

Abstract
ARID1A is a recently identified tumor suppressor gene that is mutated in approximately 50% of ovarian clear cell and 30% of ovarian endometrioid carcinomas. The mutation is associated with loss of protein expression as assessed by immunohistochemistry. In this study, we evaluated ARID1A immunoreactivity in a wide variety of carcinomas in order to determine the prevalence of ARID1A inactivation in carcinomas; mutational analysis of ARID1A was performed in selected cases. Immunoreactivity was not detected (corresponding to inactivation or mutation of ARID1A) in 36 (3.6%) of 995 tumors. Uterine low-grade endometrioid carcinomas demonstrated a relatively high frequency of loss of ARID1A expression, as 15 (26%) of 58 cases were negative. The other tumor that had a relatively high frequency loss of ARID1A expression was gastric carcinoma (11%). Mutational analysis showed 10 (40%) of 25 uterine endometrioid carcinoma, none of 12 uterine serous carcinomas and none of 56 ovarian serous and mucinous carcinomas harbored somatic ARID1A mutations. All mutations in endometrioid carcinomas were nonsense or insertion/deletion mutations and tumors with ARID1A mutations demonstrated complete loss or clonal loss of ARID1A expression. In conclusion, this study is the first large-scale analysis of a wide variety of carcinomas showing that uterine low-grade endometrioid carcinoma is the predominant tumor type harboring ARID1A mutations and frequent loss of ARID1A expression. These findings suggest that the molecular pathogenesis of low-grade uterine endometrioid carcinoma is similar to that of ovarian low-grade endometrioid and clear cell carcinoma, tumors that have previously been shown to have a high frequency of loss of expression and mutation of ARID1A.
Keywords: ARID1A, BAF250, uterine carcinoma, ovarian carcinoma
Introduction
Acquisition of somatic mutations is a molecular hallmark of neoplasia. Sequence mutations that are acquired during tumor evolution can lead to activation of oncogenes and inactivation of tumor suppressors and DNA repair genes, thereby propelling tumor development and progression (9). Identification and characterization of somatic mutations are not only fundamental in understanding the molecular pathogenesis of cancer but also can provide the rationale for the development of personalized diagnostic tests and therapy. With the use of whole exome sequencing and transcriptome sequencing, two independent studies recently reported ARID1A (also known as BAF250A) mutations in 43–56% of ovarian clear cell carcinomas and 30% of ovarian low-grade endometrioid carcinomas (6, 21) but not in matched controls, confirming the somatic nature of the mutations. Since both of these tumor types are believed to be derived from endometriosis and because one of these studies also found ARID1A mutations in adjacent atypical endometriosis it is conceivable that ARID1A loss is a relatively specific molecular event in the genesis of these tumors. Many ARID1A mutations are insertion/deletion mutations, leading to the generation of premature stop codons by frameshift that result in truncated proteins prone to degradation. It has been previously shown that loss of ARID1A expression, as assessed by immunohistochemistry, correlates closely with ARID1A mutations (11, 21).
ARID1A is located in the chromosome 1p36 region and encodes a large nuclear protein involved in chromatin remodeling. ARID1A interacts with several other proteins including the core protein, BRG or BRM with ATPase activity (4, 20). The ARID1A-BRG/BRM complex belongs to the SWI/SNF chromatin remodeling complex; remodeling activity is facilitated by ATP hydrolysis of BRG or BRM. In contrast, the non-catalytic subunits of the SWI/SNF complex such as ARID1A are responsible for modulating the target specificity and activity of the ATPase. The chromatin remodeling activity of SWI/SNF has been shown to play an integral role in controlling gene expression (19) and is critical in tissue development, cellular differentiation and tumor suppression (3, 4, 15). ARID1A is essential for SWI/SNF complexes to suppress DNA synthesis. Inactivation of ARID1A is thought to enhance cell cycle progression by potentially involving c-myc, thereby contributing to uncontrolled cellular proliferation in cancer cells (5, 13, 14).
