

Abstract
Our previous studies have indicated that chronic treatment with XNT (1-[(2-dimethylamino) ethylamino]-4-(hydroxymethyl)-7-[(4-methylphenyl) sulfonyl oxy]-9H-xanthene-9-one), an angiotensin-converting enzyme2 (ACE2) activator, reverses hypertension-induced cardiac and renal fibrosis in spontaneously hypertensive rats (SHR). Furthermore, XNT prevented pulmonary vascular remodeling and right ventricular hypertrophy and fibrosis in a rat model of monocrotaline-induced pulmonary hypertension. The aim of this study was to determine the mechanisms underlying the protective effects of XNT against cardiac fibrosis. Hydroxyproline assay was used to measure cardiac collagen content in control and XNT-treated (200ng/kg/min for 28 days) SHR. Cardiac ACE2 activity and protein levels were determined using the fluorogenic peptide assay and western blot analysis, respectively. Extracellular signal-regulated kinases (p44 and p42; ERKs) and AT1 receptor levels were quantitated by western blotting. Cardiac ACE2 protein levels were ~15% lower in SHR compared with WKY controls (1.00±0.02 vs. 0.87±0.01 ACE2/GAPDH ratio in SHR). However, treatment of SHR with XNT completely restored the decreased cardiac ACE2 levels. Also, chronic infusion of XNT significantly increased cardiac ACE2 activity in SHR. This increase in ACE2 activity was associated with decreased cardiac collagen content. Furthermore, the anti-fibrotic effect of XNT correlated with increased cardiac Ang-(1–7) immunostaining, though no change in cardiac AT1 protein levels was observed. The beneficial effects of XNT were also accompanied by a reduction in ERK phosphorylation (WKY: 1.00±0.04; Control-SHR: 1.46±0.25; SHR-treated: 0.86±0.02 phospho ERK/total ERK ratio). Our observations demonstrate that XNT activates cardiac ACE2 and inhibits fibrosis. These effects are associated with increases in Ang-(1–7) and inhibition of cardiac ERK signaling.
Keywords: XNT; Angiotensin-(1,7); Anti-fibrosis
INTRODUCTION
Development of cardiac fibrosis is a major complication of hypertensive heart disease. It contributes to progressive disruption of the normal structure of the heart resulting in increased risk for adverse cardiac events such as myocardial ischemia, infarction, arrhythmias and sudden cardiac death (Weber, 2000). Therefore, prevention and/or reversal of cardiac fibrosis becomes extremely essential in the management of hypertensive heart disease (Weber, 2000).
The renin-angiotensin system (RAS) is a pivotal regulator of cardiovascular function. While the key role of angiotensin-converting enzyme (ACE) is to generate angiotensin (Ang) II, the major vasoactive peptide of the RAS; its homologue ACE2 is responsible for metabolizing Ang II into the heptapeptide Angiotensin-(1–7) [Ang-(1–7)] (Vickers et al. 2002; Tipnis et al. 2000). Consequently, ACE2 counter-regulates the deleterious actions of Ang II (Ferreira et al. 2010), thereby promoting many beneficial effects, including tissue-specific anti-fibrotic actions (Katovich et al. 2008). As a matter of fact, cardiac overexpression of ACE2 using lentiviral gene transfer prevented hypertension-induced cardiac hypertrophy and fibrosis in spontaneously hypertensive rats (SHR) and in Ang II-infused rats (Diez-Freire et al. 2006; Huentelman et al. 2005). In addition, chronic administration of Ang-(1–7) prevented the development of ventricular fibrosis induced by DOCA-salt treatment (Grobe et al. 2006). Thus, an increased activity of the vasoconstrictive and proliferative axis of the RAS, comprising of ACE, Ang II and AT1 receptor (AT1R), is associated with the development of cardiac fibrosis (Bader at al. 2001; Leask, 2010). On the other hand, the ACE2/Ang-(1–7)/Mas receptor axis exerts protective actions against fibrosis (Ferreira et al. 2010, Katovich et al. 2008), indicating that this axis is an important cardiac protector.
More recently, we have found that chronic treatment with XNT (1-[(2-dimethylamino) ethylamino]-4-(hydroxymethyl)-7-[(4-methylphenyl) sulfonyl oxy]-9H-xanthene-9-one), an ACE2 activator discovered based on the crystal structure of this enzyme (Hernández Prada et al. 2008), prevents and reverses hypertension-induced cardiac and renal fibrosis in SHR (Hernández Prada et al. 2008). Furthermore, XNT prevents pulmonary vascular remodeling and right ventricular hypertrophy and fibrosis in a rat model of monocrotaline-induced pulmonary hypertension (Ferreira et al. 2009). However, the mechanism by which XNT exerts these beneficial actions is not fully understood. Hence, our aim in this present study was to evaluate the mechanisms underlying the cardiac anti-fibrotic effects of XNT. We hypothesized that increase in Ang-(1–7) and inhibition of extracellular signal-regulated kinases (p44 and p42; ERKs) might be associated with the protective actions of XNT.
METHODS
Ethical Approval
All animal procedures were performed in compliance with approved IACUC protocols and University of Florida regulations.
