

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder affecting predominantly motor neurons. Recent studies suggest that the disease progression of ALS is non-cell-autonomous, although the interaction between neurons and glial cells in different disease stages is not entirely clear. Here, we demonstrate that the interferon (IFN) signaling pathway is activated in human SOD1(G93A) transgenic mice, a rodent model of ALS. IFN-stimulated genes (ISGs) increased in the spinal cord of SOD1(G93A) mice at a pre-symptomatic age. In addition, the up-regulated ISGs, and most likely their transcriptional activators, were found specifically in astrocytes surrounding motor neurons, suggesting that IFN signaling in astrocytes was triggered by specific pathologic changes in motor neurons. Furthermore, induction of ISGs in cultured astrocytes was highly sensitive to IFN, especially type I IFN. ISGs in astrocytes were activated specifically by endoplasmic reticulum stress-induced neurodegeneration in vitro, implicating a similar process in the pre-symptomatic stage of SOD1 mutant mice. Finally, reduction or deletion of IFNα receptor 1 inhibited IFN signaling and increased the life-span of SOD1(G93A) mice. Thus, the activation of IFN signaling pathways represents an early “dialogue” between motor neurons and astrocytes in response to pathological changes in ALS.
Keywords: motor neuron, neurodegenerative disease, gene expression, neuron-glia interaction
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder affecting both upper and lower motor neurons, resulting in a gradual loss of motor neuron function and muscle atrophy, which ultimately leads to paralysis and death (Boillee et al. 2006a; Bruijn et al. 2004). Although 90% of all ALS cases are sporadic, there are also genetic causes, such as mutations in superoxide dismutase 1 (SOD1) (Pasinelli and Brown 2006; Rosen et al. 1993). Transgenic mouse models overexpressing mutant human SOD1 manifest symptoms of ALS (Bendotti and Carri 2004; Bruijn et al. 1997; Gurney et al. 1994). The relatively consistent disease onset and survival period of the transgenic SOD1 mouse lines makes them particularly suitable for studying temporal events linked to different disease stages.
The search for the mechanism of motor neuron degeneration in ALS originally focused on motor neurons, but non-neuronal cells and non-cell-autonomous pathways contribute to its pathogenesis. Normal non-neuronal cells can protect motor neurons that express the mutated SOD1 gene (Clement et al. 2003). Microglia and astrocytes carrying a SOD1 mutant had no effect on disease onset, but played an important role in disease progression (Boillee et al. 2006b; Yamanaka et al. 2008). Immune and inflammatory components also contribute to the pathogenesis of ALS before motor symptoms become apparent. Increased immunostaining for reactive astrocytes and microglial/macrophage markers are observed in areas of motor neuron degeneration in all forms of human ALS (Ince et al. 1996; Kawamata et al. 1992) and in symptomatic SOD1 mice (Almer et al. 1999; Bruijn et al. 1997; Hall et al. 1998, Alexianu, 2001). Pro-inflammatory cytokines such as interleukin-6 (IL6) and interleukin-1β (IL1β) are elevated in the cerebrospinal fluid (Sekizawa et al. 1998) and spinal cord (Li et al. 2000) of ALS patients. Increased levels of the macrophage-derived cytokines, IL1β and tumor necrosis factor-α (TNFα), as well as several other pro-inflammatory factors, occur in the spinal cord of SOD1 mice, even before motor neuron loss (Elliott 2001; Hensley et al. 2002; Yoshihara et al. 2002). Microglia are the resident immune cells of the CNS and the source and target of cytokines (Hanisch 2002; Streit 2002). The activation of microglia during pathogenesis will in turn activate astrocytes to release more cytokines, thus creating a vicious circle (Dong and Benveniste 2001). However, the molecular events that trigger immune/inflammatory responses in ALS and whether motor neurons participate in this process are not well understood.
Here, we found that interferon-stimulated genes (ISGs) were up-regulated in the spinal cord of SOD1(G93A) mice at a pre-symptomatic age, about 30 days before disease onset. Importantly, this up-regulation was observed specifically in astrocytes surrounding motor neurons. Furthermore, expression of ISGs in cultured astrocytes was highly inducible by type I interferon (IFN). Finally, reduction or deletion of IFNα receptor 1 (IFNAR1) inhibited IFN signaling and increased the life span of SOD1(G93A) mice. All these results suggest that the activation of IFN signaling pathway in SOD1(G93A) spinal cord may represent an early “dialogue” between motor neurons and astrocytes in response to pathological changes in ALS.
MATERIALS AND METHODS
Mice
SOD1(G93A) transgenic mice, which express the human SOD1 gene containing a G93A mutation [B6SJL-TgN(SOD1-G93A)1Gur/J line and B6.Cg-Tg(SOD1-G93A)1Gur/J line] were obtained from Jackson Laboratory. B6SJL-TgN(SOD1-G93A)1Gur/J mice were maintained as hemizygotes by breeding transgenic males with wild-type B6SJL females. The littermates were used in most experiments. B6.Cg-Tg(SOD1-G93A)1Gur/J were also maintained as hemizygotes by breeding transgenic males with wild-type C57BL/6J females. B6.Cg-Tg(SOD1-G93A)1Gur/J and C57BL/6J-IFNAR1−/− (Muller et al. 1994) mice served as founders for the cross-breeding experiment. The mice were housed in a virus-free barrier facility under a 12-h light/dark cycle, with ad libitum access to food and water. All procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the Society for Neuroscience “Guidelines for the Use of Animals in Neuroscience Research,” using protocols approved by Sanford-Burnham Medical Research Institute Animal Research Committee.
Cross-breeding of mice
Cross-breeding of B6.Cg-Tg(SOD1-G93A)1Gur/J males and C57BL/6J-IFNAR1−/− females resulted in SOD1(G93A)-IFNAR1+/− mice and IFNAR1+/− mice. We then used mice derived from breeding SOD1(G93A)-IFNAR1+/− males with IFNAR1+/− females to assess the effect of both IFNAR1 loss (IFNAR1−/−) and IFNAR1 reduction (IFNAR1+/−) on the survival of SOD1(G93A) mice. Survival time was measured by recording the date that each animal reached an endpoint where they were unable to right themselves for 10 s after being placed on their sides (Wang and Zhang 2005). At this stage, mice were killed.