Although ARID1A has emerged as a new cancer-associated gene which is frequently mutated in endometriosis-related ovarian neoplasms, it is not known whether its mutation, like FOXL2 (10, 17) and APC (8), is detected only in specific types of cancer or, like in TP53 and KRAS, occurs in a variety of neoplastic diseases. Because ARID1A mutations are randomly distributed in 20 exons and are insertion/deletion type of mutations that lead to truncated proteins, we used loss of ARID1A immunoreactivity as a surrogate marker for a mutation to screen a variety of carcinomas. Sequence analysis was then performed in the specimens that showed the highest frequency of loss of ARID1A expression.
MATERIALS AND METHODS
Tissue Material
Paraffin embedded tissue sections of normal and tumor tissues from various organs were obtained from the Department of Pathology of the National Taiwan University Hospital and Johns Hopkins Hospital, from 1994 to 2009. The normal tissues studied by IHC included esophagus, stomach, colon, salivary gland, liver, pancreas, lung, kidney, prostate, adrenal gland, testis, breast, thyroid, tonsil and placenta. The tumors included 41 hepatocellular carcinomas, 27 bile duct carcinomas, 52 pulmonary carcinomas (42 adenocarcinomas, 10 squamous carcinomas), 73 renal cell carcinomas, 91 breast invasive ductal carcinomas, 97 ovarian tumors (221 high-grade serous carcinomas, 15 low-grade serous carcinomas, and 36 mucinous carcinomas), 58 trophoblastic tumors (35 choriocarcinomas, 6 placental site trophoblastic tumors, 17 epithelioid trophoblastic tumors), 125 cervical carcinomas (114 squamous cell carcinomas, 11 adenocarcinomas), 66 uterine carcinomas (58 conventional low-grade endometrioid carcinoma, 15 serous carcinomas, 2 carcinosarcomas), 35 prostate carcinomas, 49 colon carcinomas, 45 gastric carcinomas, 48 pancreatic carcinomas and 4 oral squamous cell carcinomas. The use of the archival materials was approved by the internal review board of both institutions.
For mutation analysis, genomic DNA isolated from affinity purified tumor samples was used. Those samples included 25 uterine endometrioid carcinomas (FIGO grade 1), 12 uterine serous carcinomas, 32 ovarian high-grade serous carcinomas, 19 ovarian low-grade serous carcinomas, and 5 ovarian mucinous carcinomas. Since the ARID1A mutation status has been previously reported in ovarian clear cell and ovarian low-grade endometrioid carcinomas (6, 11, 21), these carcinomas were not included in the current study. The methodology for the isolation of tumor cells from fresh carcinoma specimens has been previously described (6).
Immunohistochemistry
Immunohistochemical analysis was performed on tissue microarrays except for 20 uterine endometrioid carcinomas and 33 renal cell carcinomas, which were done on whole tissue sections. Loss of ARID1A expression detected in the tissue microarrays was confirmed on whole tissue sections. A polyclonal rabbit anti-ARID1A antibody (Sigma-Aldrich HPA005456) was generated by immunizing a rabbit with the following peptide sequence: PGLGNVAMGPRQHYPYGGPYDRVRTEPGIGPEGNMSTGAPQPNLMPSNPDSGMYSPSRYPPQQQQQQQQRHDSYGNQFSTQGTPSGSPFPSQQTTMYQQQQQNYK. The specificity of the antibody was confirmed by Western blotting. Antigen retrieval was performed by placing sections in citrate buffer (pH 6.0), which were then placed in an autoclave at 120 °C for 10 minutes. The sections were incubated with the rabbit antibody overnight at 4 °C. A positive reaction was detected by the EnVision+System (Dako, Carpinteria, CA). Tumor stromal cells served as positive internal controls. Only nuclear staining was scored. A previous study demonstrated that loss of nuclear expression correlated with mutation of the gene. Hence, absence of nuclear staining (diffuse or focal) was considered positive for gene mutation.
Mutation analysis
A total of 93 tumor samples were analyzed for somatic ARID1A mutations. The normal tissues from the matched cases were also sequenced in parallel. Nucleotide sequences of PCR primers that amplified exon 1 to exon 20 were previously reported (6). PCR products were prepared and purified for the Sanger’s sequencing.
