

Abstract
Non-selective NSAIDs and selective cyclooxygenase-2 (COX-2) inhibitors are purported to increase adverse cardiovascular events. We hypothesized COX-2 inhibitors would alter myocardial blood flow, microvascular reactivity, oxidative stress, and prostaglandin levels. Adult Yorkshire swine were divided into three groups: no drug (control, n=7), a non-selective COX inhibitor (naproxen 400mg daily, NAP, n=7), or a selective COX-2 inhibitor (celecoxib 200mg daily, CBX, n=7). After 7 weeks physiologic measurements were taken and tissue harvested. Animals in the CBX group demonstrated significantly higher blood pressure and rate pressure product. The NAP and CBX groups demonstrated an increased microvascular contraction response to serotonin. The NAP group showed increased myocardial levels of thromboxane and lower levels of prostacyclin. Levels of protein oxidative stress were increased in the CBX group. Myocardial apoptosis was lowest in the NAP group. Immunoblotting demonstrated decreased VEGF and phospho-eNOS expression in the NAP and CBX groups. Myocardial TNFα was increased in both the NAP and CBX groups. Immunostaining for thromboxane A2 synthase and receptor demonstrated expression within the vascular smooth muscle and no observable differences between groups. Non-selective and selective COX inhibition does not alter myocardial perfusion, but results in altered myocardial and vascular physiology that may have implications regarding cardiovascular risk.
Keywords: Myocardial physiology, Cyclooxygenase inhibition, Prostaglandins, Microvascular reactivity
Introduction
Cyclooxygenase (COX) inhibitors specific for the inducible isoform (COX-2) of the enzyme were introduced in the early 1990's. This class of drug was developed to reduce the incidence of gastrointestinal complications associated with the use of non-specific non-steroidal anti-inflammatory drugs (nsNSAIDS). In a number of clinical trials COX-2 inhibitors were demonstrated to have an improved gastrointestinal safety profile.1 In the analysis of the VIGOR trial it was observed that patients with rheumatoid arthritis treated with the COX-2 inhibitor rofecoxib arm were at increased risk of myocardial infarction and stroke as compared to patients taking naproxen.2
In 2004 Merck voluntarily removed rofecoxib from the market after the APPROVe trial demonstrated a two fold increase in the risk of myocardial infarction in patients taking 25 mg of the drug after 18 months.3 The resulting scrutiny of ongoing trials showed that the risk of cardiovascular adverse events was not limited to rofecoxib, and most selective COX-2 inhibitors and nsNSAIDs were implicated.1, 4-6 At this time both selective and non-selective NSAIDS carry a black box warning from the US food and Drug Administration regarding increased cardiovascular risk.7 Further study has revealed that the risk for most COX-2 inhibitors and nsNSAIDs is dose-dependent.8
In 2007, the American Heart Association published a position paper on the use of NSAIDs and recommended a limited role for selective COX-2 inhibitors.9 However, most clinical trials examining the incidence of cardiovascular events in patients taking either selective COX-2 inhibitors or nsNSAIDS have found no difference in the incidence of adverse events.1, 4, 10
The leading theory as to why selective COX-2 inhibition may increase cardiovascular events was proposed as a result of a study by FitzGerald, and relates to differential levels of opposing prostaglandins.11 It has been hypothesized that selective COX-2 inhibition will lead to decreased endothelial prostacyclin (PGI2), a prostaglandin with vasodilatory and anti-thrombotic properties, and increased levels of thromboxane (TXA2), a platelet-derived pro-thrombotic vasoconstrictor. This shift in the myocardial prostaglandin milieu is thought to lead to increased coronary thrombosis and myocardial infarction (MI). This hypothesis was supported in a study of healthy volunteers given escalating doses of a COX-2 inhibitor (celecoxib)11. A nsNSAID (ibuprofen) suppressed TXA2-dependant platelet aggregation and celecoxib did not. Urinary concentrations of PGI2 were decreased equally by both drugs, but TXA2 concentrations were not reduced in the celecoxib group.11
To date there have not been any studies examining the effects of nsNSAIDs and COX-2 inhibitors under baseline (non-ischemic) conditions in a clinically relevant large animal model. In this study our aim was to further define the effects of NSAIDs on the non-ischemic myocardium. We hypothesized that nsNSAIDs and COX-2 inhibitors would influence myocardial perfusion, microvascular reactivity, prostaglandin levels, and the angiogenic potential of the myocardium.
Methods
Study Design
Yorkshire miniswine (Parsons Research, Amherst, MA) were divided into three groups. One received celecoxib 200 mg orally per day (CBX, n=8), another received naproxen 400 mg orally per day (NAP, n=10), and the third received no drug (control, n=8). All animals received drug treatment for seven weeks.
After seven weeks of drug therapy swine were anesthetized, the heart was exposed, and physiologic measurements were taken, followed by euthanasia. For the surgical procedure, anesthesia was induced with ketamine (10 mg/kg IM) and thiopental 2.5%, and maintained with a gas mixture of oxygen at 1.5 - 2 L/min and 3.0% isoflurane. The animals were intubated and mechanically ventilated at 16 breaths/min. After the heart was harvested, myocardial samples were rapidly frozen in liquid nitrogen (molecular studies and immunohistochemical studies) or placed in 4°C Krebs solution (microvessel reactivity studies).
