

Abstract
Exposure to environmental toxicants has been implicated as one of the causative factors for infertility in mammals. The objective of this study was to determine the amount of ingested benzo[a]pyrene (BaP), an environmental toxicant that reaches the reproductive tissues (internal dose) subsequent to a single acute exposure. Toward this end, the concentrations of BaP reactive metabolites and BaP–DNA adducts were measured throughout the course of BaP’s residence in the body. Ten-week-old female Fischer-344 rats weighing approximately 220 g were administered 5 mg BaP/kg body weight orally. 1, 7, 14, 2,1 and 28 d post BaP exposure, BaP parent compound and metabolites from plasma, ovaries, and liver tissues were extracted using liquid–liquid extraction. The extracts were analyzed by reverse-phase highperformance liquid chromatography (HPLC). DNA was isolated and analyzed for BaP-induced DNA adducts by 32P-postlabeling method. The BaP total metabolite concentrations in plasma, ovaries, and liver showed a gradual decrease from d 1 to 28 post BaP administration. The BaP–DNA adducts concentrations in ovaries and liver tissues from the treatment group demonstrated a trend similar to that observed for metabolites. Ovaries showed greater concentrations of DNA adducts compared to liver. However, with an increase in time post cessation of exposure, the adduct concentrations in liver tissue started declining rapidly, from d 1 to 28. For ovaries, the adduct concentrations demonstrated a significant decline from d 1 to 7 and a gradual fall thereafter. A concordance between BaP reactive metabolite levels and adduct concentrations indicates that the bioavailability of reactive metabolites determines the binding with DNA and consequently the formation and persistence of adducts in an acute exposure regimen.
The complex process of reproduction in mammals is highly susceptible to environmental toxicants, one of a group of legacies of industrial revolution. One of the environmental toxicants that perturbs human health is benzo[a]pyrene (BaP), a ubiquitous polycyclic aromatic hydrocarbon (PAH) compound, resulting from incomplete combustion of carbonaceous materials. Benzo[a]pyrene exists in a number of environmental products, such as soot, asphalt, tobacco smoke, petroleum, air pollutants, and cutting oils, as well as contaminated foods and drinking water. Benzo[a]pyrene therefore makes its way into the body of animals including humans mainly via inhalation of BaP-laden particulates, or orally, through various food chains (IPCS, 1998; Ramesh et al., 2004a). Several reports indicate that BaP exposure is associated with adverse health effects in humans (Melikian et al., 1999; Perera et al., 1998, 2006; Singh et al., 2007; Zanieri et al., 2007). In exposed mammals, BaP becomes activated in organs, including the ovaries, to electrophilic metabolites (Archibong et al., 2002, Harris et al., 2009) that bind covalently to nucleophilic sites of cellular macro-molecules such as DNA and may interfere with the above-mentioned gonadal function, resulting in ovarian toxicity (compromising function and viability of oocyte–granulosa cell complexes in the follicles; Zenzes et al., 1998).
In addition to biotransformation of BaP to reactive metabolites, of importance is the dose of BaP administered, as it plays an important role in the toxicity and risk assessment of this chemical. In this scenario, it is important to emphasize that toxicity is dependent not on dose administered but on the amount that actually reaches the target site where adverse effects occur, referred to as the “internal dose.” This dose may represent only a fraction of the “external dose” (Hrudey et al., 1996; Paustenbach, 2000). Another way of estimating the target tissue dose is by measuring the total delivered amount of biologically active toxicant/metabolites. This dose is termed the “biologically effective dose” (Godschalk et al., 2000).
Therefore, the aim of this study was to assess the dose of BaP reaching the target organ following oral exposure to BaP. Our earlier studies in somatic and reproductive tissues of animal models showed that BaP undergoes rapid metabolism and metabolites were quantifiable (Ramesh et al., 2001, 2002, 2008). Another objective of this study was to examine the relationship that may exist among bioavailable dose and kinetics of disposition of administered BaP in plasma and reproductive tissues. The rationale for undertaking this research is that tissue DNA acts as the internal trapping agent for reactive metabolites (Ginsberg & Atherholt, 1989; Garg et al., 1993) produced in liver and reproductive tissues postexposure. Since BaP toxicity to reproductive system is the prime focus of our research, studies on kinetics of disposition of reactive metabolites and DNA adducts will provide insight into the amount of ingested BaP reaching the target ovaries throughout the course of its residence in the body to elicit adverse effects.
MATERIAL AND METHODS
Animals and Exposure
Ten-week-old female F-344 rats weighing approximately 220 g were purchased from Harlan Laboratory, Indianapolis, IN, and allowed a 7-d acclimation period prior to the initiation of experiments. Animals were housed in a controlled environment (21 ± 2°C; humidity 50–60%) in groups of 3 per cage maintained on a 12/12-h light/dark cycle (lights on at 0600 h) and were allowed free access to rat chow (5001 Lab Meal; Purina Ralston Co.) and water. Subsequently, rats were randomly assigned to a treatment (n = 8 per time point) or control (n = 8 per time point) group. The care and husbandry of rats used in this study were in conformity with the guidelines (ILAR, 1996) that regulate the humane care and use of laboratory animals for research.
Treatment consisted of 5 mg BaP /kg body weight (97% pure, unlabeled; Sigma Chemical Co., St.Louis, MO) dissolved in tricaprilyn (vehicle; Sigma) and administered through a single oral gavage. Control rats were administered an equivalent volume of vehicle as described for rats in the treatment group.
Sample Collection and HPLC Analysis for BaP Parent Compound and Metabolites
Treated and control rats were sacrificed by decapitation following administration of a combination of ketamine and xylazine anesthetics on d 1, 7, 14, 21, or 28 postexposure. Blood samples collected for plasma extraction, ovaries, and liver were excised at each time point (8 rats/time point) and stored frozen at −70°C until analyzed. Plasma from blood samples were harvested when centrifugation at 2900 × g for 25 min and stored frozen at −70°C until analyzed.
The BaP parent compound and metabolites were resolved by a high-performance liquid chromatograph (HPLC), model 1050 (Agilent Technologies, Wilmington, DE), equipped with an HP1046 fluorescence detector. The chromatograph was operated through a 2D ChemStation (Agilent Technologies) for instrument control, data acquisition and analyses. Fifty microliters of each sample were injected onto a C18 reverse-phase column (ODS Hypersil, 5 μm, 200 × 4.6 mm; Agilent Technologies). The column (temperature 33°C) was eluted for 45 min at a flow rate of 1 ml/min with a ternary gradient of water:methanol:ethanol (40:40:20%) for 20 min, followed by the same gradient at a ratio of 30:46:24 for 10 min, 100% methanol for 10 min, and returning to the initial gradient of 40:40:20 for 5 min. The excitation and emission wavelengths for the detector were 244 and 410 nm, respectively. Benzo[a]pyrene metabolite standards were obtained from the National Cancer Institute Chemical Carcinogen Repository (Midwest Research Institute, Kansas City, MO). As BaP and its metabolite standards are potential carcinogens/mutagens, they were handled in accordance with National Institutes of Health (NIH) guidelines (NIH, 1981). Identification of the metabolites was accomplished by comparison of retention times and peak areas of the samples with that of standards.