Gene knock down and Western blot
The lentivirus expressing ARID1A shRNAs was produced using HEK293FT cells transfected with the pLKO.1-puro lentiviral plasmids (the RNAi consortium, TRC) and the second generation packaging system, pSPAX2 (Addgene plasmid 12260) and pMD2.G (Addgene plasmid 12259). The shRNA sequences were: shRNA1, GCCTGATCTATCTGGTTCAAT; shRNA2, CCTCTCTTATACACAGCAGAT; and shRNA3, CCGTTGATGAACTCATTGGTT. HeLa Cells were transduced with lentiviral particles and lysates in Laemmli sample buffer were prepared from the cells three days after transduction. For Western blots, SDS-PAGE was used to separate proteins which were then transferred onto PVDF membranes. The rabbit anti-ARID1A antibody as used in immunohistochemistry was used to hybridize the membranes (at a dilution of 1:2000) and anti-GAPDH antibody was also applied to detect the GAPDH, serving as the loading control. After incubating at room temperature for 2 hrs, the membranes were washed with TBST (0.01% Tween 20 in TBS), and were blotted with HRP-conjugated anti-mouse antibodies (Pierce, Rockford, IL), at a dilution of 1:1000 for 1 hr at room temperature. ARID1A and GAPDH bands were revealed by chemiluminescence (Amersham, Arlington Heights, IL).
RESULTS
A total of 995 carcinomas from a variety of tissue origins were studied for ARID1A expression using immunohistochemistry. In order to confirm the specificity of the anti-ARID1A antibody used in this study, we performed a gene knockdown experiment by transducing HeLa cells with three different ARID1A shRNAs. Western blot analysis demonstrated a significant decrease of ARID1A protein in HeLa cells after transfection with ARID1A specific shRNAs, especially the shRNA-2 and shRNA-3, as compared to control shRNA, indicating the specificity of the ARID1A antibody (Fig. 1). Previous reports have shown that inactivating mutations of ARID1A are associated with loss of protein expression (11, 21). Therefore, we focused our attention on those tumors with undetectable ARID1A immunoreactivity and used a scoring system to classify all cases into ARID1A negative (undetectable) and positive cases which showed any levels of ARID1A immunoreactivity. Since the ARID1A mutation status has been previously reported in ovarian clear cell and ovarian endometrioid carcinomas (6, 11, 21), these carcinomas were not included in the current study.
Fig. 1.

Specificity of the antibody in detecting ARID1A. Western blot analysis shows a predominant protein band with a molecular mass corresponding to ARID1A protein (~280kD) in HeLa cell lysate. Protein expression is significantly decreased in shRNA-2 and shRNA-3 treated HeLa cells compared to control shRNA treated cells (A). Immunohistochemistry using this anti-ARID1A antibody on an ovarian clear cell carcinoma with known bi-allelic somatic insertion/deletion mutations of ARID1A (B). The tumor cells demonstrate undetectable ARID1A immunoreactivity while the stromal cells show intense nuclear staining.
There was loss of expression of ARID1A in 36 (3.6%) of 995 cases. Stromal cells in these cases were positive for ARID1A immunoreactivity, indicating that the negative staining in tumor cells was not due to technical artifact. The immunostaining findings for each tumor type are summarized in Table 1 and representative cases illustrated in Fig. 2. Specifically, among 21 types of carcinoma in this study, uterine low-grade endometrioid carcinomas demonstrated loss of expression in 15 (26%) of 58 cases. In contrast, all the normal endometrial tissues examined in this study together with 38 normal endometrial tissues analyzed in our previous study (11) were intensely positive for ARID1A. In addition to uterine low-grade endometrioid carcinomas, gastric carcinomas were negative for ARID1A expression in 11% of cases. We also observed that some carcinomas, especially uterine low-grade endometrioid carcinomas, exhibited “clonal loss” of ARID1A immunoreactivity. That is to say that in a background of ARID1A positive tumor cells, large groups of tumor cells did not express ARID1A (Fig. 3). Although several other tumor types including carcinomas of the bile duct, lung, breast, uterine cervix, colon, and pancreas contained at least one case with negative ARID1A staining, the frequency of negative cases was very low (less than 10% of cases in each tumor type). In contrast, ARID1A was expressed in virtually all the epithelial cells in all normal adult and embryonic tissues tested which included breast, prostate, gastrointestinal tract, pancreas, bile duct, mullerian duct including endometrium (both premenopausal and postmenopausal), skin, respiratory tract, urinary tract, and trophoblast. Besides, lymphoblasts in the germinal center, smooth muscle cells, skeletal muscle cells, endothelial cells were also positive for ARID1A.