Human tissue samples
Human lumbar spinal cord segments from five patients that died of respiratory failure caused by ALS were provided by the Human Brain and Spinal Fluid Resource Center, VA West Los Angeles Healthcare Center, Los Angeles, CA, which is sponsored by NINDS/NIMH, National Multiple Sclerosis Society, and the Department of Veterans Affairs. Control samples were obtained from four individuals without evidence of ALS disease. The age, gender and post-mortem interval of the subjects are described in Table 1.
Table 1.
Patient information
| Control patients | |||
|---|---|---|---|
| Case | Sex | Age (years) | PMI (h) |
| 1 | F | 82 | 19.0 |
| 2 | M | 67 | 9.3 |
| 3 | F | 83 | 7.0 |
| 4 | F | 77 | 14.5 |
| ALS patients | |||
|---|---|---|---|
| Case | Sex | Age (years) | PMI (h) |
| 1 | F | 52 | 13.9 |
| 2 | M | 68 | 16.0 |
| 3 | M | 67 | 9.5 |
| 4 | F | 64 | 18.3 |
| 5 | M | 58 | 6.0 |
Cell Culture
Primary spinal motor neuron cultures were derived from E12.5 C57 BL/6 mouse embryos. Briefly, isolated spinal cords were digested in 0.25% papain in Hank’s balanced salt solution(HBSS) for 15 min at 37 °C. Cells were dissociated by triturating and were plated at 5 × 105 cells/cm2 on glass coverslips coated with poly-L-ornithine and laminin. Cultures were maintained in medium consisting of Neurobasal media, 2% B-27 supplement, 2% horse serum, 0.5 mM L-glutamine, 1% penicillin/streptomycin, 1 ng/ml BDNF, 100 pg/ml GDNF, and 10 ng/ml CNTF, at 37 °C and 5% CO2. All materials for cell culture were from Invitrogen except that growth factors were from Millipore. After 7 days in vitro (DIV), cultures were used for experiments. These cultures had about 31% neurons, 68% astrocytes, and 1% microglia. Neurotoxic reagents, including 100 μM glutamate, 30 nM rotenone (Sigma), and 0.2μM tunicamycin (MP Biomedicals), were added to the culture medium. Neuronal viability was assessed after neurotoxin exposure using immunostaining with an anti-SMI32 antibody and counting all surviving SMI32-positive motor neurons. Isg15 protein was examined using Western blotting.
Primary spinal cord astrocyte cultures were obtained from P1 C57/BL6 mouse pups. Spinal cord tissue was incubated for 20 min at 37 °C in HBSS plus 0.5mM EDTA and 0.5% trypsin. Cells were dissociated by trituration, plated on poly-L-lysine-coated 75 cm2 flasks, and maintained at 37 °C and 5% CO2 in DMEM with 2 mM glutamine, 10% fetal bovine serum and 100 U/ml of penicillin/streptomycin. Ten days later, cultures were shaken in an incubator at 37 °C, 250 rpm for 24 h. After rinsing with culture medium, the undetached cells were passed from the flasks into Petri dishes. The secondary cultures were grown for ≥ 8 DIV before being treated with IFNβ, IFNγ, Lipopolysaccharide (LPS, Sigma), IL1β (Sigma), TNFα (Sigma), rabbit anti-IFNα antibody, rabbit anti-IFNβ antibody, or rabbit anti-IFNγ antibody. IFNs and anti-IFN antibodies were from PBL InterferonSource.
Gene Expression Array Analysis
Total RNA was extracted individually from the lumbar spinal cord of three P60 female SOD1(G93A) mice and three wild-type female littermates using RNeasy (Qiagen). A DNA removal step was included in all RNA extraction experiments. RNA quality was examined using gel electrophoresis and optical density measurement. Total RNA was then used as a template for double-stranded cDNA synthesis (MessegeAMP II kit, Ambion), which was used as a template for biotin-labeled cRNA synthesis (High Yield RNA Transcription Labeling kit; Affymetrix). Biotin-labeled cRNA samples were submitted to the DNA Microarray Core facility at UCLA, for hybridization and scanning with a standardized protocol. Mouse Genome 430 2.0 Array chips were obtained from Affymetrix. Each chip was hybridized with the cRNA sample from one mouse. Microarray Suite 5.0 was used to generate CEL files using the default settings. dChip software (Li and Hung Wong 2001) was used for calculation of expression levels because it operates consistently in comparison to other analysis software/algorithms (Zakharkin et al. 2005). Model-based expression values were calculated by the perfect match-only method. Probe sets with at least two Present calls and an average model-based expression value of at least 30 in either SOD1(G93A) group or wild-type control group were first filtered, and then differentially regulated genes were selected by comparison analysis using a lower 90% confidence bound of a 1.5-fold change (unpaired two-tailed t - test, p < 0.05). Gene-annotation analysis was performed with the Database for Annotation, Visualization, and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/) (Dennis et al. 2003; Huang da et al. 2009). Gene ontology classes were also analyzed by DAVID. The ratio of regulated genes in SOD1 spinal cord belonging to a given gene ontology class (list hits) and the total of annotated genes present in this list (list total) were compared with the ratio of expressed, annotated genes belonging to the same ontology (population hits) and the total of expressed, annotated genes present on the array (population total). Statistical significance was analyzed using the Bonferroni multiparameter post test.
Quantitative PCR (QPCR) Analysis
Total RNA was extracted individually from the whole tissue or ventral horn of lumbar spinal cords of female SOD1(G93A) mice and their female wild-type littermates of varying ages by using RNeasy. Each group or condition had three animals. RNA quality was examined using gel electrophoresis and optical density measurement. Total RNA was used as templates for cDNA synthesis by reverse transcription (Superscript II, Invitrogen) (Xing et al. 2006). QPCR was performed with the Stratagene Mx3000p QPCR system using SYBR Green PCR Master Mix (Applied Biosystems). Each sample was analyzed in triplicate. At the end of the PCR, baseline and threshold values were set in the software and the cycle threshold values were exported to Microsoft Excel for analysis. Gene expression levels were normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to correct for RNA input variability. The primers designed using the Primer3 software (http://frodo.wi.mit.edu/primer3/) were GBP2 5′-tgtagaccaaaagttccagacaga-3′ (forward), 5′-gataaaggcatctcgcttgg-3′ (reverse); IFI27L2A, 5′-tagccacactccaatcagca-3′ (forward), 5′-agagcaaggctccaacagc-3′ (reverse); IFI44, 5′-ctgattacaaaagaagacatgacagac-3′ (forward), 5′-aggcaaaaccaaagactcca-3′ (reverse); IFIT1, 5′-ggacaaggtggagaaggtgt-3′ (forward), 5′-tcctcacagtccatctcagc-3′ (reverse); IFIT3, 5′-ggaatgcccagaacttgaat-3′ (forward), 5′-gctcgttcatttcttccaca-3′ (reverse); ISG15, 5′-gaacaagtccacgaagaccag-3′ (forward), 5′-gcagctccttgtcctccat-3′ (reverse); USP18, 5′-ggcagtgcttaggtgacaga-3′ (forward), 5′-gagagatcccatgaaccgat-3′ (reverse); and GAPDH, 5′-ggcattgctctcaatgacaa-3′ (forward), 5′-tgtgagggagatgctcagtg-3′ (reverse).
