consistent with the view that the TRIP8b core does not interact with the downstream HCN1 binding site, and that the extreme C terminus of the channel does not contribute to the effects of TRIP8b on gating. Using cell-free inside-out patches, we further found that a TRIP8bmini-HCN2 fusion protein showed a reduced voltage shift in response to cAMP, but no change in voltage-dependent gating in the absence of cAMP relative to a GFP-HCN2 fusion protein or HCN2 alone (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These effects are similar to those seen when full-length TRIP8b(1a-4) is fused to HCN2 or when TRIP8b(1a-4) and HCN2 are coexpressed as independent proteins (Zolles et al., 2009). These findings thus provide strong evidence that an interaction between the 80 aa TRIP8bmini core with the HCN channel upstream C-linker/CNBD site is necessary and sufficient for the regulatory effect that TRIP8b exerts on cyclic nucleotide gating.
Figure 8.

Characterization of a minimal TRIP8b domain that inhibits HCN1 gating using TRIP8b-HCN1 fusion proteins. Shift in ΔV1/2 produced when wild-type or mutant TRIP8b(1a-4) was fused to the N terminus of HCN1. ΔV1/2 values obtained relative to V1/2 of GFP-HCN1 fusion construct. All constructs expressed using cRNA injections in Xenopus oocytes (HCN1 fusions, 0.5 μg/μl; HCN1ΔCX fusions, 0.2 μg/μl). Population data show the difference between the V1/2 obtained when HCN1 was fused to GFP and the V1/2 when HCN1 was fused to indicated TRIP8b construct. All data points were matched by batch of oocytes, as above. Mean ΔV1/2 values ± SEM (n) are as follows: TRIP8b-HCN1, 13.8 ± 0.9 mV (n = 14, GFP-HCN1; n = 14, 1a4-HCN1); TRIP8bmini-HCN1, 14.7 ± 0.7 mV (n=14, GFP-HCN1; n = 14, mini-HCN1); TRIP8bΔint-HCN1, 4.5 ± 0.6 mV (n = 12, GFP-HCN1; n = 12, Δint-HCN1); TRIP8bmini-HCN1ΔCX, 12.6 ± 0.4 mV (n = 14, GFP-HCN1ΔCX; n = 14, mini-HCN1ΔCX); TRIP8bΔint-HCN1ΔCX, 2.1 ± 0.5 mV (n = 14, GFP-HCN1ΔCX; n = 14, Δint-HCN1ΔCX).
Upstream and downstream interaction sites differentially participate in the regulation of HCN1 channel trafficking by TRIP8b
As the upstream interaction site is both necessary and sufficient for the action of TRIP8b to inhibit HCN1 gating, we asked whether the downstream interaction site might be important in the control of HCN1 trafficking. To explore this possibility, we examined the effects of mutations in HCN1 or in three selected TRIP8b splice variants—TRIP8b(1a-4), TRIP8b(1a), and TRIP8b(1b-2). As mentioned above, these isoforms exert distinct effects on HCN1 trafficking: TRIP8b(1a-4) strongly increases HCN1 surface expression; TRIP8b(1a) produces a ∼10-fold decrease of HCN1 surface expression in Xenopus oocytes (Santoro et al., 2009) but enhances HCN1 expression in a mammalian cell line (Lewis et al., 2009); and TRIP8b(1b-2) essentially abolishes surface expression in both oocytes and mammalian cells (>50-fold decrease) by promoting channel endocytosis (Santoro et al., 2004, 2009; Lewis et al., 2009). Although both TRIP8b(1a) and TRIP8b(1b-2) decrease surface expression of HCN1 in Xenopus oocytes, the two splice variants depend on distinct trafficking sequence motifs, suggesting the possibility that their effects rely on distinct mechanisms. Thus, the effect of TRIP8b(1a) to downregulate HCN1 requires a dileucine-based trafficking motif in exon 5 whereas channel downregulation with TRIP8b(1b-2) depends on a tyrosine-based trafficking motif in exon 2 (Santoro et al., 2009).
We first examined the ability of the three wild-type TRIP8b isoforms to regulate the trafficking of the HCN1ΔSNL truncation mutant characterized above that lacks the downstream interaction site. Deletion of the SNL tripeptide caused a selective impairment in the downregulation of HCN1 surface expression by either TRIP8b(1a) or TRIP8b(1b-2). Surprisingly, the truncation had no effect on the upregulation of channel surface expression with TRIP8b(1a-4) (Fig. 9). We also found a quantitative difference in the effect of the SNL truncation on channel downregulation with TRIP8b(1a) versus TRIP8b(1b-2). Whereas the SNL deletion only partially inhibited channel downregulation with TRIP8b(1b-2), it fully blocked the effect of TRIP8b(1a) to decrease HCN1 expression. Thus, the downstream SNL interaction site of HCN1 appears selectively required for the effect of TRIP8b isoforms to downregulate HCN1 surface expression. In contrast, binding at the downstream site has little role in the effect of the TRIP8b(1a-4) isoform to upregulate HCN1 surface expression or to inhibit HCN1 gating (Figs. 6, 8).