Table 1.
Immunohistochemical study of ARID1A on 995 carcinomas.
| Tumor | Total case No. | IHC(−) | % of IHC(−) |
|---|---|---|---|
| Hepatocellular carcinoma | 41 | 0 | 0 |
| Bile duct carcinoma | 27 | 2 | 7.4 |
| Lung | |||
| adenocarcinoma | 42 | 1 | 4.2 |
| squamous carcinoma | 10 | 1 | 10 |
| Renal cell carcinoma | 73 | 0 | 0 |
| Breast carcinoma | 91 | 1 | 1.1 |
| Ovary | |||
| high-grade serous carcinoma | 221 | 0 | 0 |
| low-grade serous carcinoma | 15 | 0 | 0 |
| mucinous carcinoma | 36 | 0 | 0 |
| Uterine cervix | |||
| squamous carcinoma | 114 | 2 | 1.8 |
| adenocarcinoma | 11 | 1 | 9.1 |
| Uterine corpus | |||
| endometrioid carcinoma | 58 | 15 | 26 |
| serous carcinoma/carcinosarcoma | 17 | 0 | 0 |
| Trophoblastic tumor | |||
| choriocarcinoma | 35 | 0 | 0 |
| placental site trophoblastic tumor | 6 | 0 | 0 |
| epithelioid trophoblastic tumor | 17 | 0 | 0 |
| Prostate carcinoma | 35 | 0 | 0 |
| Colon carcinoma | 49 | 2 | 4.1 |
| Gastric carcinoma | 45 | 5 | 11 |
| Pancreatic carcinoma | 48 | 4 | 8.3 |
| Oral squamous carcinoma | 4 | 0 | 0 |
IHC: immunoreactivity of ARID1A
Fig. 2.

ARID1A immunoreactivity in representative carcinoma types (right panel) and their normal tissue counterparts (left panel). Negative staining (undetectable level) of ARID1A in a FIGO grade I endometrioid carcinoma (A), an infiltrating ductal carcinoma of the breast (B), a cervical squamous carcinoma (C), and a pancreatic carcinoma (D).
Fig. 3.
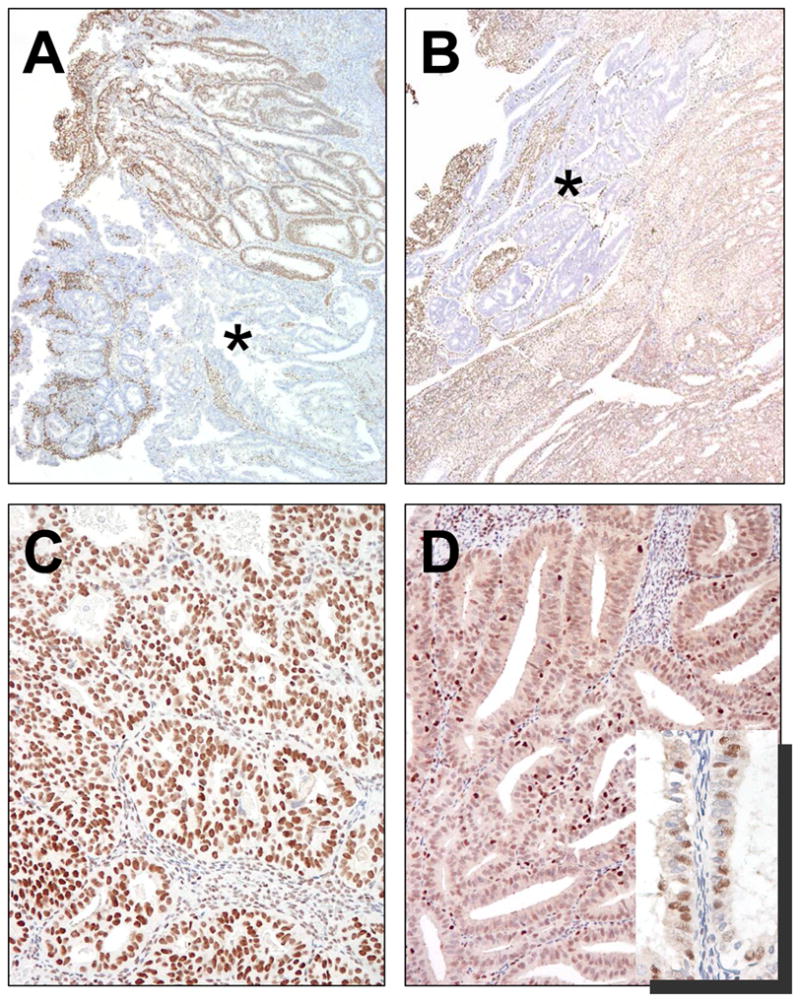
Pattern of ARID1A immunoreactivity in two uterine endometrioid carcinomas. A. The carcinoma (UEM-5) harbors biallelic ARID1A mutations (nonsense and deletion mutation) and demonstrates a clonal loss of ARID1A expression (asterisk). B. The carcinoma (UEM-3) contains monoallelic nonsense mutation show a clonal loss of ARID1A expression (asterisk). C. The carcinoma with wild-type ARID1A exhibits a diffuse and intense pattern of ARID1A staining. D. The carcinoma with wild-type ARID1A demonstrates diffuse but less intense ARID1A immunoreactivity than the tumor in C. Occasionally, patchy staining can be observed (inset).