Western Blot Analysis
The lumbar spinal cord of SOD1(G93A) mice and their wild-type littermates, spinal cord lumbar segments of ALS patients and controls, or the pellets of primary cultures, were homogenized in SDS-sample buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), and 1% protease inhibitor cocktail (Sigma). Protein samples (25 μg) were fractionated by 4% – 12% SDS-polyacrylamide gels and transferred to Hybond ECL nitrocellulose membranes (Amersham Pharmacia Biotech). The blots were probed with primary antibodies: rabbit anti-Isg15 (1:3000, from Dr. D. E. Zhang), rabbit anti-Usp18 (1:500, from Dr. D. E. Zhang), mouse anti-β-actin (1:5000, Pierce), mouse anti-GFAP (1:5000, Sigma), rabbit anti-phospho-STAT1 (Tyr701) (1:1000, Cell Signaling), or rabbit anti-phospho-STAT2 (Tyr689) (1:1000, Upstate). The blots were subsequently probed with either anti-rabbit or anti-mouse IgG peroxidase conjugate (Amersham), and the signals were detected using ECL reagents (Amersham).
Immunohistochemistry
SOD1(G93A) mice and their wild-type littermates at P45 – P120 were anesthetized with isoflurane and perfused transcardially with chilled PBS followed by phosphate-buffered 4% paraformaldehyde (PFA) solution. The spinal cord was cryoprotected in 25% sucrose. Human spinal cord lumbar segments were fixed by 4% PFA solution overnight and also cryoprotected in 25% sucrose. Transverse cryostat sections (12 μm) were processed for immunohistochemistry as described previously (Wang and Zhang 2005). In brief, sections were incubated with primary anti-Isg15 (1:1000), anti-phospho-STAT1 (Tyr701) (1:1000), or anti-phopho-STAT2 (Tyr689) (1:1000) antibodies, followed by incubation with a biotinylated secondary antibody (Vector Laboratories). The detection of immunosignals was performed using a Vectastain Elite ABC kit (Vector Laboratories) and visualized with a DAB substrate solution (Roche Applied Science). For immunofluorescence, spinal cord sections were incubated with rabbit anti-Isg15 (1:1000), mouse anti-GFAP (1:1000), mouse anti-NeuN (1:1000, Millipore), or rat anti-F4/80 (1:100, Pharmingen), followed by incubation with secondary antibodies conjugated with 555Alexafluor, 488Alexafluor, 555Alexafluor (Invitrogen), FITC (Jackson Immunoresearch), or RRX (Jackson Immunoresearch). Some sections were also incubated with FITC lectin (from tomato) (Vector Laboratories). After washing in PBS, the samples were mounted by adding Vectashield (Vector Laboratories) to the cover slips.
RESULTS
ISGs are Elevated in the Spinal Cord of Pre-symptomatic SOD1 Mice
We used a gene profiling approach to screen early molecular changes in the spinal cord of pre-symptomatic (P60) SOD1(G93A) mice. We found that 62 probe sets were >1.5-fold up-regulated and 14 probe sets were >1.5-fold down-regulated, corresponding to 72 unique genes (S Table 1). The most significant gene ontology class corresponded to the biological process of immune response (list hits = 13, list total = 46, population hits = 496, population total = 13830, Bonferroni = 2.18E-05). 15 up-regulated genes, including GBP2, GBP3, GBP6, IFI27L2A, IFI44, IFIT1, IFIT3, IIGP1, ISG15, MPA2L, OASL2, PLSCR1, RSAD2, SLC15A3, USP18, are ISGs from the IFN-signaling pathway (Bordignon et al. 2008; Der et al. 1998; Indraccolo et al. 2007; Wang et al. 2008a; Zhao et al. 2005). Some of them are preferentially induced by type I IFNs (IFNα and IFNβ), such as IFI27L2A, IFI44, IFIT1, IFIT3, ISG15, OASL2, and USP18, whereas others are preferentially induced by type II IFNγ, such as GBP2 and IIGP1.
We performed QPCR assays for seven ISGs, GBP2, IFI27L2A, IFI44, IFIT1, IFIT3, ISG15, and USP18, to confirm microarray data, and detected significantly higher levels of mRNA for all seven ISGs in SOD1 mice than that from wild-type littermate controls (Fig. 1A), consistent with microarray data. We also examined the expression levels of these ISGs in the spinal cord ventral grey matter, in which ALS-vulnerable motor neurons reside. All seven ISGs showed significantly higher mRNA levels in the ventral grey matter than in the whole spinal cord at a pre-symptomatic age (P60) and at an age (P90) around disease onset (Fig. 1B), demonstrating that the expression pattern of these genes is region-specific.
Fig. 1.

Up-regulation of ISGs in the SOD1(G93A) spinal cord. The ratio of SOD1/WT indicates the expression level of each gene. Data shown represent mean values. Both microarray and QPCR analysis showed that the seven ISGs had higher mRNA levels in P60 SOD1 mice (n = 3) than in wild-type littermates (n = 3) (A). mRNA levels were higher in the ventral grey matter than in the whole spinal cord of P60 SOD1 mice (n = 3) and P90 SOD1 mice (n = 3) (B).