Figure 9.
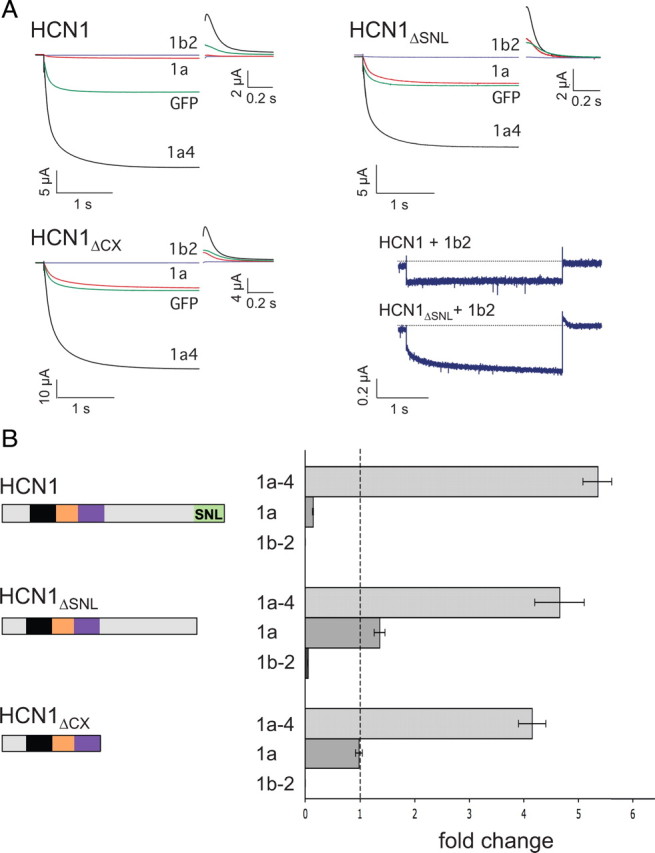
HCN1 C-terminal deletions differentially alter the ability of three TRIP8b isoforms to regulate HCN1 surface expression. Xenopus oocytes were injected with cRNA encoding wild-type HCN1 (0.5 μg/μl), HCN1ΔSNL (0.5 μg/μl) or HCN1ΔCX (0.1 μg/μl) together with cRNA encoding one of the indicated TRIP8b splice variants (0.2 μg/μl) or GFP (0.2 μg/μl) to provide a baseline control. Recordings were performed 3 d after injection; two-microelectrode voltage-clamp current traces obtained as in Figure 6. A, Sample current traces elicited from a holding potential of −30 mV to a test potential of −105 mV shown for HCN1, HCN1ΔSNL, or HCN1ΔCX (as indicated) coexpressed with GFP (green traces), TRIP8b(1a-4) (black traces), TRIP8b(1a) (red traces), or TRIP8b(1b-2) (blue traces). Tail current traces are shown at an expanded scale. Lower right, Current traces obtained at the −105 mV test potential upon coexpression of TRIP8b(1b-2) with HCN1 or HCN1ΔSNL. B, Maximal tail current amplitude (Imax) for HCN1 constructs coexpressed with a given TRIP8b isoform normalized by maximal tail current amplitude when the same channel construct was coexpressed with GFP. As in Figure 6, data points were matched by batch of oocytes. Error bars show SEM. Mean normalized Imax values ± SEM (n) are as follows: HCN1 + TRIP8b(1a-4), 5.35 ± 0.27 (n = 67, GFP; n = 70, 1a-4); HCN1 + TRIP8b(1a), 0.14 ± 0.01 (n = 56, GFP; n = 60, 1a); HCN1 + TRIP8b(1b-2), undetectable current (n = 34, GFP; n = 27, 1b-2); HCN1ΔSNL + TRIP8b(1a-4), 4.65 ± 0.45 (n = 15, GFP; n = 14, 1a-4); HCN1ΔSNL + TRIP8b(1a), 1.36 ± 0.1 (n = 15, GFP; n = 15, 1a); HCN1ΔSNL + TRIP8b(1b-2), 0.05 ± 0.005 (n = 15, GFP; n = 15, 1b-2); HCN1ΔCX + TRIP8b(1a-4), 4.15 ± 0.25 (n = 17, GFP; n = 16, 1a-4); HCN1ΔCX + TRIP8b(1a), 0.98 ± 0.06 (n = 17, GFP; n = 17, 1a); HCN1ΔCX + TRIP8b(1b-2), undetectable current (n = 13, GFP; n = 13, 1b-2).