All experiments were approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee. Animals were cared for in compliance with the Harvard Medical Area Institutional Animal Care and Use Committee and in accordance with the ‘Principles of Laboratory Animal Care’ formulated by the National Society for Medical Research and the ‘Guide for the Care and Use of Laboratory Animals’ (NIH publication no. 5377-3 1996).
Measurement of Global and Regional Myocardial Function
Indices of global and regional left ventricular (LV) function were obtained prior to animal sacrifice for 10 sequential beats using a Sonometrics system (Sonometrics Corp. London, ON, Canada), as previously described.12 Blood pressure was measured with an intra-arterial pressure catheter placed in the aorta.
Myocardial Perfusion Analysis
Myocardial perfusion was determined during each procedure with isotope-labeled microspheres (ILMs), 15 μm diameter (BioPAL Worcester, MA) using previously reported methods.13 Briefly, 1.5×107 Lutetium (rest) and Europium (pace) labeled ILMs were injected during the procedure. Following euthanasia, left ventricular sections were collected for ILM assay. The samples were exposed to neutron beams and microsphere densities were measured using a gamma counter.
Microvessel Studies
After cardiac harvest, coronary arterioles (80-180μm in diameter) from the myocardium supplied by the left anterior descending coronary artery (LAD) were dissected from the surrounding tissue and placed in an isolated microvessel chamber as described previously.14 Response to the vasoconstricting agent serotonin (5-HT, 10-9 mol/L to 10-5 mol/L) was assessed in non-precontracted vessels. Next, the microvascular response to vasorelaxing agent adenosine diphosphate (ADP, 10-9 to 10-4 mol/L, an endothelium dependent vasodilator) and sodium nitroprusside (SNP, 10-9 to 10-4 mol/L, an endothelium independent vasodilator) were evaluated after precontraction of microvessels by 30-50% of the baseline diameter with the thromboxane A2 analog U46619 (1.0 μM). Responses were defined as percent constriction of baseline diameter for serotonin and SNP, and percent relaxation of the preconstricted diameter for ADP. All drugs were applied extraluminally. Responses were defined as percent constriction of baseline diameter. All reagents were obtained from Sigma-Aldrich (St Louis, MO).
Tissue Thromboxane and Prostacyclin Levels
Tissue levels of stable thromboxane and prostacyclin breakdown products, 11-dehydrothromboxane B2 (11-d-TXB2) and 6-keto prostaglandin F1α (6-k-PGF1α), respectively were measured by ELISA (Neogen Corp, Lexington, KY). Tissue lysates underwent liquid-liquid exchange, extraction, and concentration according to the manufacturer's recommendations. The samples were put into an antibody coated plate along with HRP conjugated 6-k-PGF1α. The plate was washed and HRP substrate added. The plate was incubated and read on at 650 nm, and sample results were plotted against the standard curve.
Serum Prostacyclin Levels
Levels of 6-k-PGF1α were measured by ELISA (Neogen Corp). Serum samples underwent liquid-liquid exchange, extraction, concentration, and assay as described above.
Immunoblotting Studies
Sixty micrograms of total protein was fractionated by 4-20% gradient, SDS polyacrylamide gel electrophoresis (Invitrogen, San Diego, CA) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA). Each membrane was incubated with specific primary antibodies (Cell Signaling, Beverly, MA). Immune complexes were visualized with the enhanced chemiluminescence detection system (Amersham, Piscataway, NJ). Bands were quantified by densitometry of autoradiograph films. Ponceau staining was used to ensure equal protein loading.
Immunofluorescence Studies
Frozen sections (12μm in thickness) of myocardium from the left ventricle were formalin fixed. Antibodies against proliferation marker Ki-67 (Abcam Inc., Cambridge, MA) and endothelial marker PECAM-1 (CD-31, R&D Systems, Minneapolis, MN) were simultaneously applied to the sections and incubated. Arteriolar density was assessed by staining with smooth muscle actin (SMA, Cell Signaling). Thromboxane synthase (TS, Abcam) and receptor (TR, Cayman Chemical, Ann Arbor, MI) were assessed by co-staining with SMA. Detection was obtained using appropriate secondary antibodies (Jackson ImmunoReaserch, West Grove, PA). Sections were mounted in Vectashield with 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA). Photomicrographs were taken with a Zeiss Axiolab microscope (Carl Zeiss Inc, Thornwood, NY) equipped with a digital camera at 200× magnification (Photodoc, Upland, CA). Dividing endothelial cells, capillary density, and arteriolar cell density were counted in a blinded fashion. Data are presented as number of positive cells/mm2.
Myocardial Protein Oxidative Stress
Dinitrophenylhydrazine-derivatized myocardial tissue homogenates were separated by 10% polyacrylamide gel electrophoresis and transferred to PVFD membranes (Chemicon International, Inc. Temecula, CA). The membranes were incubated with primary antibody specific to dinitrophenylhydrazine, followed by incubation with a horseradish peroxidase-linked secondary antibody. Immune complexes were visualized with the enhanced chemiluminescence detection system (Amersham).
