

Abstract
Schizophrenia is a complex disorder, caused by both genetic and environmental factors and their interactions. Research on pathogenesis has traditionally focused on neurotransmitter systems in the brain, particularly those involving dopamine. Schizophrenia has been considered a separate disease for over a century, but in the absence of clear biological markers, diagnosis has historically been based on signs and symptoms. A fundamental message emerging from genome-wide association studies of copy number variations (CNVs) associated with the disease is that its genetic basis does not necessarily conform to classical nosological disease boundaries. Certain CNVs confer not only high relative risk of schizophrenia but also of other psychiatric disorders1–3. The structural variations associated with schizophrenia can involve several genes and the phenotypic syndromes, or the ‘genomic disorders’, have not yet been characterized4. Single nucleotide polymorphism (SNP)-based genome-wide association studies with the potential to implicate individual genes in complex diseases may reveal underlying biological pathways. Here we combined SNP data from several large genome-wide scans and followed up the most significant association signals. We found significant association with several markers spanning the major histocompatibility complex (MHC) region on chromosome 6p21.3-22.1, a marker located upstream of the neurogranin gene (NRGN) on 11q24.2 and a marker in intron four of transcription factor 4 (TCF4) on 18q21.2. Our findings implicating the MHC region are consistent with an immune component to schizophrenia risk, whereas the association with NRGN and TCF4 points to perturbation of pathways involved in brain development, memory and cognition.
To begin our search for sequence variants associated with schizophrenia, we performed a genome-wide scan of 2,663 schizophrenia cases and 13,498 controls from eight European locations (England, Finland (Helsinki), Finland (Kuusamo), Germany (Bonn), Germany (Munich), Iceland, Italy and Scotland; collectively called SGENE-plus) using the Illumina HumanHap300 and HumanHap550 BeadChips. In total, 314,868 SNPs meeting our quality control criteria were included in an allelic association analysis. To adjust for relatedness and potential population stratification, genomic control was applied to each study group.
None of the markers gave P values smaller than our genome-wide significance threshold of 0.05/314,868, or approximately 1.6 × 10−7 (see Supplementary Fig. 1 for a quantile–quantile plot and Supplementary Table 1 for markers with the smallest P values). Next, we combined findings from our top 1,500 markers with results for the same markers (or surrogates for them) from both the International Schizophrenia Consortium5 (excluding the Scottish samples overlapping with samples in our study, 2,602 cases and 2,885 controls) and the European–American portion of the Molecular Genetics of Schizophrenia6 (2,681 cases and 2,653 controls) study. Twenty-five of our top 1,500 markers (or eighteen counting very strongly correlated (r2 > 0.8) markers only once) had P values less than 1 × 10−5 in the combined results (Supplementary Table 2). These top markers were followed up in as many as 4,999 cases and 15,555 controls from four sets of additional samples from Europe (set 1, 715 cases and 3,634 controls from the Netherlands; set 2, 3,330 cases and 6,892 controls from Denmark (Aarhus), Denmark (Copenhagen), Germany (Bonn), Germany (Munich), Hungary, the Netherlands, Norway, Russia and Sweden; set 3, 287 cases and 3,987 controls from Finland; set 4, 667 cases and 1,042 controls from Spain (Santiago) and Spain (Valencia)) (Supplementary Table 3).
Three markers, all in the extended MHC region on the short arm of chromosome 6, showed genome-wide significance in the combination of SGENE-plus and the follow-up samples described above (Table 1). In addition, four other markers—two in the MHC region, one at 11q24.2 and one at 18q21.2—showed genome-wide significance when results from the International Schizophrenia Consortium and the Molecular Genetics of Schizophrenia study were included (Table 1).
Table 1.