Truncation of the entire extreme C terminus of HCN1 distal to the CNBD (HCN1ΔCX) also fully blocked the capacity of TRIP8b(1a) to decrease channel surface expression with no effect on the enhancement of channel surface expression with TRIP8b(1a-4), similar to the effects seen above with the SNL deletion (Fig. 9). However, the effects of HCN1ΔSNL and HCN1ΔCX diverged when tested against TRIP8b(1b-2); surprisingly, truncation of the entire HCN1 extreme C terminus restored the capacity of TRIP8b(1b-2) to fully abolish channel surface expression. This result reinforces the idea that TRIP8b(1a) and TRIP8b(1b-2) are likely to downregulate surface expression through distinct mechanisms (Santoro et al., 2009). Moreover, it suggests that the extreme C-terminus of HCN1, when not bound to the TPR domain of TRIP8b, may exert an inhibitory effect on the ability of TRIP8b to promote channel endocytosis.
Next, we analyzed the effects of mutations in each of the three TRIP8b isoforms on their ability to regulate the trafficking of wild-type HCN1 channels. Neither of the two N-terminal deletion mutants (TRIP8bΔNX and TRIP8bΔNter) exerted any regulatory effect on the surface expression of HCN1 (Fig. 10), consistent with the presence of key trafficking consensus sequences in the deleted regions (Santoro et al., 2009; Petrenko et al., 2010).
Figure 10.

Both downstream and upstream interaction sites in TRIP8b contribute to regulation of HCN1 trafficking by the three TRIP8b isoforms. Population data showing maximal tail current amplitudes (Imax) upon coexpression of wild-type HCN1 with the indicated mutant TRIP8b isoform, normalized to the baseline current value determined following coexpression of HCN1 with GFP control, with data points matched by batch of oocytes. As the N-terminal deletion mutants are identical for all three TRIP8b isoforms, only one set of data is shown for each mutant. Specific residue numbers for mutations introduced in the background of each isoform are provided in Materials and Methods. Mean normalized Imax values ± SEM (n) are as follows: HCN1 + TRIP8b(1a-4), 5.35 ± 0.27 (n = 67, GFP; n = 70, 1a-4); HCN1 + TRIP8b(1a), 0.14 ± 0.01 (n = 56, GFP; n = 60, 1a); HCN1 + TRIP8b(1b-2), undetectable current (n = 34, GFP; n = 27, 1b-2); HCN1 + TRIP8bΔNX, 1.24 ± 0.09 (n = 30, GFP; n = 30, ΔNX); HCN1 + TRIP8bΔNter, 1.57 ± 0.23 (n = 11, GFP; n = 11, ΔNter); HCN1 + TRIP8b(1a-4)Δint, 2.29 ± 0.18 (n = 19, GFP; n = 23, 1a-4Δint); HCN1 + TRIP8b(1a)Δint, 2.87 ± 0.18 (n = 19, GFP; n = 23, 1aΔint); HCN1 + TRIP8b(1b-2)Δint, 0.18 ± 0.02 (n = 19, GFP; n = 23, 1b-2Δint); HCN1 + TRIP8b(1a-4)NK, 5.68 ± 0.49 (n = 12, GFP; n = 12, 1a-4 NK); HCN1 + TRIP8b(1a)NK, 2.96 ± 0.2 (n = 19, GFP; n = 23, 1aNK); HCN1 + TRIP8b(1b-2)NK, 0.07 ± 0.01 (n = 15, GFP; n = 15, 1b-2NK); HCN1 + TRIP8b(1a-4)ΔTPR, 1.13 ± 0.07 (n = 18, GFP; n = 18, 1a-4ΔTPR); HCN1 + TRIP8b(1a)ΔTPR, 0.29 ± 0.02 (n = 18, GFP; n = 18, 1aΔTPR); HCN1 + TRIP8b(1b-2)ΔTPR, undetectable current (n = 15, GFP; n = 10, 1b-2ΔTPR). One-way ANOVA was used to determine that all experimental groups, except for TRIP8bΔNX, TRIP8bΔNter, and TRIP8b(1a-4)ΔTPR, significantly differ from the GFP baseline control group (p < 0.01, Tukey's multiple-comparison test).