Quantification of Apoptotic cells
Apoptotic cells in the myocardium were identified using the ApopTag detection kit according to manufacturer's specifications (Chemicon Inc.). At least one cm2 of tissue was analyzed from each animal (4 per group). The slides were counter-stained with hematoxylin allowing the morphologic identification of cell types, specifically in distinguishing cardiomyocytes from other cells. The number of TUNEL-positive cardiomyocytes is expressed as positive cells/cm2.
Data Analysis
All results are expressed as mean ± SEM. Microvessel responses are expressed as percent relaxation of the preconstricted diameter, or contraction of the baseline diameter, and were analyzed using two-way, repeated measures analysis of variance (ANOVA) with a post-hoc Bonferroni test, which was applied to interactions of treatment and dose. Western blots were analyzed after digitalization (ScanJet 4c; Hewlett-Packard, Palo Alto, CA) with NIH ImageJ 1.33 software (National Institute of Health, Bethesda, MD). For data analysis, levels of phosphorylated proteins were normalized to total expression levels. Comparisons between the three groups were analyzed by one-way, repeated measures ANOVA with Newman-Keuls Multiple Comparison post-hoc test, using GraphPad Prism 4 (GraphPad Software Inc., San Diego, CA).
Results
Functional
Both systolic and diastolic blood pressures were significantly increased in the CBX group as compared to the control and NAP groups. Calculation of rate pressure product (RPP) demonstrated significantly lower values in the NAP group as compared to the control and CBX groups. There was no difference in RPP between the control and CBX groups (Table 1). The weight of the animals increased similarly among the groups from the initiation of drug treatment until the end of the study (increase in control 10.1 ± 1.6 kg), naproxen 11 ± 1.2 kg, and celecoxib 7.7 ± 0.6 kg, p= 0.18).
Table 1.
Physiologic measurements. SBP- systolic blood pressure, RPP- rate pressure product.
| Control | NAP | CBX | p-value | |
|---|---|---|---|---|
| SBP (mmHg) | 69 ± 6.6 | 53 ± 5.4 | 96 ± 8.5 | 0.01 |
| RPP (mmHg*beats/min) | 9012 ± 1302 | 5665 ± 599 | 9029 ± 879 | 0.04 |
| Blood Flow- Rest (mL/min/gram) | 0.86 ± 0.17 | 1.22 ± 0.18 | 1.0 ± 0.18 | 0.36 |
| Blood Flow- Pacing (mL/min/gram) | 0.96 ± 0.13 | 0.86 ± 0.06 | 0.86 ± 0.18 | 0.84 |
Myocardial Perfusion
Myocardial perfusion was assessed by microsphere density. There was no difference in blood flow to the myocardial territory supplied by the LAD at rest or with pacing between the groups (Table 1).
Microvascular responses
There was no difference in the baseline diameter of the microvessels between groups (control 135 ± 9 μm, NAP 128 ± 14 μm, and CBX 144 ± 13 μm, p=0.67), or the preconstricted diameter of the microvessels when the responses to ADP or sodium nitroprusside were examined (control 95 ± 6 μm, NAP 77 ± 8 μm, and CBX 96 ± 7 μm, p= 0.16). There was no difference in the percent vessel preconstriction between the groups with U46619 (control -37 ± 2%, NAP -36 ± 3%, and CBX -34 ± 1%, p= 0.73).
The microvessel contraction response to serotonin was significantly increased in both the NAP and CBX groups as compared to the control group. There was no difference between the two drug treatment groups (Figure 1A). There was no difference in microvessel response to the endothelial dependent vasorelaxation agent ADP (Figure 1B). There was no difference in the microvascular response to endothelial independent drug SNP (control 93.4 ± 5.7%, NAP 81.5 ±5.4%, CBX 74.9 ± 7.8%, p= 0.25 at 10-5 M).
Figure 1.

Microvessel reactivity. A) Microvessels harvested from the NAP and CBX group demonstrated increased contraction responses to serotonin as compared to the control (* p= 0.003 for control v. NAP and CBX). B) There was no difference in the microvascular responses to ADP, an endothelial dependent vasorelaxation agent.
Prostaglandin levels
The level of 11-d-TXB2 in the myocardial tissue was significantly increased in the NAP group as compared to the control and CBX groups. There was no difference between the control and CBX groups (Figure 2A). The level of 6-k-PGF1α was significantly decreased in the NAP group, and there was no difference between the control and CBX groups (Figure 2B).
Figure 2.

Prostaglandin levels. A) Animals in the NAP group demonstrated increased levels of thromboxane A2 (TXA2) as compared to the control and CBX groups (* p= 0.003). The NAP group was found to have lower levels of prostacyclin I2 (PGI2) as compared to the control and CBX groups († p< 0.001). There was no difference between the control and CBX groups in TXA2 or PGI2 levels (p> 0.05 for both).
The serum levels of 6-k-PGF1α were markedly decreased in NAP and CBX groups as compared to the control (control 3457 ± 770 ng/mL, NAP 281.8 ± 81.5 ng/mL, CBX 105.6 ± 81.6 ng/mL, p< 0.001).
Immunofluorescence
Both the NAP and CBX groups had fewer myocardial arterioles than the control group (Figure 3A-D). There was a trend toward lower capillary density in the myocardium of NAP and CBX groups, but this did not reach significance (Figure 3E). The number of dividing endothelial cells, positive staining for both CD-31 and Ki67, was decreased in the NAP and CBX groups as compared to the control group (Figure 3F).