ISG15 protein levels were higher in lumbar spinal lysates from SOD1 mice showing signs of paresis than wild-type lysates (Fig. 2A). ISG15 is an ubiquitin-like modifier and is able to covalently modify target proteins to form ISG15 conjugates (Kim and Zhang 2003). In SOD1 samples the levels of ISG15 conjugates were also dramatically increased (Fig. 2A). ISG15 immunoreactivity was very weak in the spinal cords of all wild-type mice and young SOD1 mice (Fig. 2B). However, from a pre-symptomatic age (P60) to an age following disease onset (P100), strong ISG15 signals appeared consistently in the ventral horns of SOD1 spinal cords, which contain motor neurons. ISG15 induction extended to the central canal region of the SOD1 spinal cord at later stages (Fig. 2C). ISG15 was only co-localized with GFAP, an astrocyte marker, and not NeuN, a neuronal marker, F4/80, a microglia marker, or an endothelial cell/microglia marker, lectin (from tomato) (Fig. 3).
Fig. 2.

ISG15 protein expression is higher in SOD1(G93A) spinal cord. ISG15 and its conjugates were increased in spinal cord lysate from P106 SOD1 mice (A). From a pre-symptomatic age (P60) to an age (P100) following disease onset, strong ISG15 immunosignals appeared consistently in the ventral horns of the SOD1 spinal cords (B). The dotted line separates grey matter (G) from white matter (W). ISG15 induction (green) extended to the central canal region (cc) of the SOD1 spinal cord at late stage of the disease (P120) (C). Bar: 50 μm.
Fig. 3.

Specific localization of ISG15 in SOD1(G93A) spinal cord astrocytes. ISG15 was co-localized with GFAP (green), an astrocyte marker (A). Arrows indicates double-labeled cells. ISG15 was not co-localized with a neuronal marker, NeuN (red) (B), a microglia marker, F4/80 (red) (C), or an endothelial cell/microglia marker, lectin (green) (D). Bar: 50 μm.
ISG Protein Expression is also Elevated in Human ALS Spinal Cord
We next investigated whether the induction of ISGs occurred in the spinal cord of ALS patients. ALS patients showed higher levels of ISG15 in Western blots of the lumbar spinal cord than controls without evidence of motor neuron disease (Fig. 4A). Motor neurons in control lumbar spinal cord sections were large and had good morphology, but were shrunken in ALS lumbar spinal cord sections (Fig. 4B). ISG15 immunoreactivity was very weak in control lumbar spinal cord, but the abundant reactive astrocytes showed strong ISG15 immunosignals in the ventral horn of ALS spinal cord (Fig. 4B), consistent with our findings in SOD1 mouse spinal cord.
Fig. 4.

Elevation of ISG15 protein in the spinal cord of ALS patients. ISG15 protein levels in lysate samples were increased in ALS patients compared to controls (top panel) (A). Bottom panel shows densitometry quantitation of ISG15 in all ALS and control human spinal cord tissue samples normalized to β-actin. Data are presented as mean ± SEM, * P < 0.05 versus control by unpaired two tailed t-test. Control spinal cord had many large motor neurons, whereas ALS spinal cord only had few shrunken motor neurons (top panel) (B). ISG15 staining was very weak in control spinal cord ventral horn but strong in ALS spinal cord ventral horn (bottom panel) (B). Inset shows ISG15 positive reactive astrocyte in ALS sample. Bar: 20 μm.
Phosphorylation of STAT1 and STAT2 is Increased in the Spinal Cord of Pre-symptomatic SOD1 Mice
IFN signaling leads to the phosphorylation of STAT1 and STAT2 transcription factors, which are then translocated to the nucleus and induce ISG transcription. We therefore tested for pathways involved in ISG induction in SOD1 spinal cord. Spinal cord sections of P50 and P80 SOD1 mice showed strong nuclear-like labeling of both phospho-STAT1 and phospho-STAT2 in the ventral horns of all SOD1 sections, with stronger immunoreactive signals in P80 than P50 SOD1 sections (Fig. 5, S Table 2). In contrast, no such labeling was observed in wild-type sections. The numbers of phospho-STAT1-positive and phospho-STAT2-positive nuclei in the spinal cord ventral horn of SOD1 mice were similar to the number of ISG15-positive astrocytes observed around these ages. The small size of the nuclear-like labeling (~3 μm) was also consistent with the size of astrocyte nuclei. Together with the microarray data, these results suggest that IFN signaling is active in SOD1 spinal cord during presymptomatic stages.
Fig. 5.

Detection of phospho-STAT1 and phospho-STAT2 in the SOD1(G93A) spinal cord. Strong nuclear-like labeling (small arrows) of both phospho-STAT1 (A) and phospho-STAT2 (B) appeared in the ventral horns of all SOD1 sections. Immunoreactive signals were much stronger in P80 than P50 SOD1 sections. Big arrow heads indicate motor neurons. Bar: 25 μm.
Type I IFN Signaling is Highly Inducible in Spinal Cord Astrocyte Cultures
To confirm that ISG induction in astrocytes is mediated by IFN signaling, we treated primary astrocyte cultures of the spinal cord with IFN. IFNβ treatment induced ISG15 in cultured astrocytes (Fig. 6A) in a time- and concentration-dependent manner (Fig. 6B,C), with even 1 unit/ml of IFNβ sufficient to increase ISG15. Both ISG15 and USP18 were increased from two hours after IFNβ treatment, whereas phospho-STAT1 and phospho-STAT2 activation peaked within a half hour after IFNβ treatment (Fig. 6C), remained high six hours later, and had disappeared by 14 h after treatment. Thus, the activation of the type I IFN pathway in astrocytes is a highly sensitive and dynamic process.
Fig. 6.

Induction of ISGs by IFNβ in primary spinal cord astrocyte cultures. Strong ISG15 immunosignals appeared in astrocytes after 14 hr IFNβ-treatment (A). Bar: 30μm. IFNβ dose-dependently induced ISG15 in astrocytes after 14 h treatment (B) at doses as low as 1 unit/ml, and showed time-dependent changes in induction (C). The phosphorylation of STAT1 and STAT2 occurred before the induction of ISG15 and USP18.
We also tested the effect of other pro-inflammatory cytokines and an inflammation-inducing reagent, LPS, on induction of ISG15 in cultured spinal cord astrocytes. IFNγ (50 unit/ml) and LPS (1 μg/ml) increased the expression of ISG15, whereas IL1β (10 ng/ml) and TNFα (20 ng/ml) had only minimal effects (Fig. 7A). Furthermore, LPS-induced ISG15 expression was almost completely blocked by an anti-IFNβ antibody but not by an anti-IFNγ antibody or an anti-IFNα antibody (Fig. 7B). These results suggest that IFNβ is the major inflammatory trigger of ISG expression.