The conjugated metabolites were eluted via a gradient of increasing methanol in ammonium formate buffer (AFB; containing ammonium formate at 0.04 M, tetrabutyl ammonium bromide [TBAB; 0.04 M, pH 6.4; 80%], and water [20%]) for 20 min, followed by the same gradient at a ratio of 50:50 for 45 min, 30:70 for 15 min, and returning to the initial gradient of 80:20 for 15 min. The column (temparature 33°C) was eluted for 60 min at a flow rate of 1 ml/min. The excitation and emission wave-lengths were 241 and 389 nm, respectively. The water-soluble metabolites were analyzed by enzymatic hydrolysis as described below.
One-milliliter fractions of the HPLC eluate were collected and divided into equal aliquots, concentrated in a Speed-vac, and incubated with β-glucuronidase, arylsulfatase, and γ -glutamyltransferase (Sigma Chemical Co., St. Louis, MO) according to Merrick and Selkirk (1985). After incubation, each sample was extracted with an equal volume of ethyl acetate. Each ethyl acetate extract containing the released deconjugated products was concentrated under N2 gas, reconstituted in methanol, and analyzed by reversephase HPLC as outlined for organic soluble metabolites.
DNA Isolation
DNA was isolated from ovaries and liver of rats exposed to BaP. DNA isolation was performed by using the Stratagene DNA isolation kit and subjected to 32P-postlabeling. The labeled adducts were separated and quantified by thin-layer chromatography (TLC) as described later.
32P-Postlabeling
The methods of Gupta (1985) and Gupta and Randerath (1988) were used for analysis of DNA adducts, which is briefly described as follows. DNA (5 μg) from ovaries and liver was digested to 3′-dNPs (deoxyribonucleoside monophosphates) at 37°C for 2 h with 5 μg each of micrococcal nuclease and spleen phosphodiesterase. The nucleotides were enriched by adding 4 μg/μl P1 nuclease to the digest and incubated at 37°C for 60 min. The DNA digests were diluted to 50 μl (to yield 0.14 μg/μl DNA) with water. The digests were then 32P-postlabeled by incubation with 2.3 μl [γ -32P]-ATP (250 μCi/μl; Perkin Elmer, Inc., Waltham, MA) and 1.2 μl T4 polynucleotide kinase (3 U/μl) at 37°C for 30 min. Potato apyrase (20 mU/μl) was added to destroy residual [γ -32P]-ATP and the mixture was incubated at 37 °C for another 10 min.
Thin-Layer Chromatography (TLC)
The postlabeled samples were separated and quantified as previously described by Ramesh and Knuckles (2006). Briefly, samples (each 1 μl) were applied to 20 cm × 20 cm polyethylenimine-cellulose thin-layer chromatography (TLC) plates (Machery Nagel, Germany). A paper wick was stapled to the top of the TLC plates. The plates were developed overnight in 1 M NaH2PO4, pH 6. The plates were washed twice with deionized water, dried, and developed in 3.5 M lithium formate and 7 M urea, pH 3.5, from the bottom to the top of the plate. The plates were again washed in water, air-dried, and developed at a right angle to the previous direction of development in 0.8 M lithium chloride, 0.5 M Tris-HCl, and 7 M urea, pH 8. The plates were not washed, but air-dried and developed in the first direction with 1.7 M NaH2PO4, pH 6. The plates were again washed in water, dried, and wrapped in Saran wrap. The adducts were detected by autoradiography at −80°C using Kodak XAR-5 film (Sigma Chemicals, MO) and a Fisher intensifying screen as an image enhancer. After the end of exposure, the adduct spots corresponding to the autoradiograms were excised and counted in a scintillation counter (Beckman Coulter Instruments, Brea, CA). Areas adjacent to the adduct spots were counted in the same way and the background radioactivity was subtracted from the sample counts. The total nucleotides were analyzed by a diluted DNA digest (2 ng) in parallel with adducts. Subsequently, the normal nucleotides were separated by PEI-cellulose TLC in 0.3 M lithium chloride, pH 8. Adduct levels were calculated by relative adduct labeling and represented as femtomoles per microgram DNA.
Identification of BaP–DNA Adducts
The BaP metabolites, especially BaP 7,8-diol 9,10-epoxide, BaP 3,6-dione, and BaP 7,8-dione were incubated with 40 μM DNA and subjected to cochromatography with unknown adduct samples. As mentioned previously, a multidimensional TLC system was used. Those unknown adducts that exhibited equivalent mobility (comigration) with that of known standard were mapped and identified according to Walker et al. (1992).
To identify whether the adducts were those of deoxyadenosine or deoxyguanosine, solutions of respective nucleotides (40 μM each of 3′-deoxyadenosine monophosphate [dAMP], and 3′-deoxyguanosine monophosphate [dGMP]) were mixed with equal volumes of each one of the metabolite standards for 12 h at 37°C. The modified nucleotides were subjected to 32P-postlabeling analysis and run in parallel with unknown adduct samples.
Statistical Analyses of Data
Data on total metabolite concentrations in plasma, ovaries, or liver for each time point and BaP–DNA adducts concentrations were examined by one-way analysis of variance (ANOVA) and the differences among means were determined by using Bonferroni’s post hoc test. The criterion for statistical significance was set at p < 0.05.
RESULTS
For the sake of clarity and consistency the following terms are used through the rest of the article: The term “BaP organic metabolites” denotes the BaP metabolites that remain in the organic phase after liquid–liquid extraction of plasma/tissue samples. The term “BaP aqueous metabolites” denotes the BaP metabolites that remain in the aqueous phase after sample extraction.
Peak concentrations (ng/ml plasma or ng/g tissue wet weight) of unmetabolized BaP (BaP parent compound) in plasma, liver, and ovaries occurred 24 h post BaP exposure (plasma, 44 ± 2.2 ng/ml; liver, 31 ± 2.3; and ovary, 19 ± 1.5 ng/g tissue) followed by detectable low levels on d 7 (plasma, 18 ± 1.5 ng/ml; liver, 10 ± 1.3; and ovary, 5 ± 0.8 ng/g tissue); thereafter unmetabolized BaP was not detectable during the remaining time points studied.
The BaP organic metabolite concentrations in plasma, ovaries and liver on d 1, 7, 14, 21, and 28 post BaP exposure are shown in Figure 1A. The metabolite concentrations in plasma, ovaries, and liver peaked on d 1post BaP administration and decreased gradually thereafter. For all three matrices, the metabolite concentrations were statistically lower on d 14, 21, and 28 compared to d 1 post BaP exposure.
FIGURE 1.