Figure 3.
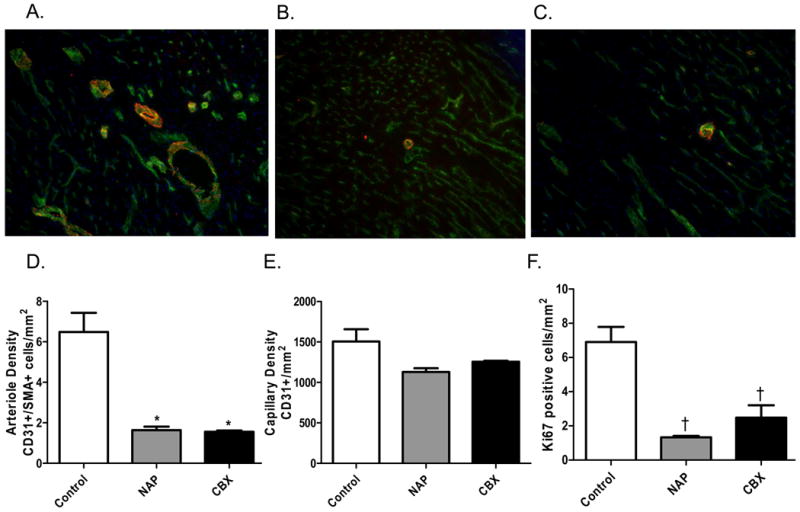
Arteriogenesis and Angiogensis. The control group demonstrated increased arteriole density by co-staining for CD-31 (an endothelial marker- green), smooth muscle actin (SMA- red), and DAPI (nuclear stain- blue) as compared to the NAP and CBX groups. A) Control group. B) NAP group. C) CBX group. All images are at 160×. D) Quantification of arteriolar density in the myocardium (p< 0.001 for control v. NAP and CBX). E) There was a trend toward decreased capillary density by CD-31 staining in the NAP and CBX groups (p= 0.12). F) Endothelial proliferation, assessed by co-staining with Ki-67 (proliferation marker) and CD-31, was significantly decreased in the NAP and CBX groups as compared to the control (p< 0.001).
Protein Expression
There was a significant downregulation of the angiogenic protein VEGF in the NAP and CBX groups as compared to the control group (Figure 4A). There was no difference in fibroblast growth factor (FGF) between the groups (p= 0.14) (not shown). The potent vasodilator and pro-angiogenic protein phospho-eNOS was significantly downregulated in the drug treatment groups (Figure 4B). There was an increase in the myocardial expression of the inflammatory cytokine TNFα in both the NAP and CBX groups as compared to the control group (Figure 4C). Levels of activated Akt and ERK (phospho-Akt (thr308) and phospho-ERK (thr218/tyr220)), normalized to total protein expression levels, were increased in the NAP group when compared to the control and CBX groups (Figure 4D and E).
Figure 4.
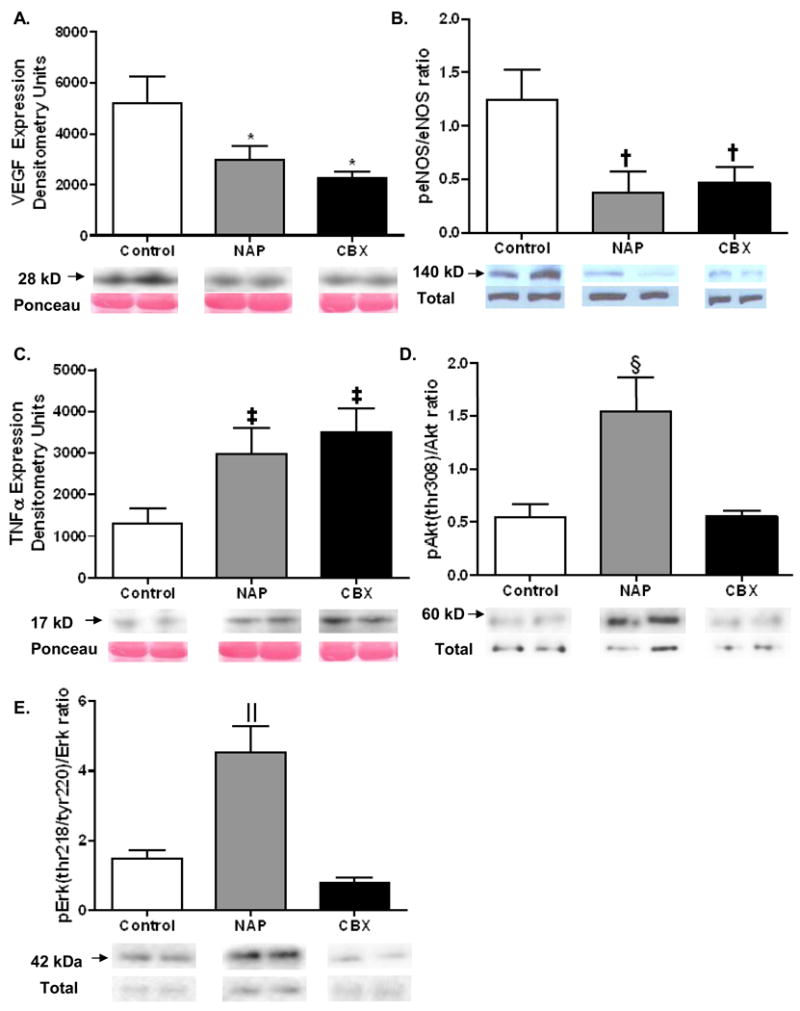
Immunoblotting. A) Expression of the angiogenic protein VEGF was highest in the control group, and significantly decreased in both the NAP and CBX groups (* p= 0.03). B) The expression of the vasodilatory, pro-angiogenic protein phospho-eNOS was highest in the control group, and significantly downregulated in the drug treatment groups († p= 0.03). C) Myocardial TNFα levels were upregulated in the NAP and CBX groups (‡ p= 0.02). D and E) Levels of activated Akt and ERK, normalized to total levels, were increased in the NAP group (§p= 0.004 and ‖ < 0.001, respectively).