Fig. 7.

Effect of pro-inflammatory cytokines and LPS on ISG15 expression in primary spinal cord astrocyte cultures. IFNγ (50 unit/ml) and LPS (1 μg/ml), but not IL1β (10 ng/ml) or TFNα (20 ng/ml), increased expression of ISG15 in astrocytes cultures (A). LPS-induced ISG15 expression was blocked by an anti-IFNβ antibody but not by an anti-IFNγ antibody or an anti-IFNα antibody (B).
ER Stress-induced Neurodegeneration Elevates Isg15 in Primary Spinal Motor Neuron Cultures
To investigate the underlying mechanisms that triggered expression of ISG in ALS mutant mice, we investigated the effect of different neurotoxins on induction of ISG15 in primary spinal motor neuron cultures. Treatment with the potent ER stress inducer, tunicamycin (0.2 μM), for 48 h dramatically decreased the number of motor neurons, with a survival rate of 24.0% ± 1.9% (Student’s t test, P < 0.01). The remaining motor neurons in the cultures had much shorter neurites than those in untreated control cultures (Fig. 8A). Tunicamycin increased ISG15 protein levels, which could be partially blocked by co-applying an anti-IFNβ antibody with tunicamycin (Fig. 8B). Baseline ISG15 levels in mixed neuron-glial cultures were more variable and higher than in pure astrocyte cultures, potentially due to baseline neuronal death. We also found a higher number of ISG15-immunopositive astrocytes in tunicamycin-treated primary spinal motor neuron cultures than in untreated controls (Fig. 8A). In contrast, exposure of glutamate (100μM, overnight), an excitotoxin, or rotenone (30 nM, 24 h), a mitochondrial damaging agent, to the same culture did not increase ISG15 expression, although both almost completely killed motor neurons (data not shown). Furthermore, direct treatment of purified astrocyte cultures with the same dose of tunicamycin, glutamate, or rotenone did not induce ISG15 expression (S Fig. 1). Thus, the induction of ISGs in astrocytes originates from neuronal injury and is insult type-dependent.
Fig. 8.

Elevation of ISG15 in primary spinal motor neuron cultures after ER stress-induced neurodegeneration. Tunicamycin (0.2 μM) exposure for 48 h dramatically decreased the number of motor neurons (top panel) and induced the expression of ISG15 in astrocytes (bottom panel) (A). Arrows indicate ISG15 immunopositive astrocytes. Bar: 50 μm. Tunicamycin exposure significantly elevated ISG15 levels in the whole cell lysates, which could be partially blocked by adding anti-IFNβ antibody along with tunicamycin (B). In the top panel, each lane contains lysate from an individual culture. Bottom panel shows densitometry quantitation of ISG15 normalized to β-actin. Data are presented as mean ± SEM.
Reduction or Deletion of IFNAR1 Prolongs the Life Span of SOD1(G93A) Mice
To assess the contribution of the IFN signaling pathway in ALS pathogenesis, we generated SOD1(G93A) mice in the context of an IFNAR1 gene knock-out in the same strain background (C57/BL6) (Muller et al. 1994). IFNAR1-deficient mice were viable and fertile, and showed no apparent phenotypic abnormalities. The type I IFN signaling pathway was severely affected in IFNAR1−/− mice, since expression of ISG15 was completely abolished in SOD1(G93A)-IFNAR1−/− spinal cord (S Fig. 2). SOD1(G93A)-IFNAR1+/+, SOD1(G93A)-IFNAR1+/−, and SOD1(G93A)-IFNAR1−/− mice had life-spans of 158.9 ± 6.8 d (n = 16), 174.1 ± 8.9 d (n = 16), and 167.1 ± 8.0 d (n = 22), respectively (Fig. 9), indicating that both reducing and deleting IFNAR1 significantly (Student’s t test, P < 0.01) prolonged survival in SOD1(G93A) mice.
Fig. 9.
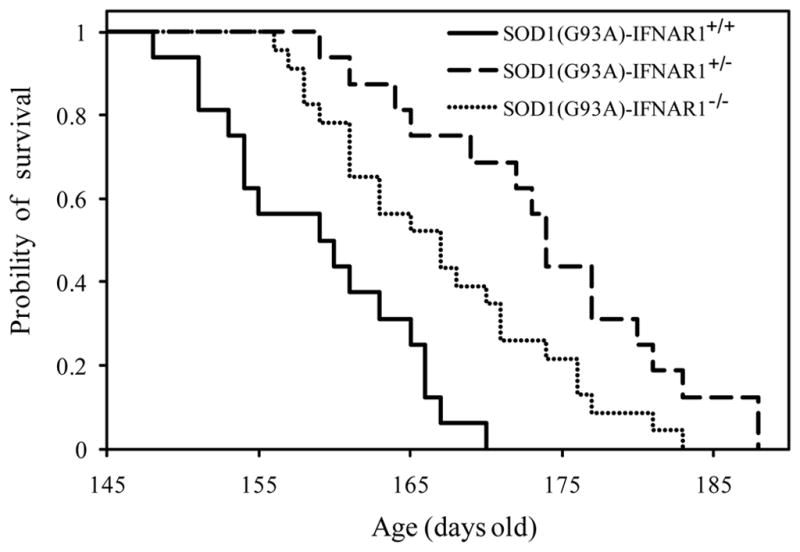
Effect of IFNAR1 on the survival of SOD1(G93A) mice. Cumulative probabilities of survival time are shown for SOD1(G93A)-IFNAR1+/+ (solid line, n = 16), SOD1(G93A)-IFNAR1+/− (dash line, n = 16), and SOD1(G93A)-IFNAR1−/− mice (dotted line, n = 22). Both absence and reduction of IFNAR1 prolonged the life span of SOD1(G93A) mice. Data were analyzed using the Kaplan–Meier method.
