

Abstract
Protein function is generated and maintained by the proteostasis network (PN) (Balch et al. (2008) Science, 319:916). The PN is a modular, yet integrated system unique to each cell type that is sensitive to signaling pathways that direct development and aging, and respond to folding stress. Mismanagement of protein folding and function triggered by genetic, epigenetic, and environmental causes poses a major challenge to human health and lifespan. Herein, we address the impact of proteostasis defined by the FoldFx model on our understanding of protein folding and function in biology. FoldFx describes how general proteostasis control (GPC) enables the polypeptide chain sequence to achieve functional balance in the context of the cellular proteome. By linking together the chemical and energetic properties of the protein fold with the composition of the PN we discuss the principle of the proteostasis boundary (PB) as a key component of GPC. The curved surface of the PB observed in 3-dimensional space suggests that the polypeptide chain sequence and the PN operate as an evolutionarily conserved functional unit to generate and sustain protein dynamics required for biology. Modeling general proteostasis provides a rational basis for tackling some of the most challenging diseases facing mankind in the 21st century.
Newly synthesized proteins must fold into a unique three-dimensional (3D) structure to become functionally active. We now appreciate that all proteins likely require the assistance of the “proteostasis network” (PN) to generate and maintain function. The PN comprises not less than a 1000 factors that regulate protein synthesis, folding, function, and degradation [1–3] (FIG. 1). These form the Yin and Yang environment that promotes what we have referred to recently as proteome balance in health [4]. Importantly, the composition of the PN is dynamically regulated by a variety of signaling pathways [5,6], and in response to developmental cues, genetic changes, epigenetic marks, environmental stress and aging; challenges that all cells encounter during their lifespan to maintain normal organismal physiology [3,7,8]. Of importance, is that a very large number of inherited diseases are caused by mutations in the sequence of a polypeptide chain, leading to loss of protein stability, misfolding and disease. While genetic changes often severely challenge the dynamics of proteostasis to retain proteome balance, the response of the PN to mutation can significantly contribute to organismal evolution [9]. Given the multiplicity of cellular PN stress responses, it is not surprising that the PN has evolved to be highly versatile in its capacity to maintain proteome balance. Herein, we discuss the role of protein energetics and kinetics in generating and maintaining proteome balance through the activity of the PN. We explore how modeling of proteostasis opens new avenues to the management of human health and disease.
Figure 1. The PN.
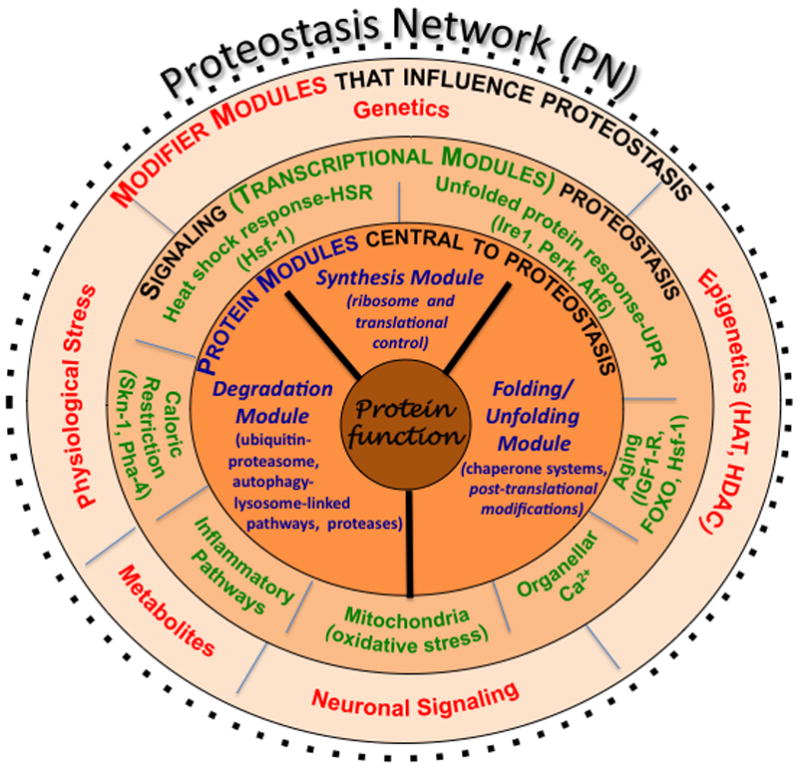
Shown are the interactions that comprise the PN, the composition of which is responsible for generating and maintaining the biological protein fold. Components comprising the PN outlined in the inner-most layer (in blue font) involve the synthesis module, the folding/unfolding module, and the degradation module (the GPC triad). A second layer shows the signaling transcriptional pathways (in green font) that influence the level and activity of the triad found in the innermost layer. The third layer (in red font) includes modifiers that influence and/or integrate the activities defined by the second and first layers. Modifiers and signaling pathways from both cell autonomous and cell non-autonomous origin. Modified figure reproduced with permission from Elsevier Press [2].