Genome-wide significant association of seven markers with schizophrenia
| Chromosome/ megabases |
SNP[allele] | Frequency | SGENE-plus* (2,663 cases; 13,498 controls) |
Follow-up (4,999 cases; 15,555 controls) |
SGENE-plus + follow-up (7,662 cases; 29,053 controls) |
SGENE-plus + follow-up + ISC + MGS (12,945 cases; 34,591 controls) |
Region/ neighbouring gene |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||||||
| OR (95% CI) |
P value | OR (95% CI) |
P value | OR (95% CI) |
P value | OR (95% CI) |
P value | ||||
| 6/27.2 | rs6913660[C]†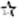
|
0.85 | 1.22 (1.10, 1.36) |
0.00023 | 1.11 (1.04, 1.19) |
0.0021 | 1.14 (1.08, 1.21) |
4.7×10−6 | 1.15 (1.10, 1.21) |
1.1×10−9 |
MHC/ HIST1H2BJ |
| 6/27.3 | rs13219354[T]‡
|
0.90 | 1.25 (1.11, 1.42) |
0.00043 | 1.19 (1.08, 1.30) |
0.00022 | 1.21 (1.12, 1.30) |
4.4×10−7 | 1.20 (1.14, 1.27) |
1.3×10−10 |
MHC/ PRSS16 |
| 6/27.4 | rs6932590[T]§
|
0.78 | 1.15 (1.05, 1.26) |
0.0024 | 1.17 (1.10, 1.25) |
4.9×10−7 | 1.17 (1.11, 1.23) |
4.4×10−9 | 1.16 (1.11, 1.21) |
1.4×10−12 |
MHC/ PRSS16 |
| 6/28.4 | rs13211507[T] ||
|
0.92 | 1.24 (1.08, 1.42) |
0.0027 | 1.27 (1.15, 1.40) |
3.1×10−6 | 1.26 (1.16, 1.36) |
3.1×10−8 | 1.24 (1.16, 1.32) |
8.3×10−11 |
MHC/ PGBD1 |
| 6/32.3 | rs3131296[G]¶
|
0.87 | 1.21 (1.08, 1.36) |
0.0011 | 1.20 (1.11, 1.30) |
5.3×10−6 | 1.21 (1.13, 1.29) |
2.1×10−8 | 1.19 (1.13, 1.25) |
2.3×10−10 |
MHC/ NOTCH4 |
| 11/124.1 | rs12807809[T] | 0.83 | 1.19 (1.08, 1.32) |
0.00045 | 1.13 (1.06, 1.21) |
0.00022 | 1.15 (1.09, 1.22) |
5.0×10−7 | 1.15 (1.10, 1.20) |
2.4×10−9 | NRGN |
| 18/51.3 | rs9960767[C]#
|
0.056 | 1.30 (1.11, 1.51) |
0.0011 | 1.20 (1.08, 1.33) |
0.00044 | 1.23 (1.13, 1.34) |
2.2×10−6 | 1.23 (1.15, 1.32) |
4.1×10−9 | TCF4 |
Allelic OR and P values (two-sided) based on the multiplicative model are shown. Frequency is the average allelic control frequency in SGENE-plus. Megabase is from the National Center for Biotechnology Information (NCBI) build 36. To combine the study groups within SGENE-plus and within the follow-up, the Mantel–Haenszel model was used. SGENE-plus and the follow-up sets were combined with the International Schizophrenia Consortium and the Molecular Genetics of Schizophrenia study by summing weighted summary statistics. Note that the combined analysis shown here differs from the one reported in the companion ISC and MGS papers; here we include the Aberdeen samples in SGENE-plus and not ISC, and we also incorporate additional follow-up samples. ISC, International Schizophrenia Consortium; MGS, Molecular Genetics of Schizophrenia.
P values were adjusted using genomic control (see Methods).
rs4452638 in International Schizophrenia Consortium (HapMap CEU r2=0.866).
rs4452638 in International Schizophrenia Consortium (HapMap CEU r2=1).
rs3800307 in International Schizophrenia Consortium (HapMap CEU r2=0.843).
rs13214023 in International Schizophrenia Consortium (HapMap CEU r2=0.915).
rs1150753 in International Schizophrenia Consortium (HapMap CEU r2=1).
rs10401120 in International Schizophrenia Consortium (HapMap CEU r2=0.867).
Imputed using MACH 1.0 in the Molecular Genetics of Schizophrenia study.
In the MHC region on chromosome 6p21.3-22.1, the five genome-wide significant markers (P ranging from 1.1 × 10−9 to 1.4 × 10−12 in all samples combined) have risk alleles with average control frequencies between 78% and 92% (Table 1). Combined odds ratios (ORs) for the markers range from 1.15 to 1.24 (Table 1) with no significant heterogeneity between the study groups (P > 0.25, Supplementary Table 4). For all of the markers, the multiplicative model for risk provides an adequate fit (P > 0.62).
Despite spanning about five megabases (Mb), the five chromosome 6p markers cover only about 1.4 centimorgans (cM) and substantial linkage disequilibrium exists between them (Supplementary Table 5). The association of rs6932590 (the most significant marker), however, cannot account for all of the association of the four remaining markers (Supplementary Table 6). Most notably, conditional on rs6932590, rs3131296 has an association P value of 3.4 × 10−6, indicating that rs3131296 may be capturing a second susceptibility variant or that both rs6932590 and rs3131296 are correlated with a third, higher risk, variant not examined here.