As our results show that the downstream SNL interaction site in HCN1 is important in the downregulation of channel surface expression by TRIP8b isoforms, we performed complementary experiments to probe the importance of the downstream interaction site in TRIP8b by introducing the equivalent of the N501K point mutation in the TPR domains of the three TRIP8b splice variants. The resulting phenotypes closely mirrored the effects seen with the HCN1 SNL truncation (compare Figs. 9, 10). Thus, the N to K mutation significantly inhibited the ability of both TRIP8b(1a) and TRIP8b(1b-2) to downregulate HCN1 surface expression, but had little effect on the ability of TRIP8b(1a-4) to upregulate channel surface expression. In fact, the TRIP8b(1a) mutant now caused a significant increase in HCN1 surface expression, similar to what we previously observed upon mutation of N-terminal trafficking consensus site in this isoform (Santoro et al., 2009).
Having established that the upstream interaction site is critical for the regulation of HCN1 channel gating by TRIP8b (Figs. 7, 8), we wondered whether it might also play a role in the control of HCN1 trafficking. We tested this hypothesis by coexpressing HCN1 with internal deletion mutants of each of the three TRIP8b isoforms lacking the central 22 aa of the core domain (TRIP8bΔint). As shown in Figure 10, disruption of the upstream interaction site clearly altered the ability of all three TRIP8b isoforms to modulate channel surface expression. Indeed, the effects of the internal deletion resembled the effects of the HCN1 SNL truncation and TRIP8b N to K mutations described above, with a significant loss of the ability of TRIP8b(1a) and TRIP8b(1b-2) to downregulate HCN1 surface expression. The ability of TRIP8b(1a-4) to enhance channel surface expression was also somewhat reduced, though still present (Fig. 10). Thus, disruption of TRIP8b/HCN1 binding at either the upstream or downstream interaction sites greatly reduces the ability of TRIP8b isoforms to downregulate HCN1 surface expression but has a limited effect on the ability of TRIP8b isoforms to enhance surface expression.
As the TPR point mutations disrupt the ability of TRIP8b(1a) and TRIP8b(1b-2) to downregulate HCN1 surface expression, we expected to observe a similar phenotype when we deleted the TPR domain of these isoforms (TRIP8bΔTPR). Surprisingly, both of these truncated TRIP8b isoforms efficiently downregulated HCN1 surface expression, very similar to the effects of the wild-type isoforms (Fig. 10). This observation is consistent with the fact that the TRIP8bΔTPR truncation mutant can efficiently bind to HCN1 at its C-linker/CNBD interaction site, and exert a normal inhibitory effect on channel gating (Figs. 3, 7). Moreover, it indicates that the presence of an unbound TPR domain can impair the ability of TRIP8b to downregulate HCN1 surface expression (see Discussion) (Fig. 11), similar to the inhibitory effect seen above with the unbound extreme C-terminus of HCN1 (Fig.9). Of interest, deletion of the TPR domain of TRIP8b(1a-4) completely blocked the capacity of this isoform to enhance HCN1 surface expression [TRIP8b(1a-4) ΔTPR] in contrast to the lack of effect of the TPR N to K point mutation on channel upregulation (Fig. 10). This suggests that, in addition to binding to HCN1 at the downstream interaction site, the TPR domain may serve to recruit specific factors important for the enhancement of channel trafficking to the surface membrane.
Figure 11.

Schematic representation of HCN1/TRIP8b interactions in the presence of intracellular trafficking factors. A, Summary of interactions between HCN1, TRIP8b, and trafficking proteins. The HCN1 channel protein is represented in gray, with C-linker helices E′ and F′ indicated by rectangles, and the C-terminal SNL sequence indicated by a circle. The TRIP8b protein is color coded as in previous figures (black, unique N-terminal region; yellow, highly conserved core domain; blue, region of homology to PEX5 with TPR repeats indicated in red). Dileucine- or tyrosine-based AP binding motifs are present in the N-terminal domain of TRIP8b, indicated by black triangles. AP complex proteins (AP-1 or AP-2) are represented in pink, positioned closely to the plasma membrane (Bonifacino and Traub, 2003), and bind to the N terminus of TRIP8b. An additional trafficking factor (potentially Rab8b), indicated in green, is shown to interact with the C-terminal TPR region of TRIP8b. Note that both of these interacting elements are hypothetical. Gating and trafficking phenotypes are indicated in each panel for their respective mutants. B, Deletion of a critical sequence within the conserved core domain of TRIP8b (Δint) results in a loss of the ability to modulate channel gating and to downregulate channel expression. A hypothetical displacement of the TRIP8b N-terminal domain is shown to impede the binding of AP complex proteins, but not of factors involved in the upregulation of HCN1 surface expression. C, Deletion of the portion of the TPR domain following the first tetratricopeptide repeat (ΔTPR) leads to a loss in the ability of TRIP8b to upregulate surface expression. We speculate that this is because required factors are unable to bind the protein's C-terminal domain. Efficient interaction at the upstream CNBD/core binding site and between the N terminus of TRIP8b and AP complex proteins would still allow for the regulation of channel gating and endocytosis. D, Deletion of the HCN1 channel's C-terminal SNL tripeptide (HCN1ΔSNL) or the N to K point mutation in the TPR domain (TRIP8bNK) results in a loss of the ability of TRIP8b to downregulate surface expression. We speculate that failure to interact at the downstream SNL/TPR binding site leads to a displacement of the C-terminal half of the TPR domain, which prevents the access of AP complex proteins to their target sequences in the TRIP8b N terminus. Interaction at the upstream CNBD/core site and with factors bound to the TPR domain that upregulate channel surface expression would not be impaired, resulting in normal gating and Imax increase phenotypes.