Principles guiding the function of the Proteostasis network
While small, single domain proteins can fold efficiently in the test tube, we now appreciate that these and multi-domain proteins generated in the crowded environment of the cell often fail to do so. This is because there are energy barriers in the landscape model (Fig. 2a, peaks and troughs) [10,11], that dictate the kinetics and thermodynamics of folding intermediates in the path(s) required to achieve the native folded state (Fig. 2a, red circles and ‘N’ in figure). The native state here is defined as the state with the lowest energy- which may or may not be the biologically important state [4]. To avoid off-pathway misfolding, degradation and/or aggregation that can occur during progress through intermediate folding steps in biology (Fig. 2a, red circles and white arrows), PN components are thought to interact with the polypeptide chain to generate and protect biological function (Fig. 2a, black and gray arrows) [1,2].
Figure 2. Coupling of the folding energy landscape with the PN.
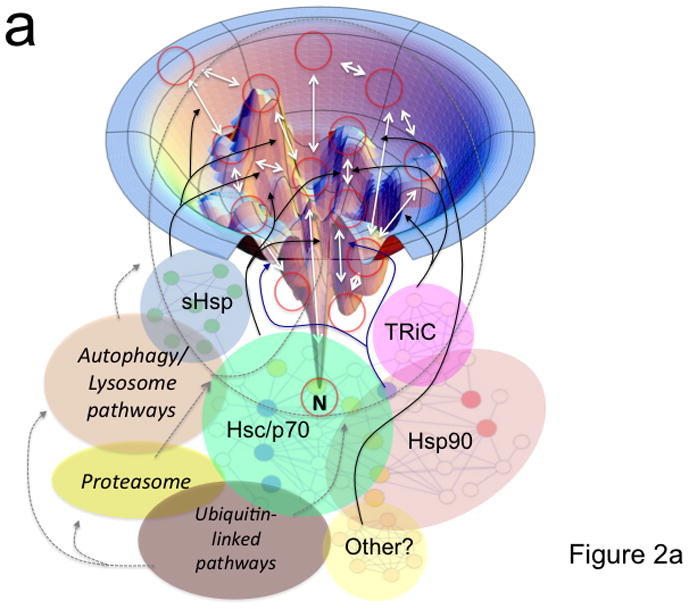
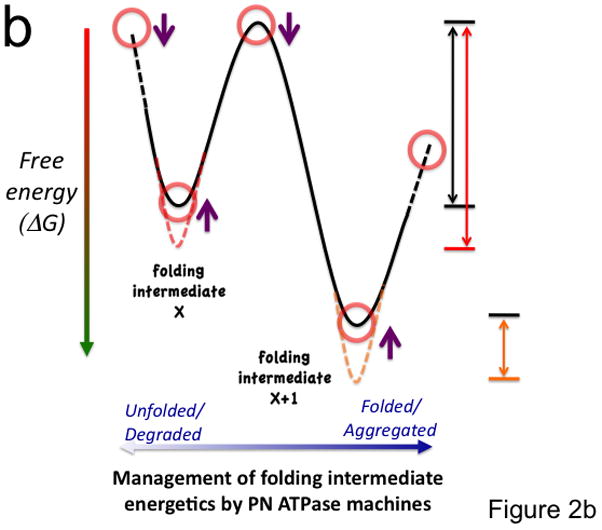
(a) Illustrated is a bumpy energy landscape funnel (http://www.dillgroup.ucsf.edu/) in which an unfolded protein proceeds along various intermediate steps (red circles) that can pose energetic barriers to achieve the native state (N) at the base of the funnel. The white arrows indicate potential pathways through various folding intermediates that the nascent protein may take to reach the native state. The solid black and blue (synthesis and folding modules) and the dashed gray (degradative module) arrows illustrate how different components of the PN may influence pathway choice. The dashed oval indicates that all intermediates are potential steps in which a protein can be targeted for degradation. Hsp90 is thought to principally facilitate late folding events (solid blue lines). (b) The energy landscape (a slice through the funnel illustrated in panel a) illustrates the central role of the ATPase cycle in synthesis, folding and degradative modules in managing the biology of the protein structure in the cell to achieve function. By coupling the energy of ATP hydrolysis (X-axis) by PN ATPase machines with the energetics imparted in the chemistry of the amino acid sequence of the polypeptide chain (Y-axis), PN ATPases maintain the protein fold in a dynamic state- essential for biology. Right side of panel illustrates energy barriers necessary to achieve a functional fold (black arrow) relative to the energetics associated with misfolding (red and orange arrows). The additional energetic demands challenging GPC through misfolding (red line) or aggregation (short orange line) are illustrated. Purple arrows indicate potential steps for GPC to alter folding kinetics and energetics to promote proteome balance and cell health. Abbreviations: sHsp (small heat shock proteins); TRiC (TCP1-ring complex).