To examine association of the genome-wide significant SNPs in the 5-Mb region on 6p21.3-22.1 with classical human leukocyte antigen (HLA) alleles, long-range phasing haplotypes7 tagging the major alleles at the HLA-A, HLA-B, HLA-C, HLA-DRB1, HLA-DQA1 and HLA-DQB1 loci in Icelanders were used. Only rs3131296 shows substantial (r2 > 0.5) correlation with any of the classical HLA alleles tested; this marker has an r2 of 0.86 with DRB1*03 and an r2 of 0.81 with HLA-B*08. Simplified tags for these two classical alleles, appropriate for the European samples of SGENE-plus, had effects that were not statistically distinguishable from the effect of rs3131296. In the case of both DRB1*03 and HLA-B*08, the classical HLA allele is paired with the protective allele of rs3131296, making the results described here consistent with the under-transmission of DRB1*03 to schizophrenic offspring reported previously8.
Many autoimmune and infectious diseases have been associated with DRB1*03 and, indeed, inspection of top MHC region SNPs from recent genome-wide association scans of three of these—type I diabetes9, coeliac disease10 and systemic lupus erythematosus11—reveals, for each disease, SNPs having a HapMap CEU (Utah residents with ancestry from northern and western Europe) r2 of at least 0.73 with rs3131296. For all of the diseases, the allele that is protective from schizophrenia is associated with the ‘at-risk’ allele for the autoimmune disease. This reciprocal association may, at least in part, explain the recently reported inverse association between type I diabetes and schizophrenia12,13. A positive association, however, has been described for coeliac disease and schizophrenia12; if this positive association has a genetic basis, it must be the result of associations at variants other than the one described here.
Schizophrenia patients are more likely, compared to the general population, to have been born in the winter or the spring. Although infections such as influenza and measles have been proposed as a possible mechanism for this distortion, a clear association between infectious agents and schizophrenia has not been demonstrated. The association with the MHC region reported here supports a role for infection but, as many non-immune-related genes are also found in the extended MHC region, it does not provide strong evidence. On the basis of the 3,130 schizophrenia patients for which month of birth information was available, no significant difference in the frequency of the top SNPs from the MHC region according to season of birth (winter/spring versus summer/autumn) was identified (P > 0.29).
The MHC region has long been postulated to harbour variants conferring risk of schizophrenia, both because of evidence for linkage in the region14 and because of the suggested involvement of infection. Association studies of variants from the MHC region to date, however, have had modest sample sizes and therefore have lacked the power to detect effects similar to those described here.
The genome-wide significant marker (P = 2.4 × 10−9) at 11q24.2, rs12807809, has an average risk allele control frequency of 83% and a combined OR of 1.15 (Table 1) with no significant OR heterogeneity between the study groups (P = 0.74, Supplementary Table 4). The multiplicative model provides an adequate fit (P = 0.18). This marker is 3,457 bases upstream of neurogranin (NRGN). NRGN has previously been reported as associated in males with schizophrenia in a small Portuguese series15, although the associated SNP in that paper, rs7113041, is not closely correlated with the SNP reported here (HapMap CEU r2 = 0.11). Furthermore, reduced NRGN immunoreactivity has been observed in prefrontal areas 9 and 32 of post-mortem schizophrenia brains16. NRGN is expressed exclusively in brain, especially in dendritic spines, with expression directly controlled by thyroid hormone17. It is therefore possible that the psychotic and cognitive features associated with thyroid dysfunction may, in part, be mediated through dysregulation of NRGN gene expression.
NRGN encodes a postsynaptic protein kinase substrate that binds calmodulin (CaM) in the absence of calcium; it is abundantly expressed in brain regions important for cognitive functions, and is especially enriched in CA1 pyramidal neurons in the hippocampus18. The main function of NRGN may be to act as a CaM reservoir, regulating its availability in the postsynaptic compartment. Glutamate stimulation of N-methyl-d-aspartate (NMDA) receptors results in Ca2+ influx to the neuron, NRGN oxidation and release of CaM19. The consequent activation of postsynaptic calcium/calmodulin-dependent protein kinase II (CaMKII) by CaM results in a sustained strengthening of synaptic connections; conversely, CaM activation of calcineurin (PP2B) weakens these connections. CaMKII has a major role in mediating the NMDA-receptor signalling involved in synaptic plasticity and formation of associative memories in the brain20. Impaired memory function is thought to be a core feature of the pathophysiology of schizophrenia21, especially affecting short term memory where CAMKII is thought to have a major role22. Glutamate stimulation of NMDA receptors results in Ca2+ influx to the neuron and in NRGN oxidation. Altered NRGN activity may therefore mediate the effects of NMDA hypofunction implicated in the pathophysiology of schizophrenia.