Discussion
Here we have mapped distinct upstream and downstream sites of interaction between HCN1 and TRIP8b, and defined the differential roles of these sites in the functional effects of three different TRIP8b splice variants on channel trafficking and cyclic nucleotide gating. The interactions between wild-type HCN1 and TRIP8b are summarized in schematic form in Figure 11, which also provides a more speculative model to explain certain phenotypes of HCN1 and TRIP8b mutants.
Structural findings
Using biochemical and molecular biological approaches, we confirmed the presence of upstream and downstream interaction sites on HCN1 and TRIP8b (Lewis et al., 2009), and refined the localization of the interaction surfaces (Fig. 11A). We found that the upstream interaction involves binding of the CNBD and last two helixes of the C-linker region of HCN1 (E′–F′/CNBD) to an 80 aa conserved core region of TRIP8b [TRIP8bmini, residues 236–316 in TRIP8b(1b-2)], immediately upstream of the TPR domain. At the downstream site, the C-terminal SNL tripeptide of the channel interacts with the TPR domain of TRIP8b, similar to the interaction of the PEX5 TPR domain with its SKL-containing targets (Gatto et al., 2000).
Using coimmunoprecipitation assays from Xenopus oocytes, we further determined that, in native conditions, both interaction sites contribute to the stability of the TRIP8b/HCN1 complex. Interestingly, the crystal structure of PEX5 in complex with the sterol carrier protein 2 (SCP2) similarly reveals a second interaction site outside the TPR domain's contact with the C-terminal AKL sequence of SCP2 (Stanley et al., 2006). The binding affinity of full-length SCP2 for PEX5 is 5- to 10-fold higher than the affinity of its C-terminal PTS1 peptide (PGNAKL), suggesting a substantial contribution of the secondary interface to overall binding. An analogous situation is found in the PEX5/alanine:glyoxylate aminotransferase complex, where an ancillary targeting domain is present ∼60 aa upstream of the C-terminal PTS1 peptide (Huber et al., 2005) (see also PDB accession 3IMZ for solved crystal structure of the complex). Indeed, it has been suggested that bipartite target binding by a second topologically distinct interaction site could be a general feature of target recognition by the PEX5 receptor (Stanley et al., 2006). Given the close evolutionary relationship between the two proteins, a similar strategy may have been adopted by TRIP8b for the recognition of its HCN channel target.
Functional findings
Our studies indicate that the two TRIP8b/HCN1 interaction sites play distinct functional roles in the effects of the different TRIP8b isoforms to inhibit channel gating compared with the effects of these isoforms to upregulate versus downregulate channel trafficking. Interaction at the upstream site between the channel CNBD and the conserved 80 aa core of TRIP8b is both necessary and sufficient to mediate the inhibitory effects of TRIP8b on HCN1 channel opening. Specifically, the ability of full-length TRIP8b to shift the channel V1/2 to more negative potentials is fully reproduced by TRIP8bmini and is unaffected by deletion of the TRIP8b TPR domain or extreme C terminus of the channel.