Mutational analysis was performed on uterine low-grade endometrioid carcinomas because they showed the highest frequency of loss of ARID1A expression. Nineteen ovarian low-grade, 32 ovarian high-grade, 5 ovarian mucinous and 12 uterine serous carcinomas were also analyzed, none of which showed loss of ARID1A expression. As shown in Table 2, somatic ARID1A mutation was detected in 10 (40%) of 25 uterine low-grade endometrioid carcinomas. As in ovarian clear cell and ovarian endometrioid carcinomas, the mutations were either insertion/deletion mutations or nonsense mutations which were widely distributed in the ARID1A gene (Table 3). Three of these 10 cases (UEM-1, -5 and -8) showed two independent ARID1A mutations, likely affecting both alleles. Correlation of ARID1A mutation status and immunoreactivity was performed in 25 uterine endometrioid carcinomas and 51 ovarian serous carcinomas. We found that 5 (50%) of 10 tumors with ARID1A mutations did not demonstrate any detectable level of ARID1A immunoreactivity. Interestingly, four ARID1A positive cases with ARID1A mutations exhibited a pattern of immunoreactivity in which areas of negative cells were present adjacent to positive areas suggesting that mutations arose in clones within the tumor (Fig. 4). In contrast, only two of 15 ARID1A wild-type endometrioid carcinomas showed complete loss of ARID1A staining while the majority of cases demonstrated diffuse ARID1A staining (>80% of tumor cells being positive). We did not observe any pattern of clonal loss in ARID1A wild-type carcinomas as in ARID1A mutated cases. All 14 ovarian low-grade serous carcinoma and 25 ovarian high-grade serous carcinomas demonstrated a diffuse ARID1A positivity and none of these as well as 5 ovarian mucinous carcinoma contained ARID1A mutations. Thus, the complete loss of ARID1A expression significantly correlated with its mutation status (p= 0.0014, Fisher’s exact test). Combining cases that were completely negative with those showing clonal loss and correlating them with ARID1A mutation was highly significant (p< 0.0001, Fisher’s exact test).
Table 2.