On 18q21.2, marker rs9960767 has a genome-wide significant P value of 4.1 × 10−9 (Table 1). The risk allele control frequency is about 6% and the OR is 1.23 with no significant OR heterogeneity between the study groups (P = 0.34, Supplementary Table 4). The multiplicative model gives an adequate fit (P = 0.81). Genome scan meta-analysis of linkage studies of schizophrenia14 ranks the 18q21.1-qter ‘bin’ around fifteenth in the genome. TCF4 is essential for normal brain development23, and mutations in the gene were recently found to be responsible for Pitt–Hopkins syndrome, an autosomal-dominant neurodevelopmental disorder characterized by severe motor and mental retardation, microcephaly, epilepsy and facial dysmorphisms24. The phenotype need not be as extreme as Pitt–Hopkins syndrome; a de novo translocation disrupting exon 4 of TCF4 was found in an individual with problems restricted to mental retardation25. Thus, it seems that variants in a single gene can be associated with a range of neuropsychiatric phenotypes including schizophrenia. This is in line with the range of phenotypes associated with CNVs recently associated with schizophrenia1–3.
In addition to these three genome-wide significant loci, further putative susceptibility variants are highlighted by this study. For instance, in the set of 18 markers taken into follow-up studies, rs2312147 achieved a P value that was not far from our genome-wide significance threshold (Supplementary Table 3). Also, markers having P values in the combined SGENE-plus, International Schizophrenia Consortium and Molecular Genetics of Schizophrenia data set (Supplementary Table 2) that are somewhat larger than those followed up here can be investigated further. Intriguingly, these markers include rs6589386, which is located in an intergenic region upstream of DRD2, a candidate gene for schizophrenia.
Our findings demonstrating association of schizophrenia with markers in the MHC region are consistent with previous reports suggesting immune system involvement in schizophrenia, whereas association with NRGN and TCF4 points more to perturbation of pathways involved in brain development and cognitive function, particularly memory. Impaired cognitive and memory functions are being recognized increasingly as core features of schizophrenia21 which are poorly addressed by current medications. The three common genetic variants we describe, which predispose to schizophrenia, have the potential to be translated into targets for the development of novel medications.
METHODS SUMMARY
Subjects
SGENE-plus, the genome-wide portion of the study, included 2,663 cases and 13,498 controls from eight European locations: England, Finland (Helsinki), Finland (Kuusamo), Germany (Bonn), Germany (Munich), Iceland, Italy and Scotland. Follow-up groups comprised 4,999 cases and 15,555 controls from twelve European locations: Denmark (Aarhus), Denmark (Copenhagen), Finland, Germany (Bonn), Germany (Munich), Hungary, the Netherlands, Norway, Russia, Spain (Santiago), Spain (Valencia) and Sweden. Cases were diagnosed with schizophrenia according to DSM-IV or ICD-10 criteria (see Supplementary Methods).
Genotyping
Genome-wide genotyping was carried out at deCODE, Duke University and the University of Bonn using either HumanHap300 or HumanHap550 BeadChips (Illumina). Individual genotyping was done via Centaurus assays at deCODE and via multiplex PCR and mini-sequencing assays, followed by MALDI-TOF mass spectrometry analysis in Spain and Finland.
Statistical analysis
A likelihood procedure described previously was used for association analysis26. To correct for relatedness and potential population stratification, genomic control was used27. Within our study, samples were combined using the Mantel–Haenszel model28. Our results were merged with those of the International Schizophrenia Consortium and the Molecular Genetics of Schizophrenia study by computing weighted averages of Z scores (see Methods).