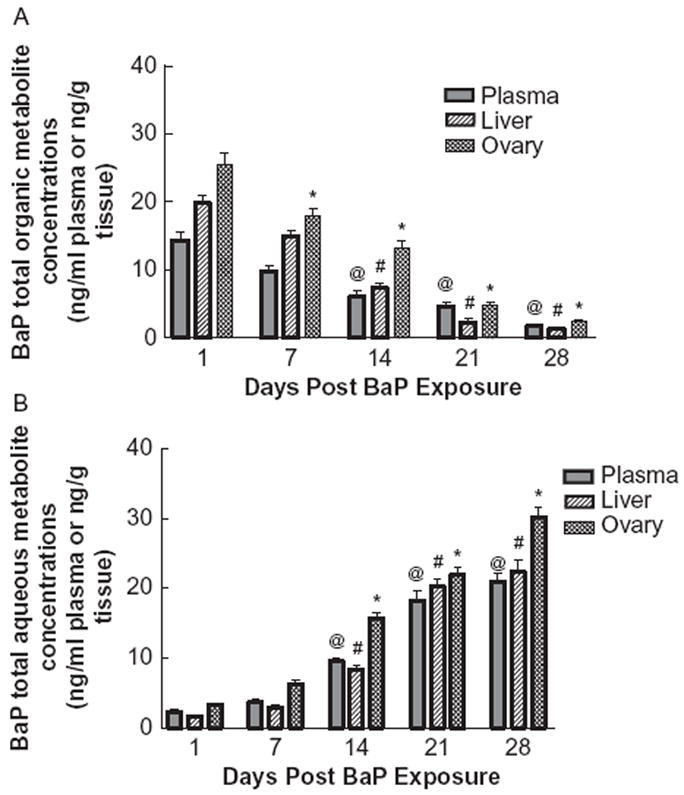
Time-course distribution of (A) total BaP organic metabolites and (B) aqueous metabolites in plasma, ovaries, and liver of F-344 rats that received BaP via oral gavage. Values represent mean ± SE (n = 6). The symbols (@, #, and *) denote statistical significance (p < .05) in metabolite concentrations in plasma or liver or ovaries at the respective time point compared to d 1 postexposure.
The BaP aqueous metabolite concentrations in plasma, ovaries, and liver are shown in Figure 1B. The aqueous metabolite concentrations showed a trend similar to that of BaP organic metabolites on d 1 and 7 post BaP exposure, but the concentrations increased on d 14, 21, and 28 postexposure. For all the three matrices, the metabolite concentrations were statistically higher on d 14, 21, and 28 compared to d 1 post BaP exposure.
An inverse relationship was observed between the organic and aqueous metabolites of BaP. As the postexposure time increased, a progressive decline in BaP organic metabolite concentrations was observed, while the reverse was true for postexposure BaP aqueous metabolite concentrations.
The concentrations of the individual BaP organic metabolites are shown in Figure 2, A, B, and C. The BaP metabolites identified in plasma, ovaries, and liver were BaP 9,10-diol, BaP 4,5-diol, BaP 7,8-diol, 3-hydroxy- and 9-hydroxy-BaP, and BaP 3,6- and 6,12-quinones. The concentrations of the individual BaP aqueous metabolites are shown in Figure 3, A, B, and C. Among the groups of aqueous metabolites, the glucuronide conjugate concentrations were higher relative to those of sulfates and glutathione conjugates. The pre-dominant BaP metabolites identified in the aqueous fraction were BaP 4,5-diol; 9,10-diol glucuronides; 3(OH) and 9(OH) sulfates; and 7,8- and 9,10-diol glutathione conjugates.
FIGURE 2.
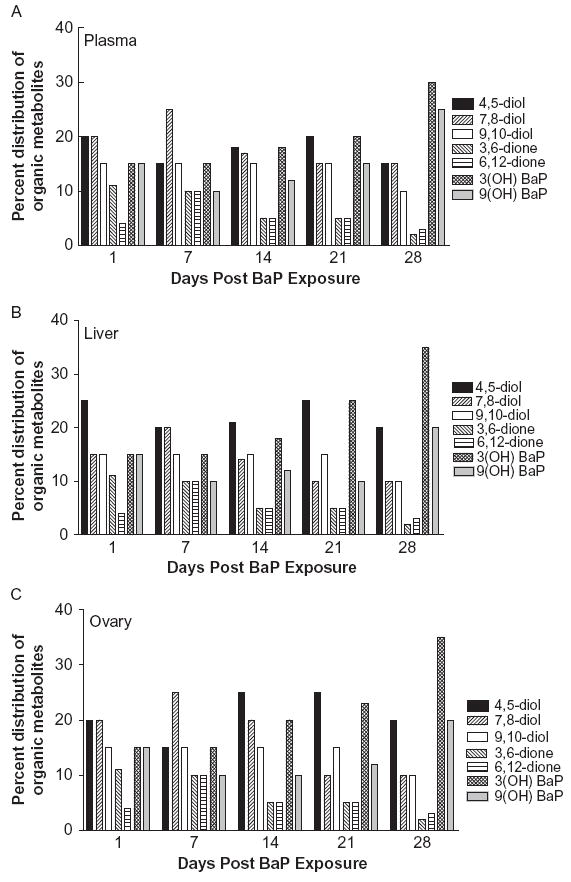
Distribution (%) of individual BaP organic metabolite types in plasma (A), ovaries (B), and liver (C) of F-344 rats that received BaP via oral gavage.
FIGURE 3.
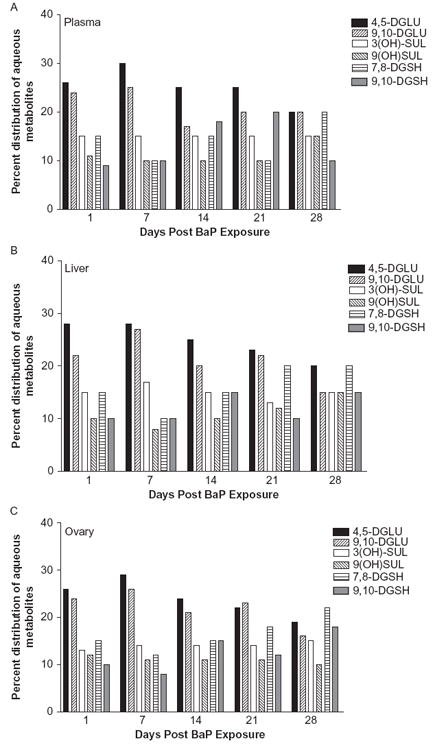
Distribution (%) of individual BaP aqueous metabolite types in plasma (A), ovaries (B), and liver (C) of F-344 rats that received BaP via oral gavage.
No BaP–DNA adducts were detected in ovaries or liver of control animals. BaP–DNA adducts in ovaries and liver were determined on 1, 7, 14, 21, and 28 d post BaP exposure (Figure 4). The ovarian tissues showed greater concentrations of DNA adducts compared to liver tissues. However, with increasing periods post cessation of exposure, the adduct levels in ovaries and liver tissue declined rapidly, from d 1 to 28. The BaP–DNA adduct concentrations were statistically lower on d 7, 14, 21, and 28 compared to d 1 post BaP exposure.
FIGURE 4.