The PN is an integrated system consisting of chaperones, folding enzymes, degradation components, and regulatory pathways that control the composition and concentration of the general proteostasis system [5,12] (Fig. 1). PN components include the molecular chaperones/co-chaperones belonging to the Hsc/Hsp (Hsc/p) 70 and 90 families [13], the GroEL/TCP1-ring complex (TRiC)/chaperonin family of folding machines [14], tetratricopeptide repeat (TRP)-domain containing proteins, proteins that modulate oxidative folding (e.g., protein disulfide isomerases [15]), and degradation components comprising both the cytosolic ubiquitin-proteasome and membrane-linked autophagy-lysosome systems [6,16] (Fig. 1). Some PN components are highly abundant (e.g., ribosome, Hsc/p70-Hsp90, proteasome/lysosome) and provide a cellular ‘buffer’ for synthesis, folding and/or degradation [12]. Most others function as specialists, either alone or together with the Hsc/p70 and Hsp90 systems, to synthesize and/or maintain specific folds for the highly evolved dynamic functions dictating extant organismal physiology. Regulation of the composition of the PN occurs through a number of signaling pathways including the unfolded protein response (UPR) [17], the heat shock response (HSR) [7,18,19], oxidative stress pathways [20], and growth factor and diet sensitive pathways [1–3], among others. A simplistic view of the PN is that components directing synthesis, (un)folding, and degradation could be considered as a triad of modules (Fig. 1, dark black lines). Triad modules recognize the chemical properties of polypeptide folding intermediates (Fig. 2), yet are integrated by the overall composition of the PN to maintain normal biological function.
It is important to recognize that the PN is unique for each type of cell and the numerous subcellular compartments within a eukaryotic cell. These environments change differentially during development, aging and in response to physiological stress [2,21]. Moreover, the PN is constantly challenged by changes in the composition of the amino acid pool, metabolites/co-factors, ion balance, genetic-epigenetic-environmental triggers, and viral/bacterial pathogens. These factors not only affect the inherited capacity of the proteostasis program, but are readily sensed by the above regulatory signaling pathways that attempt to rebalance the proteome to preserve healthspan [1,3,4]. Thus, the PN is dynamically tuned to cellular function as prescribed by cell autonomous processes and cell non-autonomous signals that optimize folding for function in complex organismal environments [7,19].
Role of ATP in maintaining proteome balance
The capacity of the PN to maintain proteome balance in the cytosol and exocytic/endocytic trafficking compartments, that is, the Yin-Yang relationship between generating and maintaining a functional fold or targeting a protein for degradation [4], is referred here as general proteostasis control (GPC). GPC emphasizes that function of a polypeptide chain is tightly linked to the local composition of the PN triad- the environment being the ultimate arbitrator of biological folding for function. What is a wild-type protein fold in one PN environment becomes a ‘mutant’ in another and can be removed and/or challenge the health status of the cell. The former is evident in the cyclical stability of proteins during cell cycle, or the transient stability observed in developmental programs. The latter is observed in, for example, numerous sporadic aggregation diseases, type II diabetes and cancer [2].
Both subtle and global challenges to protein folding energetics directly challenge the dynamics of the kinetics of protein folding and its thermodynamic stability. Thus, there is a close link between PN folding for function (Fig. 2a) and ATP-based proteostasis machines that manipulate the energy landscape dictated by the unique chemistries of amino acid sequence of each polypeptide chain (Fig. 2b). For example, during protein biogenesis, newly synthesized polypeptides are generated by the ribosome at a very high energy cost and in response to protein specific translational control programs. They emerge from the ribosome with exposed hydrophobic residues that are recognized by the folding module of the PN to prevent protein aggregation. This first step of GPC faced by nascent proteins is often regulated by members of the Hsc/p70 family [12] and/or the TRiC/chaperonin ATPase machines [22]. While TRiC ATPases appear to be dedicated to folding, the Hsc/p70 ATPases function to either promote folding/assembly of newly synthesized proteins or direct ‘non-native’ polypeptides to degradation [23], serving as a key linker between the various PN modules (Fig. 1). Thus, the Hsc/p70 system plays a critical role in proteome balance in response to energetics of the polypeptide chain [4].
Hsc/p70 family of chaperones, utilize ATP-dependent cycles of client binding and release in response to a plethora of accessory proteins, called co-chaperones. In the case of Hsc/p70 these include nucleotide exchange factors (NEFs) composed by the Bag (BCL2 associated athanogene) family of proteins, which facilitate ADP release and ATP binding to promote Hsc/p70 client substrate release, and a large Hsp40/DnaJ family of co-chaperones that stimulate the ATPase activity of Hsc/p70 and stabilize protein client-chaperone interactions [24]. Thus, Hsc/p70 co-chaperones will not only fine-tune Hsc/p70 client substrate specificity but dictate the cellular fate of the protein client [13,25,26].