METHODS
Genome-wide genotyping
SGENE was initially made up of 1,321 cases and 12,277 controls typed at deCODE Genetics using the Illumina HumanHap300 BeadChip. For SGENE-plus, an additional 859 cases and 854 controls typed at Duke University using either the Illumina HumanHap300 BeadChip or the Illumina HumanHap550 BeadChip as well as 483 cases and 367 controls typed at the University of Bonn using the HumanHap550 BeadChip were also included. Samples were excluded if they were low yield (low yield was defined as below 98% except for the samples typed at Duke, in which case low yield was below 96%), if they were duplicates of other samples included in the study, if they had a sex determined by X chromosome marker homozygosity different from their reported sex or if they were estimated to have less than 90% European ancestry by running STRUCTURE29 using the HapMap CEU, YRI and CHB/JPT individuals as reference samples. Of the 317,503 markers on the HumanHap300 BeadChip, 2,635 were deemed unusable due to lack of polymorphism, severe deviation from Hardy–Weinberg equilibrium (P < 1 × 10−10), low yield (<95% in either cases or controls) or allele frequency differences between the typing centres (P < 1 × 10−7); 314,868 markers, then, remained for analysis.
Follow-up genotyping
Follow-up set 1 (715 cases; 3,634 controls) was genotyped at UCLA on the HumanHap550 BeadChip and at deCODE Genetics on the HumanCNV370 BeadChip. Only the markers shown in Supplementary Table 3, however, were used in this study. Follow-up set 2 (3,330 cases; 6,892 controls) was genotyped at deCODE Genetics using Centaurus assays (Nanogen). Assay quality was evaluated by genotyping the CEU HapMap samples and comparing the results with the publicly released HapMap data. Assays with a greater than 1.5% mismatch rate were not used. Follow-up set 3 (287 cases; 3,987 controls) was typed in Finland using the Sequenom MassArray iPLEX genotyping system, following the manufacturer’s instructions (Sequenom Inc.). Briefly, the system involves multiplex PCR and mini-sequencing assays, followed by MALDI-TOF mass spectrometry analysis. Follow-up set 4 (667 cases; 1,042 controls) was typed at the Santiago de Compostela node of the Spanish National Genotyping Centre (http://www.cegen.org) using the Sequenom MassArray iPLEX genotyping system, following the manufacturer’s instructions (Sequenom Inc.). As a quality check, all clusters were manually inspected for accurate genotype assignment. In addition, 1,781 genotypes were successfully assayed twice, with no discordant results.
Association analysis
Association analysis was carried out using a likelihood procedure described in a previous publication implemented in NEMO software26. Allele-specific ORs and associated P values were calculated assuming a multiplicative model for the two chromosomes of an individual. Association was tested using a standard likelihood ratio statistic that, if the subjects were unrelated, would have asymptotically a chi-squared distribution with 1 degree of freedom under the null hypothesis. To correct for relatedness and potential population stratification in the genome-wide typed samples (SGENE-plus and follow-up set 1), genomic control was used27. Inflation factors, estimated by dividing the median of the 314,868 chi-squared statistics by 0.6752, were 1.01, 1.03, 1.09, 1.05, 1.05, 1.19, 1.04 and 1.09 for the SGENE-plus England, Finland (Helsinki), Finland (Kuusamo), Germany (Bonn), Germany (Munich), Iceland, Italy and Scotland groups, respectively, and 1.08 for follow-up set 1. The inflation factor was large in Iceland because of the inclusion of close relatives in that data set. Both SGENE-plus and the follow-up samples were combined using the Mantel–Haenszel model28.
Summary statistic combination
Combined P values for the SGENE-plus, International Schizophrenia Consortium and Molecular Genetics of Schizophrenia studies were calculated by summing Z scores with each group’s Z score multiplied by the inverse of that group’s standard error divided by the square root of the sum of the squared inverse standard errors. Combined ORs were calculated by summing log ORs with each log OR weighted by the inverse of its variance.
Supplementary Material
Acknowledgements
We thank the subjects and their relatives and staff at the recruitment centres. This work was sponsored by EU grants LSHM-CT-2006-037761 (Project SGENE), PIAP-GA-2008-218251 (Project PsychGene) and HEALTH-F2-2009-223423 (Project PsychCNVs). Genotyping of the Dutch samples was sponsored by NIMH funding, R01 MH078075. This work was also supported by the National Genomic Network (NGFN-2) of the German Federal Ministry of Education and Research (BMBF) and Marie Curie grant PIAP-GA-2008-218251 (PsychGene). M.M.N. received support from the Alfried Krupp von Bohlen und Halbach-Stiftung. We are grateful to S. Schreiber and M. Krawczak for providing genotype data for PopGen controls, and to K.-H. Jöckel and R. Erbel for providing control individuals from the Heinz Nixdorf Recall Study. Recruitment of the patients from Munich was partially supported by GlaxoSmithKline. We are grateful to the Genetics Research Centre GmbH, an initiative by GlaxoSmithKline and LMU. The Northern Finland Birth Cohort 1966 (NFBC66) is thanked for providing population controls for the study. The genotyping of NFBC66 was financially supported by National Institutes of Health grant 1R01HL087679-01, STAMPEED.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information Reprints and permissions information is available at www.nature.com/reprints.