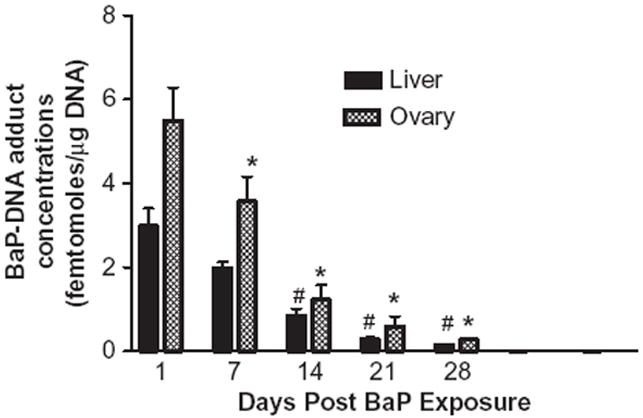
Time-course distribution (persistence) of BaP–DNA adducts in ovaries and liver of F-344 rats that received BaP via oral gavage. DNA was extracted from these samples, BaP-derived adducts were 32P-postlabeled, and TLC was performed as described in the Materials and Methods section. Films were exposed for 24 h at −80° C. Values represent mean ± SE (n = 6). The symbols (# and *) denote statistical significance (p < .05) in adduct concentrations in liver or ovaries at the respective time point compared to d 1 postexposure.
The relative distribution of BaP-DNA adduct types in the ovaries is shown in Figure 5. Data for adduct types in liver are not shown, as adduct types were similar to that observed for ovaries. The proportiosn of deoxyguanosine (dG) adducts were higher (p < 0.05) than deoxyadenosine adducts in both ovaries and liver. Some of the adducts could not be identified, and those adducts were at lesser proportions.
FIGURE 5.
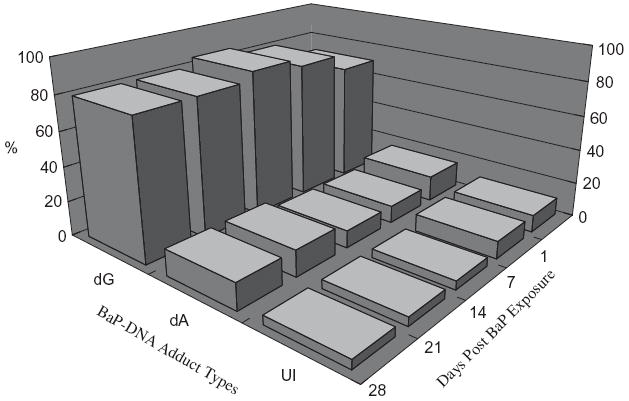
Relative distribution (%) of BaP–DNA adduct types in ovaries of F-344 rats exposed to benzo[a]pyrene. dA, deoxyadenosine adducts; dG, deoxyguanosine adducts; UI, unidentified adducts. Chi-square tests revealed a statistically significant difference (p < 0.05) between dG and dA adduct types. The dA and dG adducts were identified on the basis of comigration studies as described in the Materials and Methods section.
DISCUSSION
The BaP dose used in this study was chosen on the basis of published reports. For example, diet-related BaP intake by humans ranges from 8.4 μg/person/d (Falco et al., 2003) to 17 μg/person/d (deVos et al. 1990). For individuals who smoke a pack of cigarettes per day, additional intake of 0.1 μg/d is expected, as mainstream smoke yield of BaP per cigarette amounts to 10 ng/cigarette (Grimmer et al., 1987). Further, other occupations (industrial workers, 1.4–25 ng/m3 [Aries et al., 2008]; restaurant cooks, 6.9 ng/m3 [Pan et al., 2008]), and lifestyle habits (individuals who patronize pubs and taverns are exposed to BaP via secondhand cigarette smoke; Bolte et al., 2008) contribute to additional BaP intake. Furthermore, subjects living in the vicinity of hazardous waste sites and in unvented homes using biomass for cooking and home heating are at a risk of getting exposed to BaP. Thus, cumulative annual intake of BaP by some susceptible populations may equal the dose used in this study.
Though the lipophilic nature of BaP (Barhoumi et al., 2000) probably facilitates its absorption through the gastrointestinal tract (GIT), the detection of unchanged BaP in plasma, liver, and ovaries suggests that at 5 mg/kg dose, BaP may have been incompletely extracted by the GIT fluids, resulting in a capacity-limited absorption and biotransformation (Laher et al., 1984), contributing to unmetabolized BaP. The formation of BaP–plasma lipoprotein complex and slow metabolism of the lipoprotein molecules (Shu & Bymun, 1983) may have contributed unmetabolized BaP to cells of peripheral tissues and subsequently to the vascular circulation. Studies conducted by Ramesh et al. (2001) also documented the presence of BaP parent compound in target tissues, 3 d after oral administration of 100 mg BaP/kg to rats. A recent study involving the ability of carrier lipoproteins tomodulate the bioavailability of BaP lends credence to our postulation of a slow release of BaP from lipid-rich tissues and the likely uptake by the ovary. Carrier lipoproteins were demonstrated to react with BaP and sequester this toxicant in blood at high doses (Grova et al., 2009). The sequestered BaP was found to be redistributed and repartitioned into target tissues, altering the bioavailability of this chemical.
The detection of some unmetabolized BaP in plasma and tissues notwithstanding, a substantial portion of ingested BaP appears to have undergone metabolism. The high concentrations of BaP organic metabolites in liver relative to plasma and ovary are consistent with the report of Wall et al. (1991) that liver is the principal organ of metabolism for PAH.
Information on the composition of metabolites that arise from bioactivation of BaP is relevant to understanding the causal factors involved in toxicity. Initial oxidation of BaP catalyzed by the cytochrome P-450 (CYP450) family of enzymes (CYP1A1, CYP1A2, and CYP1B1) yields arene oxides (9-OH-BaP, 7-OH-BaP, 6-OH-BaP, 3-OH-BaP, and 1-OH-BaP). These arene oxides rearrange to phenols or undergo hydration catalyzed by epoxide hydrolase (EH), generating BaP 9,10-diol; BaP 7,8-diol; and BaP 4,5-diol (Ramesh et al., 2004a; Shimada & Guengerich, 2006). Of the drug-metabolizing enzymes that contribute to differential susceptibilities to the adverse effects of BaP, the CYP1A1 is not constitutively expressed and CYP1A2 is mostly hepatic (Guengerich, 1997). Therefore, the biotransformation of BaP in ovary may have been the result of CYP1B1, which was implicated in metabolic activation, immunotoxicity, and adduct formation in target tissues (Nebert et al., 2004). This enzyme was reported to be constitutively expressed in ovarian tissues (Otto et al., 1992).
The already-mentioned metabolites (epoxides, hydroxy metabolites, and dihydrodiols) represent phase I metabolites of BaP. Phase I metabolism introduces more polar chemical groups such as hydroxy moities into the molecule. As a result, the molecule becomes more electrophilic, leading to increased reactivity. A good example of electrophilic metabolite formation is BaP 7,8-oxide, which is inactivated by microsomal epoxide hydrolase to 7,8-dihydrodiol. The 7,8-dihydrodiol serves as a substrate for a second monooxygenation step, which introduces a further epoxide moiety leading to the formation of a dihydrodiol bay-region epoxide, termed the BaP 7,8-diol 9,10-epoxide (BPDE; Oesch, 1987). Some of the precursors of BaP reactive metabolites such as the 3-hydroxy BaP and 7,8-diol are lipophilic, resulting in an increase in uptake by plasma lipoproteins (Shu & Nichols, 1981), transportation through blood, and cellular internalization (Busbee et al., 1990). The phase I metabolites are conjugated with glutathione, sulfate or glucuronic acid to form phase II metabolites. The phase II metabolites (4,5-diol glucuronide; 9,10-diol glucuronide; 3(OH) glucuronide; 3(OH) sulfate; 9(OH) sulfate; 7,8- diol GSH; 9,10-diol GSH) are more hydrophilic and hence amenable to elimination through excretion (Ramesh et al. 2001). The phase II metabolism is considered a detoxification reaction. However, the activation of some of the phase II metabolites cannot be ruled out because of alterations in electrophilicity as BaP undergoes simultaneous and stereoselective metabolic transformations like many chemical carcinogens (Oesch, 1987).