Proteins that interact with the Hsc/p70 arm of GPC are in many cases subject to a second level of maintenance by the Hsp90 system [27]. The Hsp90 ATPase machinery appears to recognize dynamic facets of more folded substrates to modulate their activity(s) (Fig. 2a, blue lines), [27,28] (www.picard.ch/downloads/Hsp90interactors.pdf). As is seen for the Hsc/p70 system, a unique collection of co-chaperones also regulate Hsp90 ATPase activity. Hsp90 co-chaperones include the ATPase activator Aha1 and the ATPase inhibitor p23, as well as Cdc37, HOP, protein-disulfide isomerases (PPIases), and a large family of TRP-domain containing proteins. Depending on the local activity/composition of the co-chaperone environment, Hsp90 can promote either protein stability or degradation of folding intermediates- dynamically altering the proteome balance [4] (Fig. 2b).
Like the folding GPC module (Fig. 1), the degradation module involving the proteasome and the autophagy-lysosome pathways are intensely ATP-dependent [29]. While many of the regulatory factors that control function of these degradative machines remain to be determined, client targeting to multiple degradative pathways is generally regulated by the ubiquitin/sumoylation system. Targeting for degradation utilizes a highly diverse (>300) set of client specific ligases that utilize the energy of ATP to prime polypeptide targets for destruction [23]. Thus, ATP-dependent cycling of synthesis, folding, and degradation modules provides an energetic link between the functional and degradation prone states of a target protein in biology.
While GPC has a universal high level of energy demand, it is important to realize that folding/function is highly compartmentalized. For example, the cytosol is a reducing environment maintaining folded proteins in the absence of disulfide bonding. In contrast, the endoplasmic reticulum (ER), the first step in the secretory pathway, is an oxidative environment where protein folding is driven by an evolutionarily related, but distinct set of luminal PN folding components. The folding module in the ER is tightly coupled by membrane translocation pathways to the reducing cytosolic proteasomal degradation module [23,30,31]. Recent studies [32] have shown that cytosolic GPC components important for generation of newly synthesized transmembrane polypeptides in the ER also modulate protein stability at the cell surface. Likewise, the lysosome not only handles internalized cargoes from the cell surface, but is a critical partner of the autophagosome pathway that engulfs a wide range of misfolded cytosolic proteins and dysfunctional organelles [29,33]. These results suggest the importance of as yet unknown cellular proteostasis sensors that unify and balance folding throughout the cell.
In summary, it is now clear that GPC may define energetic standards for each cell type that is linked in as yet to be determined ways by the activity of PN-linked ATPase machines. By coupling the chemistry of the polypeptide chain sequence and its associated folding energetics with the ATPase activity of PN modules (Fig. 2b), a biologically dynamic GPC standard generates the proper balance between the triad of synthesis, (un)folding, and degradation modules (Fig. 1). This standard defines the proteome balance in a healthy cell and its response to stress, disease, injury and aging programs.
Modeling proteostasis
An understanding of the rules guiding GPC to achieve protein function involves integrating the chemistry of the polypeptide sequence with the activity of PN components (Fig. 2). For this purpose, we applied Michaelis-Menten formalism in the FoldEx model to describe how the inherent chemistry and energetics of the polypeptide chain can be read and manipulated by the PN for proteins trafficking through the exocytic pathway [34]. The concepts stemming from the FoldEx model were extended to describe a more encompassing model of how folding energetics and the PN work together. We refer to this new model as FoldFx [2]. FoldFx is applicable to folding of proteins in all compartments of the cell and the extracellular space in response to the composition of the local PN (Fig. 1). In FoldFx, the operational goal of the triad of protein synthesis, (un)folding, and degradation modules through GPC is to achieve ‘function’.
A key feature of the FoldFx model which rigorously defines the activity of GPC is the concept of the ‘proteostasis boundary’ or PB [2] (Fig. 3a). The PB can be used to define the minimal energetic properties that a protein must have to achieve normal function in response to the local PN. The PB is best illustrated in a 3-dimentsional (3D) space diagram as a curved surface. The position of a protein in 3D is determined by its inherent folding kinetics, misfolding kinetics, and thermodynamics (Fig. 3a). The curved shape of the PB is dictated by the variable concentration of proteostasis components. These are, in turn, defined by the genetic, epigenetic, and intrinsic and extrinsic factors that regulate PN pathways and thereby tune the PN for specific client functionality. Beneath the boundary is a normal biological network, defined by nodes (the proteins) and edges (their links to other proteins within the network) (Fig. 3a). Each node is positioned according to its folding energetics (its unique folding and misfolding rate and stability). In a healthy cell, each node and its link (the edges in Fig. 3a) are embraced by the curved space of the PB, indicating that the PN is sufficient to maintain normal function (Fig. 3a). In disease, a node falls outside the embrace of the PB, resulting in misfolding, aggregation and/or degradation (Fig. 3b).
Figure 3. The proteostasis boundary (PB).