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/nature.
References
- 1.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mefford HC, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunetti-Pierri N, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nature Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The International Schizophrenia Consortium Common polygenetic variation contributes to risk of schizophrenia and bipolar disorder. Nature. (this issue) doi: 10.1038/nature08185. doi:10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi J, et al. Common variants on chromosomes 6p22.1 are associated with schizophrenia. Nature. (this issue) doi: 10.1038/nature08192. doi:10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kong A, et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nature Genet. 2008;40:1068–1075. doi: 10.1038/ng.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li T, et al. Transmission disequilibrium analysis of HLA class II DRB1, DQA1, DQB1 and DPB1 polymorphisms in schizophrenia using family trios from a Han Chinese population. Schizophr. Res. 2001;49:73–78. doi: 10.1016/s0920-9964(00)00111-0. [DOI] [PubMed] [Google Scholar]
- 9.The Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Heel DA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nature Genet. 2007;39:827–829. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harley JB, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nature Genet. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eaton WW, et al. Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am. J. Psychiatry. 2006;163:521–528. doi: 10.1176/appi.ajp.163.3.521. [DOI] [PubMed] [Google Scholar]
- 13.Juvonen H, et al. Incidence of schizophrenia in a nationwide cohort of patients with type 1 diabetes mellitus. Arch. Gen. Psychiatry. 2007;64:894–899. doi: 10.1001/archpsyc.64.8.894. [DOI] [PubMed] [Google Scholar]
- 14.Lewis CM, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am. J. Hum. Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruano D, et al. Association of the gene encoding neurogranin with schizophrenia in males. J. Psychiatr. Res. 2008;42:125–133. doi: 10.1016/j.jpsychires.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Broadbelt K, Ramprasaud A, Jones LB. Evidence of altered neurogranin immunoreactivity in areas 9 and 32 of schizophrenic prefrontal cortex. Schizophr. Res. 2006;87:6–14. doi: 10.1016/j.schres.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 17.Bernal J, Rodriguez-Pena A, Iniguez MA, Ibarrola N, Munoz A. Influence of thyroid hormone on brain gene expression. Acta Med. Austriaca. 1992;19(suppl. 1):32–35. [PubMed] [Google Scholar]
- 18.Huang FL, Huang KP, Boucheron C. Long-term enrichment enhances the cognitive behavior of the aging neurogranin null mice without affecting their hippocampal LTP. Learn. Mem. 2007;14:512–519. doi: 10.1101/lm.636107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Pak JH, Huang FL, Huang KP. N-methyl-D-aspartate induces neurogranin/RC3 oxidation in rat brain slices. J. Biol. Chem. 1999;274:1294–1300. doi: 10.1074/jbc.274.3.1294. [DOI] [PubMed] [Google Scholar]
- 20.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 21.Toulopoulou T, et al. Substantial genetic overlap between neurocognition and schizophrenia: genetic modeling in twin samples. Arch. Gen. Psychiatry. 2007;64:1348–1355. doi: 10.1001/archpsyc.64.12.1348. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, et al. CaMKII activation state underlies synaptic labile phase of LTP and short-term memory formation. Curr. Biol. 2008;18:1546–1554. doi: 10.1016/j.cub.2008.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flora A, Garcia JJ, Thaller C, Zoghbi HY. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc. Natl Acad. Sci. USA. 2007;104:15382–15387. doi: 10.1073/pnas.0707456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitt D, Hopkins I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust. Paediatr. J. 1978;14:182–184. doi: 10.1111/jpc.1978.14.3.182. [DOI] [PubMed] [Google Scholar]
- 25.Kalscheuer VM, et al. Disruption of the TCF4 gene in a girl with mental retardation but without the classical Pitt-Hopkins syndrome. Am. J. Med. Genet. A. 2008;146A:2053–2059. doi: 10.1002/ajmg.a.32419. [DOI] [PubMed] [Google Scholar]
- 26.Gretarsdottir S, et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nature Genet. 2003;35:131–138. doi: 10.1038/ng1245. [DOI] [PubMed] [Google Scholar]
- 27.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 28.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl Cancer Inst. 1959;22:719–748. [PubMed] [Google Scholar]
- 29.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.