There were no remarkable differences among plasma, hepatic, and ovarian tissues in the BaP metabolite types formed, which indicates regular partitioning of BaP metabolites among the mentioned samples. All these samples produced considerably higher proportion of BaP 4, 5-diol and 7, 8-diol at earlier time points. However, these samples also generated a greater proportion of 3- and 9-hydroxy BaP. The shift in metabolite predominance (proportion of different metabolite types among total metabolites) over time, from diols to hydroxy metabolites, may be attributed to the lipophilicity of BaP metabolites. Shu and Nichols (1981) demonstrated that the extent of uptake by lipoproteins was more for 3-hydroxy BaP than 7,8-diol. The sequestration of hydroxy metabolites by lipoproteins during d 1 through 7 post BaP exposure and slow release of these metabolitesmay have contributed to the predominance of these metabolites during d 14 through 28 post BaP exposure. However, experimental evidence to support this hypothesis is at present not available. Overall, the pharmacokinetic behavior of individual BaP metabolite types, with their stability, lipophilicity, and uptake by tissues, may contribute to the temporal shift in their concentrations.
Our results on composition of BaP aqueous metabolites revealed a greater production of glucuronides relative to that of sulfate and glutathione conjugates in plasma, liver, and ovary. These observations are in accord with the published literature (Zheng et al., 2002; Hu & Wells, 2004; Girard et al., 2008), which suggests that glucuronidation is one of the important pathways for the detoxification of BaP, thereby avoiding alternative bioactivation to potent intermediates.
One of the mechanisms through which PAHs produce toxicity/cancer is through binding with cellular macromolecules such as proteins, and nucleic acids (Ramesh & Knuckles, 2006). Thus, measurement of BaP–DNA adducts from extracted liver and ovaries may provide a measure of the biologically effective dose of BaP. The relationship between BaP disposition and tissue damage was further examined by measuring the concentrations of DNA adducts formed by binding of BaP metabolites with DNA. A progressive decline in the adduct concentrations with increasing periods post BaP exposure is suggestive of an innate adaptive process to cope with toxicant exposure. The temporal variations in adduct concentrations might be due to enhanced detoxification by trapping metabolites of BaP, antioxidant defenses, and DNA repair processes.
The increased concentrations of DNA adducts in the ovary relative to that of liver suggest that this organ is more vulnerable to damage by BaP. The variation in adduct persistence in rat tissues at various time points post BaP exposure may have been influenced by the rate of adduct formation. The rate-limiting step for adduct formation is governed by the metabolic activation/biotransformation of BaP to electrophilic reactive metabolite BPDE (Suh et al. 1995) and BPDE binding to DNA in the vulnerable tissues (Boerrigter et al., 1995). Besides local production of BPDE metabolites in these tissues, the transport of BPDE metabolites through carrier proteins contributes to the binding of BPDE with DNA and adducts formation in metabolically quiescent tissues. Using exogenous (salmon sperm) DNA, Garg et al. (1993) demonstrated that reactive metabolites of BaP in serum could be intercepted by serum albumin to form adducts. Thus, if the pool of reactive metabolites is low in rats, a corresponding decrease could be expected in the transport of these metabolites through circulation to extrahepatic tissues where adduct formation occur.
The detection of BaP–DNA adducts in ovaries 28 d post BaP exposure, albeit at low concentrations, indicates the propensity of this lipophilic chemical to reside in target tissues and undergo biotranformation at a slow pace at high doses. Such delayed clearances for BaP metabolites and DNA adducts (persisting up to 60 d post BaP exposures) from extrahepatic tissues were also recorded in AhR wild-type mice treated orally with a single dose of 100 mg/kg BaP (Sagredo et al., 2009). A single oral dose of 10 mg/kg BaP generated BaP–DNA adduct concentrations in extrahepatic tissues of Lewis rats that were sustained for 20 d post BaP exposure (Godschalk et al., 2000). Similarly, a single oral exposure to 13 mg/kg BaP in C57BL/6J mice showed persistence of BaP–DNA adducts in lung and reproductive tissues of mice 40 d post BaP exposure (Verhofstad et al., 2010). The preponderance of dG adducts relative to those of dA are consistent with the results of studies conducted in our laboratory (Ramesh et al., 2004b; Ramesh & Knuckles, 2006) and those of others (Pelling et al., 1984; Ross et al., 1990; Arif et al., 1999).
That a sustained BaP metabolite load was registered in the ovaries even after a single acute exposure to BaP is significant from the standpoint of toxicity. Benzo[a]pyrene metabolites were found to inhibit follicular growth (Neal et al., 2007) and produce apoptosis (Mattison et al., 1989) leading to ovarian follicular atresia (Hsueh et al., 1994; Mann et al., 1999; Tuttle et al., 2009) and ovotoxicity (Borman et al., 2000). In vitro studies showed that BaP/metabolites were estrogen antagonists (Arcaro et al., 1999). Furthermore, in vivo studies conducted in our laboratory revealed that subacute exposure to BaP resulted in antiestrogenic activities (Archibong et al., 2002) that are driven by BaP metabolism. If a single acute exposure to a high dose of BaP could sustain reactive metabolites and adducts up to 1 mo after exposure, the likelihood of the damage produced by this toxicant in subacute and subchronic exposures is of concern. The possibility of sequestration of BaP in high-density lipoproteins (Polyakov et al., 1996) that are essential for steroid hormone biosynthesis in the ovary (Jefcoate et al., 2000) upon prolonged exposure to BaP cannot be ruled out. This situation may lead to altered secretion of gonadotropins such as follicle-stimulating hormone (FSH) and luteinizing hormone (LH) at proestrus in mammalian species and at the ovulatory phase of the menstrual cycle of women, with adverse outcomes in the final stages of follicular development.
In addition to toxicity, the disposition of BaP metabolites in female reproductive tissues has implications from the perspective of damage to cellular macromolecules such as DNA and contributes to carcinogenicity. Evidence subscribing to this viewpoint is furnished by studies of Zenzes et al. (1998), who reported the presence of BaP–DNA adducts in ovarian granulosa cells of women exposed to cigarette smoke. Apart from the ovary, BaP metabolites were also reported to exert their inhibitory effects on human cervical cells by enhancing cell death (Rorke et al., 1998) and BaP–DNA adduct formation (Melikian et al., 1999a, 1999b).
The findings of this study suggest that (a) BaP is sufficiently bioavailable in plasma and target tissues, (b) BaP reactive metabolites accumulate in plasma, ovaries and liver, (c) the extent of BaP–DNA adduct formation depends on BaP metabolism, and (d) distribution and persistence of BaP metabolites and adducts in the target tissue are governed by the duration post BaP exposure. The dynamics of disposition of BaP/metabolites and DNA adducts are likely to be significantly altered in a subchronic exposure regimen compared to an acute regimen. Studies are in progress in our laboratory to assess the effects of dose and duration of exposure to BaP on cellular macromolecules and how the metabolic fate of BaP modulates reproductive toxicity.