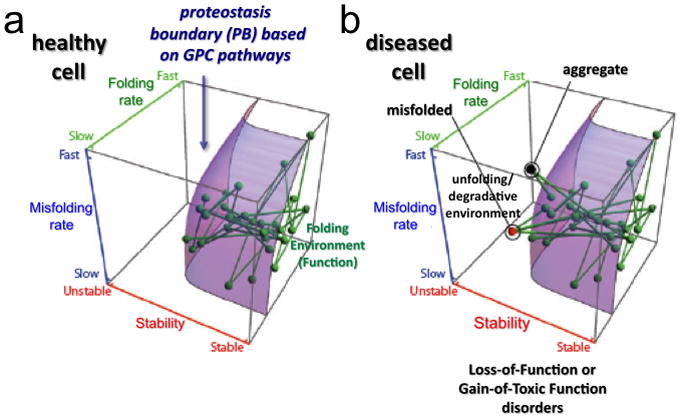
The position of each node (protein) relative to the PB (curved surface) responsible for biological function is defined by a protein’s folding properties: folding kinetics (Z-axis), misfolding kinetics (Y-axis) and thermodynamic stability (X-axis). Each line defines a physical or functional interaction between two proteins in the system. The location of the PB in 3D space is established by the composition of the PN and modulated by the GPC triad. (a) All of the nodes are within the PB boundary in a healthy cell. (b) Mutations or aberrant post-translational modifications can alter folding kinetics and energetics, making their corresponding nodes and edges fall outside (above the curved surface) of the PB. This space in the 3D plot does not support function of the energetically destabilized variant and can lead to either degradation (red node) or protein aggregation (black node). The loss of connectivity to proteins within the embrace of the PB can challenge the entire PN leading to cell, tissue, and organismal disease. Reproduced with permission from Elsevier Press [2].
Current evidence suggests that in healthy cells the PB is protective but does not have excess of proteostasis capacity [2,5,7]. To compensate for this limit threshold (Fig. 3), the PB is highly responsive to numerous signaling pathways (Fig. 1). Thus, the GPC operates as a rheostat to control the shape of the PB in time and space- it can be turned up or down to adjust the folding capacity in response to the environment to achieve biology.
GPC modeling in health and disease
Given the FoldFx model and the impact of GPC on protein function, when is a protein folded and functional from a biological perspective? Put in another way- what is disease? This remains a challenging question, as up to 30% of proteins are now thought to have some level of intrinsic ‘disorder’-that is, unfolded. One possibility suggested by FoldFx is that folding kinetics and energetics defined by the energy landscape (Fig. 2) are continuously defined and redefined by the Yin and Yang of the GPC triad in a fashion that considerably extends the chemistry imparted by the polypeptide sequence to achieve normal cellular healthspan (Fig. 4a-GPC1). Thus, we would suggest that the the polypeptide sequence per se can be of limited utility to the cell [10,11]. Rather, what it does provide is a dynamic template for the GPC to sustain and evolve biology [9]. In contrast to a healthy proteome, in inherited folding disorders, during aging and in environmentally triggered pathologies (including physical and pathological insults), the Yin-Yang of the proteome imbalance challenges GPC biology [4], exceeding the capacity of the GPC to manage the polypeptide template folding to achieve function (Fig. 4b-GPC2).
Figure 4. Modeling the Yin-Yang of proteome balance in health, disease, and in response to proteostasis therapeutics.
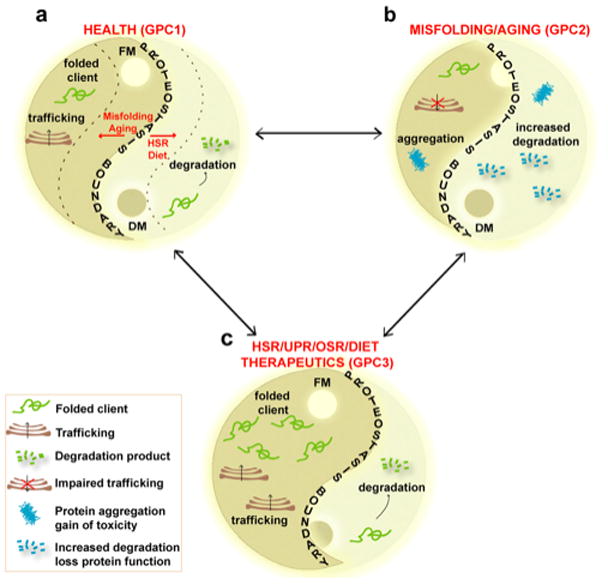
(Panel a) Proteome balance [4] in a healthy cell is determined by the composition of the synthesis and folding modules (FM) (the Yang on the left side of diagram) and degradative module (DM) (the Yin on the right side of diagram). GPC1 determines the position of the PB (the S-shaped curve) and healthspan. The dashed lines illustrate that misfolding and aging can challenge the position of the PB. (Panel b) Aging and unfolding move the PB to the left resulting in compromised proteostasis function (GPC2) and an unhealthy cell by triggering increased degradation and/or accumulation of protein aggregates. (Panel c) Biological signaling pathways including the HSR (HSF1 and IGF1-R/FOXO pathways), UPR, oxidative stress response (OSR), diet, IGF1-R and/or proteostasis targeted therapeutics can move the Yin-Yang balance defined by the PB to the right generating GPC3. GPC3 provides an environment that protects the cell from physiological stress, misfolding and aging, allowing the cell to return to GPC1.