The structural bases for the effects of the three TRIP8b isoforms on trafficking are complex. Interaction at the upstream site is necessary (TRIP8bΔint) (Fig. 11B) and also sufficient (TRIP8bΔTPR) (Fig. 11C) for TRIP8b(1a) or TRIP8b(1b-2) to downregulate HCN1 surface expression, as long as the trafficking motifs in the N-terminal region of TRIP8b are present. Interaction at the downstream site between the TPR domain of TRIP8b and the SNL tripeptide of HCN1 is not strictly required for the downregulation of HCN1 surface expression with TRIP8b(1a) or TRIP8b(1b-2), given that normal downregulation is observed in the absence of the TRIP8b TPR domain (though see discussion below). In contrast, deletion of the TPR domain fully blocks the ability of TRIP8b(1a-4) to upregulate HCN1 surface expression (TRIP8bΔTPR) (Fig. 11C). This effect cannot be explained by the simple loss of binding at the downstream interaction site because channel upregulation by TRIP8b(1a-4) is unaffected in the TRIP8bNK and HCN1ΔSNL mutants, which both have disrupted downstream binding (Fig. 11D). Rather, we suggest that the TPR region may recruit some cytoplasmic trafficking factor independent of the TPR domain's role in binding to the SNL site of HCN1 (e.g., Rab8b) (Chen et al., 2001a; Fransen et al., 2008). To upregulate HCN1 surface expression, the recruitment of such a factor must act in combination with sequences present in the N-terminal region of TRIP8b, as upregulation is not observed when this region is deleted (TRIP8bΔNX and TRIP8bΔNter).
One surprising result is that relatively small perturbations in the downstream binding site of either HCN1 (e.g., HCN1ΔSNL) or TRIP8b (e.g., TRIP8bNK) are more effective in disrupting the downregulation of channel surface expression than are larger truncations of either the C-terminal TPR domain of TRIP8b (TRIP8bΔTPR) or the extreme C terminus of HCN1 (HCN1ΔCX). We hypothesize that the unbound TPR domain of TRIP8b may exert an inhibitory effect to prevent the assembly of macromolecular complexes involved in channel trafficking at the N terminus of TRIP8b (Fig. 11D). This implies that the normal interaction of HCN1 and TRIP8b at the downstream binding site may induce a conformational change in the TPR domain that allows for assembly of the trafficking complex.
The idea that the downstream interaction of the HCN1 SNL tripeptide with TRIP8b triggers a conformational change in the TPR domain is consistent with x-ray crystal structures of the PEX5 TPR region when bound and unbound to its cargo target peptide. Such studies reveal that cargo binding causes the TPR region to undergo a conformational change from a more open to a more closed structure (Stanley et al., 2006, 2007). Based on the high degree of amino acid similarity between the TPR regions of PEX5 and TRIP8b (see supplemental Fig. 3, available at www.jneurosci.org as supplemental material), we generated a structural homology model for TRIP8b with its SNL peptide bound or absent (Fig. 12). In the model, unbinding of the cargo peptide causes the C-terminal half of the TRIP8b TPR domain to swing away from the N-terminal half, which remains relatively immobile (Fig. 12B), similar to the PEX5 results. Thus, the downstream TPR/SNL interaction site might primarily act to stabilize the C-terminal structure of TRIP8b. A displaced TPR domain could interfere with clathrin-mediated endocytosis of the HCN1/TRIP8b complex; deletion of the C-terminal portion of the TPR domain would remove this inhibitory effect, rescuing the ability of TRIP8b to mediate HCN channel endocytosis.
Figure 12.

Homology model of the TRIP8b tetratricopeptide repeat domain in bound and unbound form. A, Top view (left) and bottom view (right) of homology model of TRIP8b TPR domain in complex with the HCN channel C-terminal tripeptide (−SNL, yellow stick figure representation) based on PEX5 structure. The side chains of four TRIP8b amino acid residues predicted to make critical contacts with the target peptide are shown in green stick figure representations (V391, N395, N501, R532) (Fig. 4C). Note that the N-terminal two helices, corresponding to the first tetratricopeptide repeat (TPR1, in red) do not contact the SNL target peptide. These helices are preserved in TRIP8bΔTPR (only the gray portion of the TPR domain is deleted in this construct). The N-terminal end of the structure is colored in blue, and corresponds to the C-terminal tail of the TRIP8bmini construct (no structural information is available N-terminal to this point). B, Homology models of different conformations assumed by the TPR domain of TRIP8b upon binding or unbinding of the SNL target peptide. The holo (bound) structure (black) is superimposed on two alternative conformations assumed by the apo (unbound) structure (light blue and green). Note that the C-terminal TPR segments rotate away from the N-terminal TPR segments, with the two halves of the TPR domain behaving as near-rigid bodies (Stanley et al., 2007). Models were constructed using the program MODELLER based on Protein Data Bank accessions 1FCH, 2C0M, and 2J9Q. Molecular figures were generated using PyMOL (DeLano, 2002). The view of the structure in B is rotated ∼90° with respect to the view shown in A, as indicated by the arrow. For orientation purposes, the holo structure is reproduced on the right, showing the position of the −SNL peptide. Structure in A corresponds to residues 297–615 of TRIP8b(1b-2), and structures in B to residues 304–615 (see supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The loop between TPR3 and TPR4 (Gatto et al., 2000), as well as the loop between TPR7 and the C-terminal three-helical bundle (7C loop) (Stanley et al., 2006), have been substituted by a dotted line, as these elements are not resolved in the PEX5 structures.