Acknowledgments
This publication was made possible by grants G12 RR03032 from the National Center for Research Resources (NCRR), 5 S11 ES014156 02 from the National Institute of Environmental Health Sciences (NIEHS), 5 U54 HD044315 05 from the National Institute of Child Health and Human Development (NICHD), and 1R01CA142845-01A1 from the National Cancer Institute (NCI), all, of which are components of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
References
- Arcaro KF, O’Keefe PW, Yang Y, Clayton W, Gierthy JF. Antiestrogenicity of environmental polycyclic aromatic hydrocarbons in human breast cancer cells. Toxicology. 1999;133:115–127. doi: 10.1016/s0300-483x(99)00018-9. [DOI] [PubMed] [Google Scholar]
- Archibong AE, Inyang F, Ramesh A, Greenwood M, Nayyar T, Kopsombut P, Hood DB, Nyanda AM. Alteration of pregnancy related hormones and fetal survival in F-344 rats exposed by inhalation to benzo(a)pyrene. Reprod Toxicol. 2002;16:801–808. doi: 10.1016/s0890-6238(02)00058-8. [DOI] [PubMed] [Google Scholar]
- Aries E, Anderson DR, Fischer R. Exposure assessment of workers to airborne PCDD/Fs, PCBs and PAHs at an electric arc furnace steelmaking plant in the United Kingdom. Ann Occup Hyg. 2008;52:213–225. doi: 10.1093/annhyg/men011. [DOI] [PubMed] [Google Scholar]
- Arif JM, Shappell N, Sikka HC, Kumar S, Gupta RC. 32P-postlabeling analysis of lipophilic DNA adducts resulting from interaction with (+/-)-3-hydroxy-trans-7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo(a)pyrene. Chem Biol Interact. 1999;118:87–97. doi: 10.1016/s0009-2797(98)00116-1. [DOI] [PubMed] [Google Scholar]
- Barhoumi R, Mouneimne Y, Ramos KS, Safe SH, Phillips TD, Centonze VE, Ainley C, Gupta MS, Burghardt RC. Analysis of benzo[a]pyrene partitioning and cellular homeostasis in a rat liver cell line. Toxicol Sci. 2000;53:264–270. doi: 10.1093/toxsci/53.2.264. [DOI] [PubMed] [Google Scholar]
- Boerrigter METI, Wei JY, Vijg J. Induction and repair of benzo(a)pyrene-DNA adducts in C57BL/6 and BALB/c mice: Association with ageing and longevity. Mech Ageing Dev. 1995;82:31–50. doi: 10.1016/0047-6374(95)01603-w. [DOI] [PubMed] [Google Scholar]
- Bolte G, Heitmann D, Kiranoglu M, Schierl R, Diemer J, Koerner W, Fromme H. Exposure to environmental tobacco smoke in German restaurants, pubs and discotheques. J Expos Sci Environ Epidemiol. 2008;18:262–271. doi: 10.1038/sj.jes.7500590. [DOI] [PubMed] [Google Scholar]
- Borman SM, Christian PJ, Sipes IG, Hoyer PB. Ovotoxicity in female Fischer rats and B6 mice induced by low-dose exposure to three polycyclic aromatic hydrocarbons; Comparison through calculation of an ovotoxic index. Toxicol Appl Pharmacol. 2000;167:191–198. doi: 10.1006/taap.2000.9006. [DOI] [PubMed] [Google Scholar]
- Busbee DL, Norman JO, Ziprin RL. Comparative uptake, vascular transport, and cellular internalization of aflatoxin-B1 and benzo(a)pyrene. Arch Toxicol. 1990;64:285–290. doi: 10.1007/BF01972988. [DOI] [PubMed] [Google Scholar]
- de Vos RH, van Dokkum W, Schouten A, deJong-Berkhout P. Polycyc aromatic hydrocarbons in Dutch total diet samples (1984–1986) Food Chem Toxicol. 1990;28:263–268. doi: 10.1016/0278-6915(90)90038-o. [DOI] [PubMed] [Google Scholar]
- Falco G, Domingo JL, Llobet JM, Teixido C, Muller L. Polycyclic aromatic hydrocarbons in foods: Human exposure through the diet in Catalonia, Spain. J Food Prot. 2003;60:2325–2331. doi: 10.4315/0362-028x-66.12.2325. [DOI] [PubMed] [Google Scholar]
- Garg A, Beach AC, Gupta RC. Interception of reactive, DNA adduct-forming metabolites present in rodent serum following carcinogen exposure: Implications in biomonitoring. Teratogen Carcinogen Mutagen. 1993;13:151–166. doi: 10.1002/tcm.1770130402. [DOI] [PubMed] [Google Scholar]
- Ginsberg GL, Atherholt TB. Transport of DNA-adducting metabolites in mouse serum following benzo[a]pyrene administration. Carcinogenesis. 1989;10:673–679. doi: 10.1093/carcin/10.4.673. [DOI] [PubMed] [Google Scholar]
- Girard H, Butler LM, Villeneuve L, Millikan RC, Sinha R, Sandler RS, Guillemette C. UGT1A1 and UGT1A9 functional variants, meat intake, and colon cancer, among Caucasians and African Americans. Mutat Res. 2008;26:56–63. doi: 10.1016/j.mrfmmm.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godschalk RWL, Moonen EJC, Schilderman PAEL, Broekmans WMR, Kleinjans JCS, van Schooten FJ. Exposure-route-dependent DNA adduct formation by polycyclic aromatic hydrocarbons. Carcinogenesis. 2000;1:87–92. [PubMed] [Google Scholar]
- Grimmer G, Naujack KW, Dettbarn G. Gas chromatographic determination of polycyclic aromatic hydrocarbon, aza-arenes, aromatic amines in the particles and vapor phase of mainstream smoke of cigarettes. Toxicol Lett. 1987;35:117–124. doi: 10.1016/0378-4274(87)90095-6. [DOI] [PubMed] [Google Scholar]
- Grova N, Prodhomme EJF, Schellenberger MT, Farinelle S, Muller CP. Modulation of carcinogen bioavailability by immunization with benzo(a)pyrene-conjugate vaccines. Vaccine. 2009;27:4142–4151. doi: 10.1016/j.vaccine.2009.04.052. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Comparisons of catalytic selectivity of cytochrome P450 subfamily enzymes from different species. Chem Biol Interact. 1997;106:161–182. doi: 10.1016/s0009-2797(97)00068-9. [DOI] [PubMed] [Google Scholar]
- Gupta RC. Enhanced sensitivity of 32P-postlabeling analysis of aromatic carcinogen: DNA adducts. Cancer Res. 1985;45:5656–5662. [PubMed] [Google Scholar]
- Gupta RC, Randerath K. Analysis of DNA adducts by 32P labeling and thin layer chromatography. In: Friedberg EC, Hanawalt PC, editors. DNA repair. Vol. 3. New York: Marcel Dekker; 1988. pp. 399–418. [Google Scholar]
- Harris DL, Huderson AC, Niaz MS, Ford JJ, Archibong AE, Ramesh A. Comparative metabolism of benzo(a)pyrene by ovarian microsomes of various species. Environ Toxicol. 2009;24:603–609. doi: 10.1002/tox.20461. [DOI] [PubMed] [Google Scholar]