Of particular contemporary interest is the aging-related decline in the functionality of the GPC triad, triggering systemic and neurodegenerative disease [3,35,36]. Increased protein ‘wear and tear’, transcriptional and translational dysfunction, and altered signaling pathways associated with aging can significantly challenge the PB, reducing the effective capacity of GPC triad and thereby triggering proteome imbalance (Fig. 3b, Fig. 4b). It is now recognized that aging related decay of the PN can be remediated by IGF1-R (FOXO and HSF-1 transcription factors), diet, and oxidative stress pathways. For example, the levels of expression of the Hsc/p70 chaperone system decrease in senescent fibroblasts and in the human brain during aging [3,35]. Likewise, the DNA binding activity of the HSF-1 transcription factor is diminished with aging in rat hepatocytes, causing an imbalance in the expression of the genes encoding chaperones during the stress response [37]. Studies have demonstrated that delaying the aging process by improving the activity of the Hsc/p70 system in both Huntington disease (HD) and Alzheimer disease (AD) models prevent disease onset [38]. During aging, protein degradation by autophagy is enhanced and Bag3 (an Hsc/p70 ATPase regulator), has been implicated as the main player linking the Hsc/p70 chaperone module and misfolded clients to the autophagy system [39]. Interestingly, senescent cells switch expression from the Bag1 to Bag3 co-chaperone [25], possibly as a compensatory mechanism to clear all the misfolded proteins that increasingly accumulate in aged cells. Similar to genetic and aging related changes in the GPC triad, environmental triggered pathologies are more cumulative and hugely impact the capacity of the PN to maintain proteome balance during aging as illustrated by both systemic (type II diabetes) and neurodegenerative (amyloid) disease.
In summary, it is becoming increasingly evident that we will need to further develop the FoldFx model in the context of genetic, epigenetic, and environmental factors that directly affect the status of the GPC triad during aging and its effect on proteome balance (Fig. 4a) to improve human healthspan.
Therapeutics in modeling of the GPC triad
Multiple lines of evidence suggest that protection to misfolding disease and aging can be boasted through multiple pathways that regulate the expression of PN components (e.g., HSR, IGF1-R signaling, diet restriction and pathways that protect against oxidative stress mentioned above) [1,3] (Fig. 1, Fig. 4c -GPC3). Modulation of components of the PN biologically by targeting individual PN components with siRNA implicated in these pathways can dramatically affect the outcome of disease. For example, depletion of Aha1 (an Hsp90 ATPase regulator) or E3ligase RMA1/CHIP, partially restores functionality in cystic fibrosis (CF) models [40,41]. These represent changes in distinct arms of the Yin-Yang balancing act, involving both cytosolic and exocytic/endocytic membrane trafficking pathways managed by the GPC triad (Fig. 4c). Moreover, overexpression of Hsc/p70 and its co-chaperones has been shown to reduce aggregation and toxicity in models of neurodegenerative/misfolding diseases, such as AD [42], prion disease [43], and HD [24]. Overexpression of Hsp40 reduces polyQ inclusion formation and toxicity [24] while the co-chaperone CHIP suppresses the toxicity of α-synuclein and polyQ proteins [44], and reduces accumulation of tau and Aβ [45], possibly through removal of aggregated misfolded proteins via the proteasome. The cofactor Bag1 also has been shown to reduce toxicity caused by polyQ Huntington aggregates [46]. Indeed, the FoldFx model predicts that bolstering the operation of the PN along specific axis’s is likely to not only improve healthspan, but also simultaneously improve longevity- the ultimate test of a therapeutic approach [1,2,7,8,47].
An increasing number of small molecules are now recognized to impact these pathways and provide protective function to human disease [4,48,49] (Fig. 4c). For example, the inhibition of the proteasome arrests myeloma disease [50], kinetic stabilizers arrest onset of TTR [51], histone deacetylases function to correct CF [52], Friedreich Ataxia [53], HD [54] and poly-glutamine (polyQ) disease [26] and have a strong like to GPC [18].