DISCUSSION
Gene expression studies with microarrays provide an experimental approach to the molecular events that occur in ALS. Two early studies examined the entire spinal cord of the transgenic SOD1(G93A) mouse model (Gurney et al. 1994) and both found changes in gene expression only after symptom onset (Olsen et al. 2001; Yoshihara et al. 2002). Not surprisingly, our microarray samples from before disease onset did not completely overlap with these findings. More recently, a study using laser-capture microdissected spinal motor neurons from active and inactive SOD1 mutants (G93A, G37R, and G85R) showed dysregulation of the d/l-serine biosynthetic pathway and induction of neuronal components of the classic complement system (Lobsiger et al. 2007). However, another study using a similar approach found a different list of genes regulated during the disease stage of transgenic SOD1(G93A) mice (Ferraiuolo et al. 2007). In fact, no studies agree on the major regulated genes, suggesting that disease-induced changes in the transcriptome are complex and subtle. Certain genes identified here, particularly ATF3, C1QA, C1QB, GRN, IFIT3, LAPTM5, and LY86, were also up-regulated in other studies (Lobsiger et al. 2007). However, we are the first to show ISG up-regulation in an ALS mouse model, a novel finding for neurodegenerative models.
We found that a number of ISGs, including GBP2, IFI27L2A, IFI44, IFIT1, IFIT3, ISG15, and USP18, are up-regulated within the grey matter of the lumbar spinal cord of SOD1(G93A) mice at an early pre-symptomatic stage (P60). Most of these ISGs are preferentially induced by type I IFN. Type I and type II IFNs initiate two similar but distinct signaling pathways (Borden et al. 2007). Type I IFNs bind to their heterodimeric receptor and trigger phosphorylation of JAK1 and TYK2 tyrosine kinases, which in turn phosphorylate STAT1 and STAT2 transcription factors. Phospho-STAT1 and phospho-STAT2 move into the nucleus and form a complex with ISGF3G, which activates the transcription of many ISGs. In contrast, type II IFNγ acts on a tetrameric receptor to activate JAK1 and JAK2 kinases, which subsequently phosphorylate STAT1. The phospho-STAT1 forms a homodimer, translocates to the nucleus, and induces the expression of IFNγ-stimulated genes. We detected both phospho-STAT1 and phospho-STAT2 in the spinal cord of pre-symptomatic transgenic SOD1 mice and IFNβ-treated spinal astrocyte cultures, providing further evidence IFN signaling, especially the type I IFN pathway, in the spinal cord of SOD1 mice. Activation of IFN signaling in the brain is usually associated with inflammation triggered by viral infection or autoimmunization (Dafny and Yang 2005). However, SOD1 mice show little sign of inflammation at the early pre-symptomatic stages, making sensitive and robust activation of IFN signaling pathways at this stage a surprising but intriguing finding. We found increased mRNA levels of IFNβ, but not IFNα, in the spinal cord of SOD1 mice in the early disease stage (S Fig. 3). However, the type I IFNs could not be detected directly in lysates from the spinal cord of SOD1 mice by a sensitive Enzyme-Linked Immunosorbent Assay kit (PBL Interferon Source) (data not shown), suggesting the expression of type I IFNs under disease conditions is restricted in both concentration and distribution. It is perhaps not unusual for signaling molecules like IFNs to show such low expression when associated with degenerative or chronic diseases, otherwise the expression of downstream molecules would not be spatially restricted. Developing techniques for detecting low levels of IFNs in vivo will be a challenging task for the future.
The earliest pathological changes seen in SOD1(G93A) mice are mitochondrial swelling, first detectable around postnatal day 30 (P30) by electron microscopy and then around day P50–60 by light microscopy (Bendotti et al. 2001; Chiu et al. 1995; Dal Canto and Gurney 1994; Kong and Xu 1998). Another detectable early change is degeneration of motor neuron axons, with the largest axons innervating fast muscle fibers the earliest to degenerate at around P50 (Frey et al. 2000; Pun et al. 2006). Degeneration of motor axons in ventral roots can be seen from P80 (Fischer et al. 2004). Consistent with axonal degeneration, abnormal muscle activities are also detected by electromyograms (EMG) as early as P60 (Kennel et al. 1996; Miana-Mena et al. 2005). Besides these morphological and physiological changes, expression of several molecules also increases in the pre-symptomatic stage (P60), including p38 MAP kinase, neuronal nitric oxide synthase, AKT MAP kinase, and caspase-3, as well as cytokines like TNFα, transforming growth factor β, and macrophage colony-stimulating factor (Elliott 2001; Wengenack et al. 2004). On the other hand, activation of astrocytes and microglia occurs at disease onset around P90 and becomes more prevalent at later stages (Hall et al. 1998; Wengenack et al. 2004). Interestingly, the expression of ISGs in the spinal cord occurs long before motor neuron degeneration and any clinical sign of the disease in SOD1 mice. For example, ISGs and their transcriptional activators in SOD1 spinal cord ventral horn were higher from P50 to P60, which paralleled the first sign of motor axon terminal damage and is much earlier than gliosis. Furthermore, increased ISG protein was detected in the spinal cord of ALS patients, despite the fact that most control samples were from an aging population and might have higher background ISG levels. Given their robust expression, ISGs may be reliable biomarkers for detecting early pathological changes in the ALS spinal cord.
We also showed that ISGs are transcribed and expressed at much higher levels in the pre-symptomatic ventral horn, where the motor neurons reside, than the whole spinal cord. For example, ISG15 protein was robustly induced only in the ventral horn of SOD1 spinal cord at P60. This temporal and spatial expression pattern of ISGs in SOD1 mice suggests that the activation of IFN signaling is triggered by pathological changes in motor neurons. Because we could not directly detect the source of type I IFNs in the spinal cord of SOD1 mice due to technical limitations, we can only speculate that early pathological changes in motor neurons induce the release of either interferon directly or other signaling molecules that can trigger interferon release from glial cells such as microglia and astrocytes. Interestingly, the expression of ISG15 in primary motor neuron cultures was specifically triggered by ER stress-induced neuronal injury, implicating a similar process in the pre-symptomatic stage of SOD1 mice. ER stress is one early pathogenic mechanism of motor neuron death in SOD1 mice (Kanekura et al. 2009; Saxena et al. 2009), which fits well with the fact that ALS etiology is tightly linked to an increased propensity for protein aggregation and decreased protein stability seen in over one hundred single SOD1 mutations (Wang et al. 2008b). Many ER stress-related molecules are up-regulated in SOD1 mice with a similar temporal pattern as ISGs (Atkin et al. 2006; Kikuchi et al. 2006; Nagata et al. 2007; Saxena et al. 2009; Tobisawa et al. 2003). Further elucidation of the molecular events in motor neurons that trigger the activation of IFN signaling pathways will provide important clues to understand the early stages of ALS.