- Hrudey SE, Chen W, Rousseaux CG. Bioavailability in environmental risk assessment. Boca Raton, FL: CRC Press; 1996. [Google Scholar]
- Hsueh AJW, Billig H, Tsafriri A. Ovarian follicle atresia: A hormonally controlled apoptotic process. Endocr Rev. 1994;15:707–724. doi: 10.1210/edrv-15-6-707. [DOI] [PubMed] [Google Scholar]
- Hu Z, Wells PG. Human interindividual variation in lymphocyte UDP-glucuronosyltransferases as a determinant of in vitro benzo(a)pyrene covalent binding and cytotoxicity. Toxicol Sci. 2004;78:32–40. doi: 10.1093/toxsci/kfh010. [DOI] [PubMed] [Google Scholar]
- Institute for Laboratory Animal Research. Guide for the care and use of laboratory animals. Washington DC: National Academies Press; 1996. [PubMed] [Google Scholar]
- Inyang F, Ramesh A, Kopsombut P, Niaz MS, Hood DB, Nyanda AM, Archibong AE. Disruption of testicular steroidogenesis and epididymal function by inhaled benzo(a)pyrene. Reprod Toxicol. 2003;17:527–537. doi: 10.1016/s0890-6238(03)00071-6. [DOI] [PubMed] [Google Scholar]
- International Programme on Chemical Safety. Environmental health criteria 202: Selected non-heterocyclic polycyclic aromatic hydrocarbons. Lyon, France: International Programme on Chemical Safety, World Health Organization; 1998. [Google Scholar]
- Jefcoate CR, Liehr JG, Santen RJ, Sutter TR, Yager JD, Yue W, Santner SJ, Tekmal R, Demers L, Pauley R, Naftolin F, Mor G, Berstein L. Tissue-specific synthesis and oxidative metabolism of estrogens. JNCI Monogr. 2000;27:95–112. doi: 10.1093/oxfordjournals.jncimonographs.a024248. [DOI] [PubMed] [Google Scholar]
- Laher JM, Rigler MW, Vetter RD, Barrowman JA, Patton JS. Similar bioavailability and lymphatic transport of benzo(a)pyrene when administered to rats in different amounts of dietary fat. J Lipid Res. 1984;25:1337–1342. [PubMed] [Google Scholar]
- Mann KK, Matulka RA, Hahn ME, Trombino AF, Lawrence BP, Kerkvliet NI, Sherr DH. The role of polycyclic aromatic hydrocarbon metabolism in dimethylbenz(a)anthracene-induced pre-β lymphocyte apoptosis. Toxicol Appl Pharmacol. 1999;161:10–22. doi: 10.1006/taap.1999.8778. [DOI] [PubMed] [Google Scholar]
- Mattison DR, Singh H, Takizawa K, Thomford PJ. Ovarian toxicity of benzo(a)pyrene and metabolites in mice. Reprod Toxicol. 1989;3:115–126. doi: 10.1016/0890-6238(89)90045-2. [DOI] [PubMed] [Google Scholar]
- Melikian AA, Sun P, Prokopczyk B, El Bayoumy K, Hoffman D, Wang X, Waggoner S. Identification of benzo(a)pyrene metabolites in cervical mucus and DNA adducts in cervical tissues in humans by gas chromatographymass spectrometry. Cancer Lett. 1999a;146:127–134. doi: 10.1016/s0304-3835(99)00203-7. [DOI] [PubMed] [Google Scholar]
- Melikian AA, Wang X, Waggoner S, Hoffman D, El-Bayoumy K. Comparative response of normal and of human papilloma virus-16 immortalized human epithelial cervical cells to benzo(a)pyrene. Oncol Rep. 1999b;6:1371–1376. doi: 10.3892/or.6.6.1371. [DOI] [PubMed] [Google Scholar]
- Merrick BA, Selkirk JK. HPLC of benzo(a)pyrene glucuronide, sulfate, and glutathione conjugates and water-soluble metabolites from hamster embryo cells. J Chromatogr. 1985;6:1303–1307. doi: 10.1093/carcin/6.9.1303. [DOI] [PubMed] [Google Scholar]
- Neal MS, Zhu J, Holloway AC, Foster WG. Follicle growth is inhibited by benzo(a)pyrene, at concentrations representative of human exposure, in an isolated rat follicle culture assay. Hum Reprod. 2007;22:961–967. doi: 10.1093/humrep/del487. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- National Institutes of Health. NIH guidelines for the laboratory use of chemical carcinogens. Washington, DC: U.S. Government Printing Office; 1981. NIH publication no. 81-2385. [Google Scholar]
- Oesch F. Significance of various enzymes in the control of reactive metabolites. Arch Toxicol. 1987;60:174–178. doi: 10.1007/BF00296975. [DOI] [PubMed] [Google Scholar]
- Otto S, Bhattacharyya KK, Jefcoate CR. Polycyclic aromatic hydrocarbon metabolism in rat adrenal, ovary, and testis microsomes is catalyzed by the same novel cytochrome P450 (P450RAP) Endocrinology. 1992;131:3067–3076. doi: 10.1210/endo.131.6.1332854. [DOI] [PubMed] [Google Scholar]
- Pan CH, Chan C-C, Wu K-Y. Effects on Chinese restaurant workers of exposure to cooking oil fumes: A cautionary note on urinary 8-hydroxy-2′-deoxyguanosine. Cancer Epidemiol Biomarkers Prev. 2008;17:3351–3357. doi: 10.1158/1055-9965.EPI-08-0075. [DOI] [PubMed] [Google Scholar]
- Paustenbach DJ. The practice of exposure assessment. A state of the art review. J Toxicol Environ Health B. 2000;3:179–291. doi: 10.1080/10937400050045264. [DOI] [PubMed] [Google Scholar]
- Pelling JC, Slaga TJ, DiGiovanni J. Formation and persistence of DNA, RNA, and protein adducts in mouse skin exposed to pure optical enantiomers of 7 beta, 8 alpha-dihydroxy-9 alpha, 10 alpha-epoxy-7,8,9,10-tetrahydrobenzo(a)pyrene in vivo. Cancer Res. 1984;44:1081–1086. [PubMed] [Google Scholar]
- Perera FP, Whyatt RM, Jedrychowski W, Rauh V, Manchester D, Santella RM, Ottoman R. Adverse reproductive outcomes from exposure to environmental polycyclic aromatic hydrocarbons on birth outcomes in Poland. Am J Epidemiol. 1998;147:309–314. doi: 10.1093/oxfordjournals.aje.a009451. [DOI] [PubMed] [Google Scholar]
- Perera FP, Rauh V, Whyatt RM, Tsai WY, Tang D, Diaz D, Hoepner L, Barr D, Tu YH, Camann D, Kinney P. Effect of prenatal exposure to airborne polycyclic aromatic hydrocarbons on neurodevelopment in the first 3 years of life among inner-city children. Environ Health Perspect. 2006;114:1287–1292. doi: 10.1289/ehp.9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyakov LM, Chasovskikh MI, Panin LE. Binding and treatment of benzo(a)pyrene by blood plasma lipoproteins: The possible role of apolipoprotein B in this process. Bioconjugate Chem. 1996;7:396–400. doi: 10.1021/bc960005e. [DOI] [PubMed] [Google Scholar]