It is now clear that the ultimate goal for FoldFx modeling will be to utilize it as framework for further understanding of human biology and for development of small molecule therapeutics that manipulate GPC triad to maintain and restore human health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- **2.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. This article develops in detail the concept of the FoldFx model for understanding the impact of proteostasis on human biology. [DOI] [PubMed] [Google Scholar]
- 3.Douglas PM, Dillin A. Protein homeostasis and aging in neurodegeneration. J Cell Biol. 2010;190:719–729. doi: 10.1083/jcb.201005144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *4.Hutt D, Balch WE. Cell Biology. The proteome in balance. Science. 2010;329:766–767. doi: 10.1126/science.1194160. A short perspective on the article by Okiyoneda et al. (2010) (reference 32) that establishes the importance of proteostasis pathways affecting both synthesis and maintenance of the protein fold for function leading to proteome balance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **7.Prahlad V, Morimoto RI. Integrating the stress response: lessons for neurodegenerative diseases from C. elegans. Trends Cell Biol. 2009;19:52–61. doi: 10.1016/j.tcb.2008.11.002. Excellent review on proteostasis, aging and human neurodegenerative disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **8.Prahlad V, Cornelius T, Morimoto RI. Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science. 2008;320:811–814. doi: 10.1126/science.1156093. A key paper that strongly impacts our understanding on cell non-automous (neuronal) signaling pathways that regulate proteostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindquist S. Protein folding sculpting evolutionary change. Cold Spring Harb Symp Quant Biol. 2009;74:103–108. doi: 10.1101/sqb.2009.74.043. [DOI] [PubMed] [Google Scholar]
- 10.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys. 2005;38:245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 11.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 12.Broadley SA, Hartl FU. The role of molecular chaperones in human misfolding diseases. FEBS Lett. 2009;583:2647–2653. doi: 10.1016/j.febslet.2009.04.029. [DOI] [PubMed] [Google Scholar]
- *13.Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry. 2008;47:7001–7011. doi: 10.1021/bi800639z. Comprehensive review on major proteostasis components belonging to the Hsc/p 70 and heat shock families of chaperones/co-chaperones. [DOI] [PubMed] [Google Scholar]
- 14.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 15.Shamovsky I, Nudler E. New insights into the mechanism of heat shock response activation. Cell Mol Life Sci. 2008;65:855–861. doi: 10.1007/s00018-008-7458-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konstantinova IM, Tsimokha AS, Mittenberg AG. Role of proteasomes in cellular regulation. Int Rev Cell Mol Biol. 2008;267:59–124. doi: 10.1016/S1937-6448(08)00602-3. [DOI] [PubMed] [Google Scholar]
- *17.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. A comprehensive review of the UPR and its role in type II diabetes, a model and pervasive disease in the human population for understanding the impact of the proteostasis program on human (patho)physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **18.Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress- inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. An important paper that links HDAC activity to the proteostasis program. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng L, Chen R, Liang F, Tsuchiya H, Murai H, Nakahashi T, Iwai K, Takahashi T, Kanda T, Morimoto S. Silent information regulator, Sirtuin 1, and age-related diseases. Geriatr Gerontol Int. 2009;9:7–15. doi: 10.1111/j.1447-0594.2008.00504.x. [DOI] [PubMed] [Google Scholar]
- 20.Boutten A, Goven D, Boczkowski J, Bonay M. Oxidative stress targets in pulmonary emphysema: focus on the Nrf2 pathway. Expert Opin Ther Targets. 2010;14:329–346. doi: 10.1517/14728221003629750. [DOI] [PubMed] [Google Scholar]
- *21.Hutt DM, Powers ET, Balch WE. The proteostasis boundary in misfolding diseases of membrane traffic. FEBS Lett. 2009;583:2639–2646. doi: 10.1016/j.febslet.2009.07.014. A review that emphasizes proteostasis component compartmentalization and its impact on generating and maintaining proteome balance at the cell, tissue and organismal levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Baker ML, Schroder GF, Douglas NR, Reissmann S, Jakana J, Dougherty M, Fu CJ, Levitt M, Ludtke SJ, et al. Mechanism of folding chamber closure in a group II chaperonin. Nature. 2010;463:379–383. doi: 10.1038/nature08701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *24.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. An excellent review on the role of the Hsp40 family of proteostasis components in managing protein folding in the cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009;28:889–901. doi: 10.1038/emboj.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **26.Wang AM, Morishima Y, Clapp KM, Peng HM, Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP. Inhibition of hsp70 by methylene blue affects signaling protein function and ubiquitination and modulates polyglutamine protein degradation. J Biol Chem. 2010;285:15714–15723. doi: 10.1074/jbc.M109.098806. A key paper that describes that utility of proteostasis regulators to modulate the function of the Hsp70 interaction with Hsp40 to impact the onset and duration of neurodegenerative polyQ disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 28.Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J Biol Chem. 2008;283:18473–18477. doi: 10.1074/jbc.R800007200. [DOI] [PubMed] [Google Scholar]