Protein Oxidative Stress Levels
The levels of protein peroxidation were increased in the CBX as compared to the control and NAP groups. There was no difference between the control and NAP groups (Figure 5A).
Figure 5.

Levels of Oxidative stress and Apoptosis. Protein peroxidation was increased in the CBX group as compared to the control and NAP groups (* p< 0.001). Apoptotic cell death was decreased in the NAP groups vs. control and CBX groups († p= 0.007).
Levels of Apoptosis
The level of apoptosis in the myocardium at the time of tissue harvest was lowest in the NAP group. There was no difference in the number of TUNEL positive cells between the control and CBX groups (Figure 5B).
Discussion
NSAIDs have been purported to increase the risk of adverse cardiovascular events in patients. Although this has been described in a number of large clinical trials, other trials have found both non-selective and selective COX-2 inhibitors to be safe. The potential underlying basic pathophysiology and molecular influences of the purported increased incidence of adverse events with COX inhibition have not been clearly defined. Previous studies in cultured cell models have suggested that NSAIDs will impact the cardiovascular system.15, 16 We sought to define the effects of these drugs on the cardiovascular physiology, prostaglandin levels, and the angiogenic potential in a large animal model that mimics human cardiovascular physiology in most respects. There were a number of significant findings in this work that may give insight into how nsNSAIDs and selective COX-2 inhibitors affect the myocardium. Some of the effects are similar and others are divergent between the groups. Celebrex led to increased blood pressure and protein peroxidation, while naproxen caused a decrease in levels of prostacyclin and an increase in thromboxane, as well as decreased apoptosis. Both drugs altered the microvascular contractile response to serotonin, decreased serum PGI2 levels, and diminished vascular capillary density in this model during which the cardiac weight increases considerably during the course of the experiment. However, in the present study, myocardial perfusion surprisingly was not significantly affected by treatment with either naproxen or celecoxib.
Most NSAIDs have been implicated in causing systemic hypertension. In the current study only the selective COX-2 inhibitor was associated with increased blood pressure. The etiology of NSIAD induced hypertension is multifactorial and likely involves retention of sodium, increased renal vascular constriction, and microvascular dysfunction.17, 18 The increased microvascular contraction response to serotonin, and presumably other vasoconstrictive agents, may offer one potential mechanism for the increased blood pressure. In the current work we examined the effect of three molecules on the coronary microvasculature. In vivo there are many molecules influencing microvascular tone, and dysfunction has been linked to the development of hypertension.19 Additionally, while increased expression of thromboxane receptor can lead to hypertension14, we did not observe differences in thromboxane receptor or thromboxane synthase in the vascular smooth muscle of either treatment group, nor was the microvascular response to the TXA2 analog U46619 different between groups.
The decrease in rate pressure product in the NAP group was unexpected. While a decreased RPP may have the potential to decrease cardiovascular risk, the effects of the nsNSAID on levels of prostacyclin could increase risk. According to the FitzGerald hypothesis, the increase in cardiovascular events with selective COX-2 treatment may be due to an imbalance of pro- and anti-thrombotic prostaglandins.11 In our study naproxen treatment led to increased thromboxane and decreased prostacyclin, while the selective COX-2 inhibitor, celebrex, did not, though both drugs significantly decreased the serum levels of prostacyclin. This finding is contrary to what would be predicted by the FitzGerald hypothesis, which would predict that a selective COX-2 inhibitor would produce more TXA2 and less PGI2 compared to a non-selective COX inhibitor.
Both naproxen and celecoxib affected molecular and cellular indices of angiogenesis although perfusion was similar between treatment groups. Both drugs decreased arteriolar and to a lesser extent, capillary density in the young pigs undergoing significant myocardial growth during the course of the experiment. In addition, endothelial cell proliferation was diminished by both naproxen and celecoxib as compared to the control. VEGF, phospho-eNOS, and COX-2 are all integral parts of the angiogenic process. The importance of COX-2 as an essential component of angiogenesis has been well described. In one study, the formation of vessels in Matrigel was decreased in endothelial cells transfected with COX-2 silencing RNA, while cells overexpressing COX-2 led to increased vascular density.21 Collateral development in ischemic myocardium can bypass coronary vessels affected by occlusive disease. The presence of a well-developed network of collaterals has been associated with a reduced risk of death and myocardial injury after an MI.23 Recently, we reported that both naproxen and celecoxib alter the vascular and myocardial homeostasis under conditions of chronic myocardial ischemia.24 These changes did not lead to a reduction in collateral dependent perfusion despite diminished capillary density as compared to non-treated control animals. In fact, naproxen was actually associated with a significant increase in collateral dependent perfusion, while celecoxib was associated with no difference in collateral dependent perfusion compared to the non-treatment control. Interestingly, the decrease in arteriolar density did not translate to reduced blood flow to the myocardium in either study, as perfusion at rest and during ventricular pacing was not significantly different between groups.