- Ramesh A, Inyang F, Hood DB, Archibong AE, Knuckles ME, Nyanda AM. Metabolism, bioavailability, and toxicokinetics of benzo(a)pyrene in F344 rats following oral administration. Exp Toxicol Pathol. 2001;53:275–290. doi: 10.1078/0940-2993-00192. [DOI] [PubMed] [Google Scholar]
- Ramesh A, Hood DB, Inyang F, Greenwood M, Archibong AE, Knuckles ME, Nyanda AM. Comparative metabolism, bioavailability and toxicokinetics of benzo(a)pyrene in rats after acute oral, inhalation, and intravenous administration. Polycyclic Aromatic Compounds. 2002;22:969–980. [Google Scholar]
- Ramesh A, Walker SA, Hood DB, Guillén MD, Schneider K, Weyand EH. Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. Int J Toxicol. 2004a;23:301–333. doi: 10.1080/10915810490517063. [DOI] [PubMed] [Google Scholar]
- Ramesh A, Inyang F, Knuckles ME. Modulation of adult rat benzo(a)pyrene metabolism and DNA adduct formation by neonatal diethylstilbestrol exposure. Exp Toxicol Pathol. 2004b;56:129–138. doi: 10.1016/j.etp.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Ramesh A, Knuckles ME. Dose-dependent benzo(a)pyrene [B(a)P]–DNA adduct levels and persistence in F-344 rats following subchronic dietary exposure to B(a)P. Cancer Lett. 2006;240:268–278. doi: 10.1016/j.canlet.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Ramesh A, Inyang F, Lunstra DD, Niaz MS, Kopsombut P, Jones KM, Hood DB, Hills ER, Archibong AE. Alteration of fertility endpoints in adult male F-344 rats by subchronic exposure to inhaled benzo(a)pyrene. Exp Toxicol Pathol. 2008;60:269–280. doi: 10.1016/j.etp.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorke EA, Sizemore N, Mukhtar H, Couch LH, Howard PC. Polycyclic aromatic hydrocarbons enhance terminal cell death of human ectocervical cells. Int J Oncol. 1998;13:557–563. doi: 10.3892/ijo.13.3.557. [DOI] [PubMed] [Google Scholar]
- Ross J, Nelson G, Kligerman A, Erexson G, Bryant M, Earley K, Gupta RC, Nesnow S. Formation and persistence of novel benzo(a)pyrene adducts in rat lung, liver, and peripheral blood lymphocyte DNA. Cancer Res. 1990;50:5088–5094. [PubMed] [Google Scholar]
- Sagredo C, Mollerup S, Cole KJ, Phillips DH, Uppstad H, Øvrebø S. Biotransformation of benzo(a)pyrene in AhR knockout mice is dependent on time and route of exposure. Chem Res Toxicol. 2009;22:584–591. doi: 10.1021/tx8003664. [DOI] [PubMed] [Google Scholar]
- Shimada T, Guengerich FP. Inhibition of human cytochrome P450 1A1-, 1A2-, and 1B1-mediated activation of procarcinogens to genotoxic metabolites by polycyclic aromatic hydrocarbons. Chem Res Toxicol. 2006;19:288–294. doi: 10.1021/tx050291v. [DOI] [PubMed] [Google Scholar]
- Singh R, Sram RJ, Binkova B, Kalina I, Popov TA, Georgieva T, Garte S, Taioli E, Farmer PB. The relationship between biomarkers of oxidative DNA damage, polycyclic aromatic hydrocarbon–DNA adducts, antioxidant status and genetic susceptibility following exposure to environmental air pollution in humans. Mutat Res. 2007;620:83–92. doi: 10.1016/j.mrfmmm.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Suh M, Ariese F, Small GJ, Jankowiak R, Hewer A, Phillips DH. Formation and persistence of benzo[a]pyrene-DNA adducts in mouse epidermis in vivo: Importance of adduct conformation. Carcinogenesis. 1995;16:2561–2569. doi: 10.1093/carcin/16.10.2561. [DOI] [PubMed] [Google Scholar]
- Shu HP, Nichols AV. Uptake of lipophilic carcinogens by plasma lipoproteins: Structure–activity studies. Biochim Biophys Acta. 1981;665:376–384. doi: 10.1016/0005-2760(81)90249-6. [DOI] [PubMed] [Google Scholar]
- Shu HP, Bymun EN. Systemic excretion of benzo(a)pyrene in the control and microsomally induced rat: The influence of plasma lipoproteins and albumin as carrier molecules. Cancer Res. 1983;43:485–490. [PubMed] [Google Scholar]
- Tuttle AM, Stämpfli M, Foster WG. Cigarette smoke causes follicle loss in mice ovaries at concentrations representative of human exposure. Hum Reprod. 2009;24:1452–1459. doi: 10.1093/humrep/dep023. [DOI] [PubMed] [Google Scholar]
- Verhofstad N, van Oostrom CTM, van Benthem J, van Schooten FJ, van Steeg H, Godschalk RWL. DNA adduct kinetics in reproductive tissues of DNA repair proficient and deficient male mice after oral exposure to benzo(a)pyrene. Environ Mol Mutagen. 2010;51:123–159. doi: 10.1002/em.20516. [DOI] [PubMed] [Google Scholar]
- Wall KL, Gao WS, te Koppele JM, Kwei GY, Kauffman FC, Thurman RG. The liver plays a central role in the mechanism of chemical carcinogenesis due to polycyclic aromatic hydrocarbons. Carcinogenesis. 1991;12:783–786. doi: 10.1093/carcin/12.5.783. [DOI] [PubMed] [Google Scholar]
- Walker MP, Jahnke GD, Snedeker SM, Gladen BC, Lucier GW, DiAugustine RP. 32P-postlabeling analysis of the formation and persistence of DNA adducts in mammary glands of parous and nulliparous mice treated with benzo(a)pyrene. Carcinogenesis. 1992;13:2009–2015. doi: 10.1093/carcin/13.11.2009. [DOI] [PubMed] [Google Scholar]
- Zanieri L, Galvan P, Checchini L, Cincinelli A, Lepri L, Donzelli GP, Del Bubba M. Polycyclic aromatic hydrocarbons (PAHs) in human milk from Italian women: Influence of cigarette smoke and residential area. Chemosphere. 2007;67:1265–1274. doi: 10.1016/j.chemosphere.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Zenzes MT, Puy LA, Bielecki R. Immunodetection of benzo(a)pyrene adducts in ovarian cells of women exposed to cigarette smoke. Mol Hum Reprod. 1998;4:159–165. doi: 10.1093/molehr/4.2.159. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Fang JL, Lazarus P. Glucuronidation: An important mechanism for detoxification of benzo(a)pyrene metabolites in aerodigestive tract tissues. Drug Metab Dispos. 2002;30:397–403. doi: 10.1124/dmd.30.4.397. [DOI] [PubMed] [Google Scholar]