Given that the conserved core region of TRIP8b is contiguous with the TPR domain, a similar scenario may explain how an internal deletion in the upstream site (TRIP8bΔint) interferes with the downregulation of HCN1 surface expression. Loss of the upstream interaction may lead to the improper positioning of the N-terminal domain of TRIP8b, preventing it from interacting with its normal protein trafficking partners (Fig. 11B). Alternatively, the upstream site could help anchor the SNL/TPR complex to the rest of the channel to prevent it from interfering with the assembly of protein complexes required for membrane trafficking.
Physiological consequences of the bipartite TRIP8b-HCN1 interaction
Our findings that mutations that perturb the downstream interaction between TRIP8b and HCN1 differentially alter the functional consequences of residual binding at the upstream site have interesting implications for the dynamic regulation of the TRIP8b/HCN1 interaction in vivo. For example, our results with HCN1ΔSNL suggest that a post-translational modification that weakens the downstream interaction might prevent downregulation of channel expression with TRIP8b(1a) without changing the upregulation of channel expression with TRIP8b(1a-4) (Fig. 9) or the effect of either isoform to regulate cyclic nucleotide-mediated channel gating (Fig. 6).
Given this potential for regulation, it is of interest that two proteomic screens of phosphopeptides from mouse brain synapses identified in vivo phosphorylation sites within TRIP8b (Trinidad et al., 2006; Munton et al., 2007). One such phosphorylation site, located in the loop between TPR3 and TPR4, is poised to regulate the downstream interaction between TRIP8b and HCN1. A second phosphorylation site, located in the conserved core region (TRIP8bmini), could potentially affect binding at the upstream site and thus disrupt the gating and trafficking effects of all TRIP8b isoforms (Fig. 10). Differential phosphorylation may also explain why TRIP8b(1a) downregulates HCN1 surface expression in Xenopus oocytes (Santoro et al., 2009), whereas it upregulates HCN1 in a mammalian cell line (Lewis et al., 2009).
As the proper assembly of trafficking complexes appears to depend on the specific conformation of TRIP8b N-terminal and C-terminal domains, might the large cytoplasmic N-terminal and C-terminal domains of the HCN channels play a similar regulatory function? Indeed, we find that whereas the extreme N and C termini of HCN1 (outside of the SNL sequence) do not directly bind TRIP8b, these domains appear to modulate the functional association between HCN1 and TRIP8b. Thus, the decreased ability of TRIP8b(1b-2) to abolish HCN1 surface expression seen with truncation of the SNL tripeptide of the channel is rescued upon truncation of the entire nonconserved C terminus of HCN1 (Fig. 9). Moreover, deletion of the extreme N terminus of HCN1 strongly enhances the ability of TRIP8b(1a) to downregulate channel surface expression (A. Kushnir and B. Santoro, unpublished observation). Given that the extreme N and C termini of the four HCN channel isoforms diverge in length and sequence, the association of TRIP8b with different channel isoforms could result in different functional phenotypes. Indeed, whereas TRIP8b(1a-4) strongly increases HCN1 surface expression (Lewis et al., 2009; Santoro et al., 2009), this isoform decreases the surface expression of HCN2 (Zolles et al., 2009) (L. Hu, unpublished observation). Such variations could add a further layer of complexity and versatility to the regulatory roles played by the HCN channel TRIP8b auxiliary subunit within the nervous system.
Footnotes
This work was partially supported by National Institutes of Health Grant NS36658 (S.A.S.), a Fellowship from the Italian Academy for Advanced Studies in America (B.S.), and a Research Grant from the Epilepsy Foundation (B.S.), as well as Award F32NS064732 from National Institute of Neurological Disorders and Stroke (R.A.P.). We thank Alexander Kushnir for his contributions during the development of this study, Lei Zhou and Yuhang Chen for help with homology modeling and figure construction, and John Riley for excellent technical assistance. We are also indebted to Justine Barry for her advice and critical reading of the manuscript.