ARID1A somatic mutations in uterine and ovarian carcinomas.
| Uterine Endometrioid | Uterine Serous | Ovarian HG | Ovarian LG | Ovarian mucinous | |
|---|---|---|---|---|---|
| Cases with ARID1A mutation | 10 | 0 | 0 | 0 | 0 |
| Total cases | 25 | 12 | 32 | 19 | 5 |
HG: high-grade; LG: low-grade
Table 3.
Correlation of ARID1A mutations and Immunoreactivity.
| Case | Tumor type | Nucleotide change (allele one) | Nucleotide change (allele two) | Amino Acid | Mutation type | ARID1A immunoreactivity |
|---|---|---|---|---|---|---|
| UEM-1 | Uterine EMCA | 6393delC | 5553insG | frameshift | ins/del | negative |
| UEM-2 | Uterine EMCA | 2352_2353insG | wild-type | frameshift | ins/del | diffusely positive |
| UEM-3 | Uterine EMCA | 5701G>T | wild-type | 1901G>X | nonsense | clonal loss; 20% negative |
| UEM-4 | Uterine EMCA | 1010delG | wild-type | frameshift | ins/del | negative |
| UEM-5 | Uterine EMCA | 6298C>T | 1996_2000delATTTC | 2100Q>X | nonsense | clonal loss; 50% negative |
| UEM-6 | Uterine EMCA | 3211delA | wild-type | frameshift | ins/del | clonal and mixed; 50% neg |
| UEM-7 | Uterine EMCA | 4683_4684insC | wild-type | frameshift | ins/del | clonal; 20% negative |
| UEM-8 | Uterine EMCA | 6446_6447insA | 6446_6447insA | frameshift | ins/del | negative |
| UEM-9 | Uterine EMCA | 2023_2032delAATCCAGCTC | wild-type | frameshift | ins/del | negative |
| UEM-10 | Uterine EMCA | 2368C>T | wild-type | 790Q>X | nonsense | negative |
| UEM-11 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | mixed; 10% negative |
| UEM-12 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | diffusely but weakly positive |
| UEM-13 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | mixed; 10% negative |
| UEM-14 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | negative |
| UEM-15 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | diffusely positive |
| UEM-16 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | negative |
| UEM-17 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | diffusely but weakly positive |
| UEM-18 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | diffusely positive |
| UEM-19 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | mixed; 20% negative |
| UEM-20 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | diffusely positive |
| UEM-21 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | mixed; 20% negative |
| UEM-22 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | diffusely positive |
| UEM-23 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | n/a |
| UEM-24 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | n/a |
| UEM-25 | Uterine EMCA | wild-type | wild-type | wild-type | wild-type | n/a |
| SMBT-1 | Seromucinous BT | 3216delA (homo) | 3216delA (homo) | frameshift | ins/del | negative |
| SMBT-2 | Seromucinous BT | 2165_2171delACCAGAT (homo) | 2165_2171delACCAG AT (homo) | frameshift | ins/del | negative |
| OVEM | Ovarian EMCA | Exon20 5′ intron1G>T (homo) | Exon20 5′ intron1G>T (homo) | frameshift | splicing | negative |
| OVCC | Ovarian CC | 4247insCAGC | 6026_6052del | frameshift | ins/del | negative |
Discussion
In the past few years, substantial progress has been made in cataloging molecular genetic alterations at a genome wide scale. One of the main findings has been the identification of somatic mutations of several chromatin remodeling genes in certain types of human cancer. These genes include JARID1C in renal cell carcinoma (1), BRG1 (SMARCA4) in lung carcinoma (12, 16) and, most recently, ARID1A in ovarian clear cell and ovarian low-grade endometrioid carcinoma (6, 21). The findings in the present study extend previous observations and provide cogent evidence that epigenetic changes, like genetic alteration, is a “driver” rather than a “passenger” that is directly involved in tumor development of uterine low-grade endometrioid carcinoma (7).