- 29.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brodsky JL, Wojcikiewicz RJ. Substrate-specific mediators of ER associated degradation (ERAD) Curr Opin Cell Biol. 2009;21:516–521. doi: 10.1016/j.ceb.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakatsukasa K, Brodsky JL. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic. 2008;9:861–870. doi: 10.1111/j.1600-0854.2008.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **32.Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329:805–810. doi: 10.1126/science.1191542. A critical paper that describes the role of proteostasis components in maintaining the activity of proteins throughout the cell. See also Hutt and Balch (2010) (reference 4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab. 2010;21:142–150. doi: 10.1016/j.tem.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **34.Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, Balch WE. An adaptable standard for protein export from the endoplasmic reticulum. Cell. 2007;131:809–821. doi: 10.1016/j.cell.2007.10.025. A Michaelis-Menton based formalism that establishes the utility of the FoldEx model to understand the impact proteostasis on trafficking of protein through the eukaryotic exocytic pathway. [DOI] [PubMed] [Google Scholar]
- 35.Morimoto RI, Cuervo AM. Protein homeostasis and aging: taking care of proteins from the cradle to the grave. J Gerontol A Biol Sci Med Sci. 2009;64:167–170. doi: 10.1093/gerona/gln071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamanaka T, Miyazaki H, Oyama F, Kurosawa M, Washizu C, Doi H, Nukina N. Mutant Huntingtin reduces HSP70 expression through the sequestration of NF-Y transcription factor. EMBO J. 2008;27:827–839. doi: 10.1038/emboj.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **38.Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. A critical paper that describes the role of the IGF1-R signaling pathway in regulating the proteostasis program to increase longevity and program mammalian physiology from neurodegenerative disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carra S, Seguin SJ, Landry J. HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy. 2008;4:237–239. doi: 10.4161/auto.5407. [DOI] [PubMed] [Google Scholar]
- **40.Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. A paper that describes a system-based proteomic approach using mass spectrometry to understand the role of the proteostasis network in the manifestation of inherited human misfolding disease. [DOI] [PubMed] [Google Scholar]
- 41.Grove DE, Rosser MF, Ren HY, Naren AP, Cyr DM. Mechanisms for Rescue of Correctable Folding Defects in CFTR{Delta}F508. Molecular Biology of the Cell. 2009 doi: 10.1091/mbc.E08-09-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Y, Cao Z, Klein WL, Luo Y. Heat shock treatment reduces beta amyloid toxicity in vivo by diminishing oligomers. Neurobiol Aging. 2010;31:1055–1058. doi: 10.1016/j.neurobiolaging.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rambold AS, Miesbauer M, Rapaport D, Bartke T, Baier M, Winklhofer KF, Tatzelt J. Association of Bcl-2 with misfolded prion protein is linked to the toxic potential of cytosolic PrP. Mol Biol Cell. 2006;17:3356–3368. doi: 10.1091/mbc.E06-01-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tetzlaff JE, Putcha P, Outeiro TF, Ivanov A, Berezovska O, Hyman BT, McLean PJ. CHIP targets toxic alpha-Synuclein oligomers for degradation. J Biol Chem. 2008;283:17962–17968. doi: 10.1074/jbc.M802283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar P, Ambasta RK, Veereshwarayya V, Rosen KM, Kosik KS, Band H, Mestril R, Patterson C, Querfurth HW. CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence Abeta metabolism. Hum Mol Genet. 2007;16:848–864. doi: 10.1093/hmg/ddm030. [DOI] [PubMed] [Google Scholar]
- 46.Sroka K, Voigt A, Deeg S, Reed JC, Schulz JB, Bahr M, Kermer P. BAG1 modulates huntingtin toxicity, aggregation, degradation, and subcellular distribution. J Neurochem. 2009;111:801–807. doi: 10.1111/j.1471-4159.2009.06363.x. [DOI] [PubMed] [Google Scholar]
- 47.Kirstein-Miles J, Morimoto RI. Caenorhabditis elegans as a model system to study intercompartmental proteostasis: Interrelation of mitochondrial function, longevity, and neurodegenerative diseases. Dev Dyn. 2010;239:1529–1538. doi: 10.1002/dvdy.22292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mu TW, Ong DST, Segatori L, Wang YJ, Balch WE, Yates JR, IIIrd, Kelly JW. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chauhan D, Hideshima T, Anderson KC. Targeting proteasomes as therapy in multiple myeloma. Adv Exp Med Biol. 2008;615:251–260. doi: 10.1007/978-1-4020-6554-5_12. [DOI] [PubMed] [Google Scholar]
- *51.Connelly S, Choi S, Johnson SM, Kelly JW, Wilson IA. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr Opin Struct Biol. 2010;20:54–62. doi: 10.1016/j.sbi.2009.12.009. A contemporary review that describes the role of protein folding stabilizers and approaches necessary to alleviate human systemic amyloid disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *52.Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 6:25–33. doi: 10.1038/nchembio.275. A paper that describes how the epigenetic environment modulated by histone deacetylases (HDAC) may play a critical role in managing the fold of inherited disease variants to mitigate that onset and impact of disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rai M, Soragni E, Jenssen K, Burnett R, Herman D, Coppola G, Geschwind DH, Gottesfeld JM, Pandolfo M. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One. 2008;3:e1958. doi: 10.1371/journal.pone.0001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci U S A. 2008;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]