References
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Chen S, Liang MC, Chia JN, Ngsee JK, Ting AE. Rab8b and its interacting partner TRIP8b are involved in regulated secretion in AtT20 cells. J Biol Chem. 2001a;276:13209–13216. doi: 10.1074/jbc.M010798200. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001b;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. DeLano Scientific; 2002. The PyMOL molecular graphics system. Available at http://www.pymol.org. [Google Scholar]
- Fransen M, Amery L, Hartig A, Brees C, Rabijns A, Mannaerts GP, Van Veldhoven PP. Comparison of the PTS1- and Rab8b-binding properties of Pex5p and Pex5Rp/TRIP8b. Biochim Biophys Acta. 2008;1783:864–873. doi: 10.1016/j.bbamcr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- Gatto GJ, Jr, Geisbrecht BV, Gould SJ, Berg JM. Peroxisomal targeting signal-1 recognition by the TPR domains of human PEX5. Nat Struct Biol. 2000;7:1091–1095. doi: 10.1038/81930. [DOI] [PubMed] [Google Scholar]
- Huber PA, Birdsey GM, Lumb MJ, Prowse DT, Perkins TJ, Knight DR, Danpure CJ. Peroxisomal import of human alanine:glyoxylate aminotransferase requires ancillary targeting information remote from its C terminus. J Biol Chem. 2005;280:27111–27120. doi: 10.1074/jbc.M502719200. [DOI] [PubMed] [Google Scholar]
- Klein AT, Barnett P, Bottger G, Konings D, Tabak HF, Distel B. Recognition of peroxisomal targeting signal type 1 by the import receptor Pex5p. J Biol Chem. 2001;276:15034–15041. doi: 10.1074/jbc.M010776200. [DOI] [PubMed] [Google Scholar]
- Lewis AS, Schwartz E, Chan CS, Noam Y, Shin M, Wadman WJ, Surmeier DJ, Baram TZ, Macdonald RL, Chetkovich DM. Alternatively spliced isoforms of TRIP8b differentially control h channel trafficking and function. J Neurosci. 2009;29:6250–6265. doi: 10.1523/JNEUROSCI.0856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, Biel M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999;18:2323–2329. doi: 10.1093/emboj/18.9.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munton RP, Tweedie-Cullen R, Livingstone-Zatchej M, Weinandy F, Waidelich M, Longo D, Gehrig P, Potthast F, Rutishauser D, Gerrits B, Panse C, Schlapbach R, Mansuy IM. Qualitative and quantitative analyses of protein phosphorylation in naive and stimulated mouse synaptosomal preparations. Mol Cell Proteomics. 2007;6:283–293. doi: 10.1074/mcp.M600046-MCP200. [DOI] [PubMed] [Google Scholar]
- Pascale MC, Malagolini N, Serafini-Cessi F, Migliaccio G, Leone A, Bonatti S. Biosynthesis and oligosaccharide structure of human CD8 glycoprotein expressed in a rat epithelial cell line. J Biol Chem. 1992;267:9940–9947. [PubMed] [Google Scholar]
- Petrenko AG, Popova NV, Deyev IE. Association of brain-specific adapter protein TRIP8b with clathrin. Soc Neusci Abstr. 2010;36:41–10. [Google Scholar]
- Popova NV, Plotnikov AN, Ziganshin RKh, Deyev IE, Petrenko AG. Analysis of proteins interacting with TRIP8b adapter. Biochemistry (Moscow) 2008;73:644–651. doi: 10.1134/s0006297908060035. [DOI] [PubMed] [Google Scholar]
- Santoro B, Wainger BJ, Siegelbaum SA. Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J Neurosci. 2004;24:10750–10762. doi: 10.1523/JNEUROSCI.3300-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Piskorowski RA, Pian P, Hu L, Liu H, Siegelbaum SA. TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron. 2009;62:802–813. doi: 10.1016/j.neuron.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley WA, Filipp FV, Kursula P, Schüller N, Erdmann R, Schliebs W, Sattler M, Wilmanns M. Recognition of a functional peroxisome type 1 target by the dynamic import receptor pex5p. Mol Cell. 2006;24:653–663. doi: 10.1016/j.molcel.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley WA, Pursiainen NV, Garman EF, Juffer AH, Wilmanns M, Kursula P. A previously unobserved conformation for the human Pex5p receptor suggests roles for intrinsic flexibility and rigid domain motions in ligand binding. BMC Struct Biol. 2007;7:24. doi: 10.1186/1472-6807-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinidad JC, Specht CG, Thalhammer A, Schoepfer R, Burlingame AL. Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Mol Cell Proteomics. 2006;5:914–922. doi: 10.1074/mcp.T500041-MCP200. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–810. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- Zolles G, Wenzel D, Bildl W, Schulte U, Hofmann A, Müller CS, Thumfart JO, Vlachos A, Deller T, Pfeifer A, Fleischmann BK, Roeper J, Fakler B, Klöcker N. Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron. 2009;62:814–825. doi: 10.1016/j.neuron.2009.05.008. [DOI] [PubMed] [Google Scholar]