In this study a large number and variety of normal and tumor tissues were evaluated. The main findings were the loss of ARID1A immunoreactivity and mutation of ARDIA in low-grade uterine endometrioid carcinoma as compared to other types of carcinomas. Although loss of ARID1A expression also occurs in other tumor types, the frequency of loss of expression is low, indicating that ARID1A mutations are associated with specific types of carcinoma. Mutational analysis demonstrated somatic ARID1A mutations in 40% of uterine endometrioid carcinoma, a finding that has not been previously reported.
Endometrial carcinomas are divided into two broad groups designated type I and type II. Type I tumors are composed of endometrioid carcinomas which frequently harbor sequence mutations in CCNB1, PTEN and PIK3CA while type II tumors are largely serous carcinomas that contain TP53 mutations in the majority of cases (2). Patients with type I tumors are usually younger, present at an earlier clinical stage and have a more indolent clinical course compared to women with type II tumors. Type I tumors arise from endometrial hyperplasia and type II tumors develop from endometrial intraepithelial carcinoma that is frequently associated with endometrial polyps. Thus the relatively frequent loss of ARID1A expression and ARID1A mutations in uterine endometrioid carcinoma but not in uterine serous carcinoma further supports their distinct pathogenesis. Given the well established roles of Pten-AKT pathway and Wnt pathway in the development of low-grade uterine endometrioid carcinoma (type I tumor), it will be important to determine if the ARID1A pathway cross-talks with those signaling pathways and to assess how inactivation of ARID1A contributes to tumor initiation and progression in this type of carcinoma.
Although ARID1A mutations were most often associated with complete loss of its protein expression in low-grade uterine endometrioid carcinoma, we observed several uterine endometrioid carcinomas with ARID1A mutations that showed a heterogeneous staining pattern. In these cases there were significantly large areas that were negative in what were otherwise positive cases. This geographic distribution of loss of ARID1A expression strongly suggests that ARID1A mutation occurred in clones of cells within the tumor. This type of clonal loss of ARID1A immunoreactivity was only detected in uterine endometrioid carcinomas with ARID1A mutations but not in those without mutations or in ovarian clear cell carcinomas. It is likely that ARID1A mutations occur after tumor initiation in endometrioid carcinoma, creating tumor subclones during evolution of the carcinoma. It is of great interest to study ARID1A immunostaining patterns in endometrial hyperplasia to determine how early ARID1A protein is lost during tumor progression of endometrioid carcinoma.
Like endometrial carcinoma, ovarian epithelial carcinomas have been divided into type I and type II categories based on their distinctive clinicopathologic and molecular features (18). Ovarian clear cell carcinoma and low-grade ovarian endometrioid carcinoma comprise the majority of type I ovarian tumors and are frequently associated with endometriosis. The results from this study along with our previous reports (6, 11, 21) strongly suggest that loss of ARID1A expression and/or its mutations are confined to ovarian clear cell and ovarian endometrioid carcinomas, because the high-grade and low-grade serous carcinomas as well as mucinous carcinomas did not show ARID1A mutations or loss of expression. It is, therefore, conceivable that ARID1A mutation plays an important role in the development of ovarian tumors derived from endometriosis. Since it is generally thought that endometriosis develops from retrograde menstruation, the underlying critical molecular event for the development of uterine low-grade endometrioid, ovarian endometrioid, and clear cell carcinomas in some cases is mutation of ARIDIA in endometrial tissue. Although we did not detect ARID1A mutation in ovarian mucinous carcinomas, the small number of mucinous carcinomas analyzed in this study precludes a definitive conclusion regarding them.
In conclusion, based on ARID1A immunohistochemistry and mutational analysis, we found that ARID1A inactivation, either by somatic mutations or by loss of expression, frequently occurs in uterine low-grade endometrioid carcinomas in addition to ovarian low-grade endometrioid and ovarian clear cell carcinomas. Thus, it appears that ARID1A inactivation is mainly confined to certain types of gynecologic cancers that arise from endometrial tissue, either from the uterine cavity or from an ectopic site, i.e., endometriosis and, therefore, leads to the conclusion that these endometrium-related tumors share a similar molecular pathogenesis. Our findings also support the utility of complete loss or clonal loss of ARID1A immunoreactivity as a surrogate marker to detect ARID1A mutations in tissues; however, further studies are necessary to determine the role of ARID1A in differential diagnosis. It would be also interesting to determine the mutation status and immunoreactivity of ARID1A in uterine high-grade endometrioid carcinoma and clear cell carcinoma.