The disease progression of ALS is non-cell-autonomous (Boillee et al. 2006a; Clement et al. 2003) and astrocytes play a crucial role in the degeneration of spinal motor neurons in ALS (Di Giorgio et al. 2007; Julien 2007; Nagai et al. 2007; Yamanaka et al. 2008). Our study demonstrated that astrocytes showed the most robust ISG expression upon stimulation, and ISG15 was selectively co-localized with GFAP, an astrocyte marker, in the ventral horn of SOD1 spinal cord. Phosphorylation of STAT1 and STAT2, two major components of the type I IFN signaling pathway, also occurred prior to the increase of ISG15 in SOD1 mice. Furthermore, no induction of ISG15 was detected in IFNAR1-deficient SOD1(G93A) mice that lack type I IFN signaling. The dependence of ISG15 signaling on type I IFN and the astrocyte-specific expression of ISG15 suggest that the activation of type I IFN signaling occurs specifically in astrocytes. Interestingly, the life span of SOD1(G93A)-IFNAR1+/− was longer than SOD1(G93A)-IFNAR1−/− mice, indicating that reduced IFNAR1 is more effective than its absence to promote the survival of SOD1 mice. Type I IFN-activated ISGF3-dependent signal transduction protects against the ISGF3-independent pathophysiological actions of the type I IFNs in the CNS (Hofer et al. 2010), suggesting a pleotropic role of type I IFNs in neuronal survival (van Boxel-Dezaire et al. 2006). Our findings further suggest that the ISGF3-dependent and independent actions of type I IFNs in CNS may be “dose-dependent”, each requiring a different optimal level of IFNAR1 expression. We speculate that low levels of type I IFNs in the early stage of ALS may be protective, whereas chronic, higher levels of type I IFNs in the later stages of ALS are harmful. Thus, further work is needed to understand the role of type I IFNs in astrocyte function during ALS pathogenesis.
Astrocytes expressing a mutant SOD1 accelerate disease progression but not disease onset in SOD1 mutant mice (Boillee et al. 2006b; Yamanaka et al. 2008), suggesting that the detrimental effect of astrocytes is induced most likely by pathological changes in motor neurons, although the induction process is not understood. Most of the ISGs identified here have not been studied in detail, but some are components of the immune system: ISG15 (Lenschow et al. 2005), USP18 (Malakhov et al. 2002; Malakhova et al. 2006), IFI27 (Labrada et al. 2002), and GBP2 (Balasubramanian et al. 2006). Although the role of these ISGs in astrocytes is not clear, their up-regulation may result in altered astrocyte function in the spinal cord of SOD1 mice. Thus, our findings open a new avenue for investigating the early “dialogue” between motor neurons and astrocytes in response to pathological changes in ALS. The biological functions of this molecular events in the disease progression of ALS warrant further investigation.
Supplementary Material
Acknowledgments
We thank Dr. Dong-Er Zhang, University of California, San Diego, CA, USA for IFNAR1 null mice and antibodies against mouse ISG15 and USP18. We also thank Dr. Zugen Chen, DNA Microarray Core facility, University of California, Los Angeles, USA for the microarray analysis. This work was supported by NIH grant R21 DE015129.
References
- Almer G, Vukosavic S, Romero N, Przedborski S. Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1999;72(6):2415–25. doi: 10.1046/j.1471-4159.1999.0722415.x. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281(40):30152–65. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S, Nada S, Vestal D. The interferon-induced GTPase, mGBP-2, confers resistance to paclitaxel-induced cytotoxicity without inhibiting multinucleation. Cell Mol Biol (Noisy-le-grand) 2006;52(1):43–9. [PubMed] [Google Scholar]
- Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M, Silani V, De Biasi S. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci. 2001;191(1–2):25–33. doi: 10.1016/s0022-510x(01)00627-x. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Carri MT. Lessons from models of SOD1-linked familial ALS. Trends Mol Med. 2004;10(8):393–400. doi: 10.1016/j.molmed.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006a;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science. 2006b;312(5778):1389–92. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6(12):975–90. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordignon J, Probst CM, Mosimann AL, Pavoni DP, Stella V, Buck GA, Satproedprai N, Fawcett P, Zanata SM, de Noronha L, et al. Expression profile of interferon stimulated genes in central nervous system of mice infected with dengue virus Type-1. Virology. 2008;377(2):319–29. doi: 10.1016/j.virol.2008.04.033. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–38. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–49. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6(4):349–62. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302(5642):113–7. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Dafny N, Yang PB. Interferon and the central nervous system. Eur J Pharmacol. 2005;523(1–3):1–15. doi: 10.1016/j.ejphar.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. American Journal of Pathology. 1994;145(6):1271–9. [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):P3. [PubMed] [Google Scholar]
- Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95(26):15623–8. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10(5):608–14. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36(2):180–90. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Elliott JL. Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Brain Res Mol Brain Res. 2001;95(1–2):172–8. doi: 10.1016/s0169-328x(01)00242-x. [DOI] [PubMed] [Google Scholar]
- Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci. 2007;27(34):9201–19. doi: 10.1523/JNEUROSCI.1470-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185(2):232–40. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20(7):2534–42. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–5. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23(3):249–56. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–55. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Hensley K, Floyd RA, Gordon B, Mou S, Pye QN, Stewart C, West M, Williamson K. Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J Neurochem. 2002;82(2):365–74. doi: 10.1046/j.1471-4159.2002.00968.x. [DOI] [PubMed] [Google Scholar]
- Hofer MJ, Li W, Lim SL, Campbell IL. The type I interferon-alpha mediates a more severe neurological disease in the absence of the canonical signaling molecule interferon regulatory factor 9. J Neurosci. 2010;30(3):1149–57. doi: 10.1523/JNEUROSCI.3711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ince PG, Shaw PJ, Slade JY, Jones C, Hudgson P. Familial amyotrophic lateral sclerosis with a mutation in exon 4 of the Cu/Zn superoxide dismutase gene: pathological and immunocytochemical changes. Acta Neuropathol. 1996;92(4):395–403. doi: 10.1007/s004010050535. [DOI] [PubMed] [Google Scholar]