Vascular density is one determinate of blood flow, but other mechanisms may have compensated including neurohumoral influences and autoregulation. Coronary autoregulation and metabolic control of perfusion are powerful mediators of coronary blood flow that ensure constant blood flow to meet the heart's metabolic demands.25 The mechanism by which autoregulation works is not completely understood, but likely involves vasoactive metabolites such as those listed above and autonomic control of myogenic responses.26 We assessed a limited number of these potential mediators of blood flow, though they likely played a role in maintaining myocardial perfusion. The implications of the above described differences between groups in adult patients not undergoing significant myocardial growth or ischemia are not known.
While myocardial perfusion was not different between groups in this study, levels of protein oxidative stress were increased in the CBX group as compared to the other groups. Increased oxidative stress is often related to an increase in reactive oxygen species (ROS). NSAIDs have been shown in cell culture studies to either increase or decrease ROS. A prolonged increase in ROS can lead to cardiac remodeling and ultimately heart failure via cardiomyocyte hypertrophy, induction of interstitial fibrosis, and apoptosis.27 Upregulation of TNFα has been associated with elevated ROS.28 In our study the reason for elevated TNFα in the drug treatment groups was not clear, though it may have been compensatory for the generally anti-inflammatory action of NSAIDs. While increased ROS and TNFα are associated with apoptosis, surprisingly we did not observe this, and apoptosis at the time of tissue harvest was actually decreased in the NAP group.
Control of apoptosis is complex, but it has been demonstrated that both Akt and ERK decrease this process.29 In a recent study it was shown that phospho-ERK, in association with IL-10, decreases TNFα induced apoptosis.30 The increased expression of these activated proteins may have contributed to the reduced level of apoptosis in the naproxen treated group.
Limitations
There are several limitations of this study. First, in most situations the porcine coronary circulation closely mimics the physiology and pathophysiology of the human coronary circulation, though this may not necessarily be the case for COX inhibition. Second, while we were able to assess a number of physiologic and molecular pathways, we did not measure platelet activity or the production of thromboxane by platelets. Platelet-derived thromboxane will contribute to a pro-thrombotic, vasconstrictive milieu in the myocardium. Prostaglandins are not only synthesized in vascular tissue, but also in the lung, gut, prostate gland and other organs, which may affect the conclusion that can be drawn from this study. Finally, only a single dose of each drug was investigated, and different dosages would likely result in different effects on the myocardium. Further, a number of significant findings related to nsNSAIDs and selective COX-2 inhibitors have been described here, but these results cannot be considered to represent the effects of other NSAIDs. The NSAIDs are a complex class of drugs that are often dose-dependent and act along a spectrum on multiple pathways.
Conclusion
In this study we attempted to describe the effects of non-selective COX and selective COX-2 inhibitors on the normally perfused myocardium. Celecoxib caused increased blood pressure and protein peroxidation, while naproxen caused decreased tissue prostacyclin and increased thromboxane, as well as decreased numbers of apoptotic cells. Both drugs altered the microvascular response to serotonin, reduced serum prostaglandin levels, and decreased arteriogenesis. Importantly, neither drug lead to an alteration of baseline myocardial perfusion or perfusion during rapid pacing. Overall, both non-selective and selective COX inhibition result in altered systemic and myocardial physiology. The impact of these drugs appears to be quite complex, and may have implications regarding cardiovascular risk.
Figure 6.
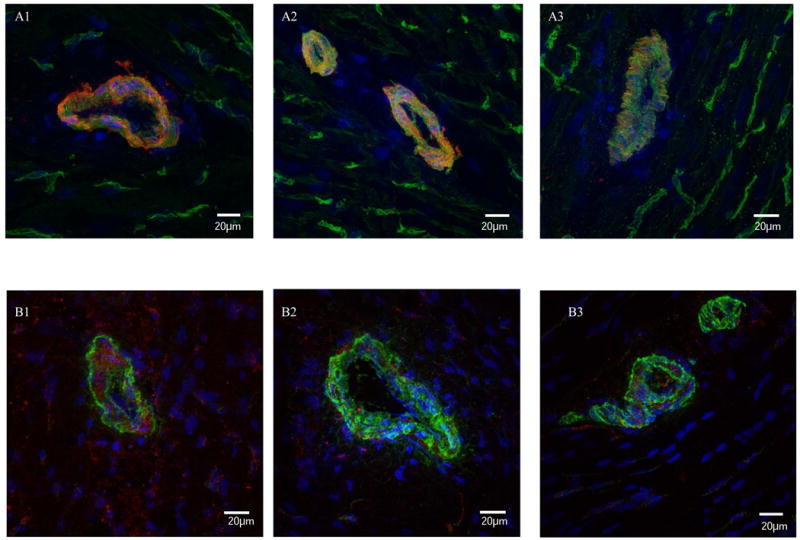
Immunostaining for thromboxane synthase (TS) and receptor (TR). Co-staining for TS and SMA (A) demonstrated localization in the vascular smooth muscle, and no obvious differences between groups; 1) control, 2) NAP, and 3) CBX. Immunostaining for TR and SMA (B) also showed localization in the vascular smooth muscle and no major differences in expression levels between groups; 1) control, 2) NAP, and 3) CBX.