Acknowledgments
This study is supported by NIH/NCI grants, CA129080, CA103937 and CA116184. BG is partly supported by the grant, 2010HREAOSB1, from the HERA Women’s Cancer Foundation.
References
- 1.Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Cristofano A, Ellenson LH. Endometrial carcinoma. Annu Rev Pathol. 2007;2:57–85. doi: 10.1146/annurev.pathol.2.010506.091905. [DOI] [PubMed] [Google Scholar]
- 3.Gao X, Tate P, Hu P, et al. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci U S A. 2008;105:6656–6661. doi: 10.1073/pnas.0801802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang J, Zhao YL, Li Y, et al. Genomic and functional evidence for an ARID1A tumor suppressor role. Genes Chromosomes Cancer. 2007;46:745–750. doi: 10.1002/gcc.20459. [DOI] [PubMed] [Google Scholar]
- 6.Jones S, Wang TL, Shih IM, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaiser J. Genes link epigenetics and cancer. Science. 2010;330:577. doi: 10.1126/science.330.6004.577. [DOI] [PubMed] [Google Scholar]
- 8.Kinzler KW, Nilbert MC, Vogelstein B, et al. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers. Science. 1991;251:1366–1370. doi: 10.1126/science.1848370. [DOI] [PubMed] [Google Scholar]
- 9.Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761, 763. doi: 10.1038/386761a0. [news; comment] [DOI] [PubMed] [Google Scholar]
- 10.Kobel M, Gilks CB, Huntsman DG. Adult-type granulosa cell tumors and FOXL2 mutation. Cancer Res. 2009;69:9160–9162. doi: 10.1158/0008-5472.CAN-09-2669. [DOI] [PubMed] [Google Scholar]
- 11.Maeda D, Mao T-L, Fukayama M, et al. Clinicopathological Significance of Loss of ARID1A Immunoreactivity in Ovarian Clear Cell Carcinoma. International Journal of Molecular Sciences. 2010;11:5120–5128. doi: 10.3390/ijms11125120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medina PP, Romero OA, Kohno T, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat. 2008;29:617–622. doi: 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- 13.Nagl NG, Jr, Patsialou A, Haines DS, et al. The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res. 2005;65:9236–9244. doi: 10.1158/0008-5472.CAN-05-1225. [DOI] [PubMed] [Google Scholar]
- 14.Nagl NG, Jr, Zweitzig DR, Thimmapaya B, et al. The c-myc gene is a direct target of mammalian SWI/SNF-related complexes during differentiation-associated cell cycle arrest. Cancer Res. 2006;66:1289–1293. doi: 10.1158/0008-5472.CAN-05-3427. [DOI] [PubMed] [Google Scholar]
- 15.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez-Nieto S, Canada A, Pros E, et al. Massive parallel DNA pyrosequencing analysis of the tumor suppressor BRG1/SMARCA4 in lung primary tumors. Hum Mutat. 2010 doi: 10.1002/humu.21415. [DOI] [PubMed] [Google Scholar]
- 17.Schrader KA, Gorbatcheva B, Senz J, et al. The specificity of the FOXL2 c.402C>G somatic mutation: a survey of solid tumors. PLoS One. 2009;4:e7988. doi: 10.1371/journal.pone.0007988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih I-M, Kurman RJ. Ovarian tumorigenesis- a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511–1518. doi: 10.1016/s0002-9440(10)63708-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Rechem C, Boulay G, Leprince D. HIC1 interacts with a specific subunit of SWI/SNF complexes, ARID1A/BAF250A. Biochem Biophys Res Commun. 2009;385:586–590. doi: 10.1016/j.bbrc.2009.05.115. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Nagl NG, Wilsker D, et al. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem J. 2004;383:319–325. doi: 10.1042/BJ20040524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]