- Indraccolo S, Pfeffer U, Minuzzo S, Esposito G, Roni V, Mandruzzato S, Ferrari N, Anfosso L, Dell’Eva R, Noonan DM, et al. Identification of genes selectively regulated by IFNs in endothelial cells. J Immunol. 2007;178(2):1122–35. doi: 10.4049/jimmunol.178.2.1122. [DOI] [PubMed] [Google Scholar]
- Julien JP. ALS: astrocytes move in as deadly neighbors. Nat Neurosci. 2007;10(5):535–7. doi: 10.1038/nn0507-535. [DOI] [PubMed] [Google Scholar]
- Kanekura K, Suzuki H, Aiso S, Matsuoka M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol Neurobiol. 2009;39(2):81–9. doi: 10.1007/s12035-009-8054-3. [DOI] [PubMed] [Google Scholar]
- Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140(3):691–707. [PMC free article] [PubMed] [Google Scholar]
- Kennel PF, Finiels F, Revah F, Mallet J. Neuromuscular function impairment is not caused by motor neurone loss in FALS mice: an electromyographic study. Neuroreport. 1996;7(8):1427–31. doi: 10.1097/00001756-199605310-00021. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Almer G, Yamashita S, Guegan C, Nagai M, Xu Z, Sosunov AA, McKhann GM, 2nd, Przedborski S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103(15):6025–30. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KI, Zhang DE. ISG15, not just another ubiquitin-like protein. Biochem Biophys Res Commun. 2003;307(3):431–4. doi: 10.1016/s0006-291x(03)01216-6. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18(9):3241–50. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrada L, Liang XH, Zheng W, Johnston C, Levine B. Age-dependent resistance to lethal alphavirus encephalitis in mice: analysis of gene expression in the central nervous system and identification of a novel interferon-inducible protective gene, mouse ISG12. J Virol. 2002;76(22):11688–703. doi: 10.1128/JVI.76.22.11688-11703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenschow DJ, Giannakopoulos NV, Gunn LJ, Johnston C, O’Guin AK, Schmidt RE, Levine B, Virgin HWt. Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J Virol. 2005;79(22):13974–83. doi: 10.1128/JVI.79.22.13974-13983.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2(8):RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, et al. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288(5464):335–9. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- Lobsiger CS, Boillee S, Cleveland DW. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc Natl Acad Sci U S A. 2007;104(18):7319–26. doi: 10.1073/pnas.0702230104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277(12):9976–81. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, Shuai K, Zhang DE. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. Embo J. 2006;25(11):2358–67. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miana-Mena FJ, Munoz MJ, Yague G, Mendez M, Moreno M, Ciriza J, Zaragoza P, Osta R. Optimal methods to characterize the G93A mouse model of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6(1):55–62. doi: 10.1080/14660820510026162. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615–22. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata T, Ilieva H, Murakami T, Shiote M, Narai H, Ohta Y, Hayashi T, Shoji M, Abe K. Increased ER stress during motor neuron degeneration in a transgenic mouse model of amyotrophic lateral sclerosis. Neurol Res. 2007;29(8):767–71. doi: 10.1179/016164107X229803. [DOI] [PubMed] [Google Scholar]
- Olsen MK, Roberds SL, Ellerbrock BR, Fleck TJ, McKinley DK, Gurney ME. Disease mechanisms revealed by transcription profiling in SOD1-G93A transgenic mouse spinal cord. Annals of Neurology. 2001;50(6):730–40. doi: 10.1002/ana.1252. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7(9):710–23. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9(3):408–19. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis [published erratum appears in Nature 1993 Jul 22;364(6435):362] [see comments] Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12(5):627–36. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Sekizawa T, Openshaw H, Ohbo K, Sugamura K, Itoyama Y, Niland JC. Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases. J Neurol Sci. 1998;154(2):194–9. doi: 10.1016/s0022-510x(97)00228-1. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40(2):133–9. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Tobisawa S, Hozumi Y, Arawaka S, Koyama S, Wada M, Nagai M, Aoki M, Itoyama Y, Goto K, Kato T. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem Biophys Res Commun. 2003;303(2):496–503. doi: 10.1016/s0006-291x(03)00353-x. [DOI] [PubMed] [Google Scholar]
- van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25(3):361–72. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Wang J, Campbell IL, Zhang H. Systemic interferon-alpha regulates interferon-stimulated genes in the central nervous system. Mol Psychiatry. 2008a;13(3):293–301. doi: 10.1038/sj.mp.4002013. [DOI] [PubMed] [Google Scholar]
- Wang Q, Johnson JL, Agar NY, Agar JN. Protein aggregation and protein instability govern familial amyotrophic lateral sclerosis patient survival. PLoS Biol. 2008b;6(7):e170. doi: 10.1371/journal.pbio.0060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zhang D. Memantine prolongs survival in an amyotrophic lateral sclerosis mouse model. Eur J Neurosci. 2005;22(9):2376–80. doi: 10.1111/j.1460-9568.2005.04431.x. [DOI] [PubMed] [Google Scholar]
- Wengenack TM, Holasek SS, Montano CM, Gregor D, Curran GL, Poduslo JF. Activation of programmed cell death markers in ventral horn motor neurons during early presymptomatic stages of amyotrophic lateral sclerosis in a transgenic mouse model. Brain Res. 2004;1027(1–2):73–86. doi: 10.1016/j.brainres.2004.08.054. [DOI] [PubMed] [Google Scholar]
- Xing GG, Wang R, Yang B, Zhang D. Postnatal switching of NMDA receptor subunits from NR2B to NR2A in rat facial motor neurons. Eur J Neurosci. 2006;24(11):2987–92. doi: 10.1111/j.1460-9568.2006.05188.x. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara T, Ishigaki S, Yamamoto M, Liang Y, Niwa J, Takeuchi H, Doyu M, Sobue G. Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 2002;80(1):158–67. doi: 10.1046/j.0022-3042.2001.00683.x. [DOI] [PubMed] [Google Scholar]
- Zakharkin SO, Kim K, Mehta T, Chen L, Barnes S, Scheirer KE, Parrish RS, Allison DB, Page GP. Sources of variation in Affymetrix microarray experiments. BMC Bioinformatics. 2005;6:214. doi: 10.1186/1471-2105-6-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao KW, Li D, Zhao Q, Huang Y, Silverman RH, Sims PJ, Chen GQ. Interferon-alpha-induced expression of phospholipid scramblase 1 through STAT1 requires the sequential activation of protein kinase Cdelta and JNK. J Biol Chem. 2005;280(52):42707–14. doi: 10.1074/jbc.M506178200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.