Acknowledgments
Grants: Funding for this project was provided to F.W.S. by NHLBI (RO1HL46716, RO1HL69024, and RO1HL85647), NIH 5T32-HL0074 (M.P.R.) and the Irving Bard Memorial Fellowship (M.P.R., L.M.C.).
Abbreviations
- ADP
adenosine diphosphate
- AAR
myocardial area at risk
- COX
cyclooxygenase
- FGF
fibroblast growth factor
- LAD
left anterior descending coronary artery
- LCx
left circumflex coronary artery
- nsNSAID
non-selective non-steriodal anti-inflammatory drug
- PGI2
prostacyclin
- RPP
rate pressure pruduct
- SMA
smooth muscle actin
- SNP
sodium nitroprusside
- TXA2
thromboxane
- VEGF
vascular endothelial growth factor
Footnotes
Disclosures: Dr. Frank W. Sellke has research support from Ikaria (Clinton, NJ) and Orthologic (Tempe, AZ), and is a consultant for Novo Nordisk (Princeton, NJ), CSL Behring, and Cubist Pharmaceutical (Lexington, MA). Dr. Sellke was a consultant for the law firms representing Pfizer (Princeton, NJ) in the Bextra/Celebrex litigation, but no funding was received for this study, and there was no consultation or notification regarding this study.
References
- 1.Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, Makuch R, Eisen G, Agrawal NM, Stenson WF, Burr AM, Zhao WW, Kent JD, Lefkowith JB, Verburg KM, Geis GS. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284(10):1247–1255. doi: 10.1001/jama.284.10.1247. [DOI] [PubMed] [Google Scholar]
- 2.Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, Day R, Ferraz MB, Hawkey CJ, Hochberg MC, Kvien TK, Schnitzer TJ. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343(21):1520–1528. doi: 10.1056/NEJM200011233432103. 1522 p following 1528. [DOI] [PubMed] [Google Scholar]
- 3.Farkouh ME, Greenberg BP. An evidence-based review of the cardiovascular risks of nonsteroidal anti-inflammatory drugs. Am J Cardiol. 2009;103(9):1227–1237. doi: 10.1016/j.amjcard.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Singh G, Fort JG, Goldstein JL, Levy RA, Hanrahan PS, Bello AE, Andrade-Ortega L, Wallemark C, Agrawal NM, Eisen GM, Stenson WF, Triadafilopoulos G. Celecoxib versus naproxen and diclofenac in osteoarthritis patients: SUCCESS-I Study. Am J Med. 2006;119(3):255–266. doi: 10.1016/j.amjmed.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 5.Hernandez-Diaz S, Varas-Lorenzo C, Garcia Rodriguez LA. Non-steroidal antiinflammatory drugs and the risk of acute myocardial infarction. Basic Clin Pharmacol Toxicol. 2006;98(3):266–274. doi: 10.1111/j.1742-7843.2006.pto_302.x. [DOI] [PubMed] [Google Scholar]
- 6.Patrono C, Garcia Rodriguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353(22):2373–2383. doi: 10.1056/NEJMra052717. [DOI] [PubMed] [Google Scholar]
- 7.Dogne JM, Supuran CT, Pratico D. Adverse cardiovascular effects of the coxibs. J Med Chem. 2005;48(7):2251–2257. doi: 10.1021/jm0402059. [DOI] [PubMed] [Google Scholar]
- 8.Solomon SD, Wittes J, Finn PV, Fowler R, Viner J, Bertagnolli MM, Arber N, Levin B, Meinert CL, Martin B, Pater JL, Goss PE, Lance P, Obara S, Chew EY, Kim J, Arndt G, Hawk E. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation. 2008;117(16):2104–2113. doi: 10.1161/CIRCULATIONAHA.108.764530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA. Use of Nonsteroidal Antiinflammatory Drugs: An Update for Clinicians: A Scientific Statement From the American Heart Association. Circulation. 2007;115(12):1634–1642. doi: 10.1161/CIRCULATIONAHA.106.181424. [DOI] [PubMed] [Google Scholar]
- 10.Schnitzer TJ, Burmester GR, Mysler E, Hochberg MC, Doherty M, Ehrsam E, Gitton X, Krammer G, Mellein B, Matchaba P, Gimona A, Hawkey CJ. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet. 2004;364(9435):665–674. doi: 10.1016/S0140-6736(04)16893-1. [DOI] [PubMed] [Google Scholar]
- 11.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96(1):272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osipov RM, Robich MP, Feng J, Liu Y, Clements RT, Glazer HP, Sodha NR, Szabo C, Bianchi C, Sellke FW. Effect of hydrogen sulfide in a porcine model of myocardial ischemia-reperfusion: comparison of different administration regimens and characterization of the cellular mechanisms of protection. J Cardiovasc Pharmacol. 2009;54(4):287–297. doi: 10.1097/FJC.0b013e3181b2b72b. [DOI] [PubMed] [Google Scholar]
- 13.Boodhwani M, Voisine P, Ruel M, Sodha NR, Feng J, Xu SH, Bianchi C, Sellke FW. Comparison of vascular endothelial growth factor and fibroblast growth factor-2 in a swine model of endothelial dysfunction. Eur J Cardiothorac Surg. 2008;33(4):645–650. doi: 10.1016/j.ejcts.2007.12.016. discussion 251-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robich MP, Araujo EG, Feng J, Osipov RM, Clements RT, Bianchi C, Sellke FW. Altered coronary microvascular serotonin receptor expression after coronary artery bypass grafting with cardiopulmonary bypass. J Thorac Cardiovasc Surg. 139(4):1033–1040. doi: 10.1016/j.jtcvs.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Hortmann M, Daiber A, Oelze M, Ostad MA, Schwarz PM, Xu H, Xia N, Kleschyov AL, Mang C, Warnholtz A, Munzel T, Forstermann U. Cyclooxygenase 2-selective and nonselective nonsteroidal anti-inflammatory drugs induce oxidative stress by up-regulating vascular NADPH oxidases. J Pharmacol Exp Ther. 2008;326(3):745–753. doi: 10.1124/jpet.108.139030. [DOI] [PubMed] [Google Scholar]
- 16.Tsoyi K, Kim HJ, Shin JS, Kim DH, Cho HJ, Lee SS, Ahn SK, Yun-Choi HS, Lee JH, Seo HG, Chang KC. HO-1 and JAK-2/STAT-1 signals are involved in preferential inhibition of iNOS over COX-2 gene expression by newly synthesized tetrahydroisoquinoline alkaloid, CKD712, in cells activated with lipopolysacchride. Cell Signal. 2008;20(10):1839–1847. doi: 10.1016/j.cellsig.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Johnson AG, Nguyen TV, Owe-Young R, Williamson DJ, Day RO. Potential mechanisms by which nonsteroidal anti-inflammatory drugs elevate blood pressure: the role of endothelin-1. J Hum Hypertens. 1996;10(4):257–261. [PubMed] [Google Scholar]
- 18.Zhang J, Ding EL, Song Y. Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: meta-analysis of randomized trials. JAMA. 2006;296(13):1619–1632. doi: 10.1001/jama.296.13.jrv60015. [DOI] [PubMed] [Google Scholar]
- 19.Hoenig MR, Bianchi C, Rosenzweig A, Sellke FW. The cardiac microvasculature in hypertension, cardiac hypertrophy and diastolic heart failure. Curr Vasc Pharmacol. 2008;6(4):292–300. doi: 10.2174/157016108785909779. [DOI] [PubMed] [Google Scholar]
- 20.White WB. Heart rate and the rate-pressure product as determinants of cardiovascular risk in patients with hypertension. Am J Hypertens. 1999;12(2 Pt 2):50S–55S. doi: 10.1016/s0895-7061(98)00280-5. [DOI] [PubMed] [Google Scholar]
- 21.Wu G, Mannam AP, Wu J, Kirbis S, Shie JL, Chen C, Laham RJ, Sellke FW, Li J. Hypoxia induces myocyte-dependent COX-2 regulation in endothelial cells: role of VEGF. Am J Physiol Heart Circ Physiol. 2003;285(6):H2420–2429. doi: 10.1152/ajpheart.00187.2003. [DOI] [PubMed] [Google Scholar]
- 22.Gasparini G, Longo R, Sarmiento R, Morabito A. Inhibitors of cyclo-oxygenase 2: a new class of anticancer agents? Lancet Oncol. 2003;4(10):605–615. doi: 10.1016/s1470-2045(03)01220-8. [DOI] [PubMed] [Google Scholar]
- 23.Meier P, Gloekler S, Zbinden R, Beckh S, de Marchi SF, Zbinden S, Wustmann K, Billinger M, Vogel R, Cook S, Wenaweser P, Togni M, Windecker S, Meier B, Seiler C. Beneficial effect of recruitable collaterals: a 10-year follow-up study in patients with stable coronary artery disease undergoing quantitative collateral measurements. Circulation. 2007;116(9):975–983. doi: 10.1161/CIRCULATIONAHA.107.703959. [DOI] [PubMed] [Google Scholar]
- 24.Robich MP, Chu LM, Feng J, Burgess TA, Laham RJ, Bianchi C, Sellke FW. Effects of selective cyclooxygenase-2 and nonselective cyclooxygenase inhibition on ischemic myocardium. J Thorac Cardiovasc Surg. doi: 10.1016/j.jtcvs.2010.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feigl EO. Coronary autoregulation. J Hypertens Suppl. 1989;7(4):S55–58. discussion S59. [PubMed] [Google Scholar]
- 26.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88(3):1009–1086. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- 27.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81(3):449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 28.Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, Glembotski CC, Quintana PJ, Sabbadini RA. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98(12):2854–2865. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22(56):8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 30.Dhingra S, Sharma AK, Singla DK, Singal PK. p38 and ERK1/2 MAPKs mediate the interplay of TNF-alpha and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007;293(6):H3524–3531. doi: 10.1152/ajpheart.00919.2007. [DOI] [PubMed] [Google Scholar]