Animals
Male Wistar-Kyoto (WKY) rats and SHR, aged 12 weeks, were purchased from Charles River Laboratories (Wilmington, MA, USA). Five-day-old SHR pups were used to obtain primary cell cultures of cardiac fibroblasts. Osmotic minipumps (Alzet, model 2004) containing either 10mg/mL of XNT (60μg/day; 28 days) or vehicle (saline pH 2.0 to 2.5) were implanted subcutaneously after allowing them to equilibrate in sterile saline at 37°C for 24 hours. XNT was delivered at an infusion rate of 260ng/kg per minute. We have observed that, under these conditions, this vehicle does not change arterial pressure, heart rate, responses to Ang II, bradykinin and losartan, heart function, myocardial and renal structures and ACE2 activity in SHR (Hernandez Prada et al. 2008).
Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from the left ventricles of SHR and WKY rats with RNaqueous-4RCP Kit (Ambion). cDNA samples (2μl) of reverse transcription reactions were amplified by quantitative real-time PCR (qPCR) using primers and probes for ACE2 from Applied Biosystems (Cat# Mm01159013_m1) and an ABI PRISM 7000 HT Detection system. ACE2 mRNA levels were normalized to 18S RNA from the same samples.
Western Blotting
Cardiac ACE2, AT1 receptor and phosphorylated ERKs (p44 and p42) were quantitated by western blotting. Forty micrograms of protein extracted from the left ventricles of WKY rats and SHR were electrophoresed on 12% SDS-PAGE gel and then transferred onto a nitrocellulose membrane. After 1 hour of blocking with 5% milk in Tris-buffered saline-Tween, the membranes were probed with one of the following primary antibodies: anti-ACE2 (1:400, Cat#sc-20998, Santa Cruz Biotechnology, Inc., Santa Cruz, CA); anti-AT1 (1:400, Cat#sc-579, Santa Cruz Biotechnology, Inc., Santa Cruz, CA); anti-phospho-p44/42 MAPK (ERK1/2) (1:200, Cat#9101, Cell Signaling Technology, Inc., Danvers, MA); and anti-total-p44/42 MAPK (ERK1/2) (1:200, Cat#9102, Cell Signaling Technology, Inc., Danvers, MA). Membranes were washed 3 times for 10 minutes in Tris-buffered saline-Tween and incubated with anti-rabbit IgG-horseradish peroxidase-conjugated secondary antibody (1:5000) for 1 hour at room temperature. After final washes, the membrane was incubated with chemiluminescent agent for 1 minute and then exposed to X-ray film to visualize protein bands. Protein levels were expressed as a ratio of optical densities. GAPDH (ACE2 and AT1 receptor) and total ERK (phosphorylation of ERKs - p44 and p42) bands served as loading controls to correct for inaccuracies in protein loading.
Cardiac ACE2 Activity
ACE2 activity was determined in the left ventricles of WKY rats and SHR using a fluorogenic peptide substrate [ACE2 substrate, fluorogenic peptide VI (FPS VI), Mca-YVADAPK(Dnp)-OH, catalog. ID: ES007, where Mca is 7-(methoxycoumarin-4-yl) acetyl and Dnp is 2,4-dinitrophenyl; R&D Systems, Minneapolis, MN], as described previously (Huentelman et al. 2004). The reaction buffer [75 mmol/l Tris/HCl (pH 7.5), 1 mol/l NaCl and 0.5 mmol/l ZnCl2] was added to the tissues for protein extraction. Protein concentration was determined using a standard Bradford assay. The enzyme removes the C-terminal dinitrophenyl moiety that quenches the inherent fluorescence of the 7-methoxycoumarin group, resulting in increased fluorescence at excitation and emission spectra of 320 nm and 405 nm, respectively. Enzymatic activity was measured with a Spectra Max Gemini EM Fluorescence Reader (Molecular Devices). All assays were performed at least in triplicate and samples were read every minute for at least 120 minutes immediately after the addition of fluorogenic peptide substrates at 37°C.
Collagen Content Assay
Total collagen content was determined by hydroxyproline assay. Cardiac tissues (left ventricles) were collected, trimmed and homogenized in 1.5ml 0.5 mol/l glacial acetic acid. This mixture was dried in a speed vacuum and weighed. The dried samples were redissolved in 1.5 ml of 6 N HCl and hydrolyzed at 110°C overnight. 7.5 μl-samples were transferred to 96-well plates, dried and ressuspended in 7.5 μl of citrate-acetate buffer (5% citric acid, 1.2% glacial acetic acid, 7.24% sodium acetate and 3.4% NaOH dissolved in distilled water and adjusted to pH 6.0). One hundred microliters of freshly prepared chloramine-T solution (0.282 g chloramine-T, 2 ml n-propanol, 2 ml distilled water QS to 20 ml with citrate-acetate buffer), was added and the reaction mixture was allowed to stand at room temperature for 20 minutes. Afterward, 100μl of freshly prepared Ehrlich’s solution (4.5 g 4-dimethylaminobenzaldehyde dissolved in 18.6 ml of n-propanol and 7.8 ml of 70% perchoric acid), was added and the mixture was heated to 65°C for 15 minutes. The absorbance was recorded at 550 nm. A standard curve of samples with known quantity of hydroxyproline was generated for each assay. Absorbance of unknown sample was compared to the standard curve and hydroxyproline content per sample was calculated.
Cell Culture
Cardiac fibroblasts were isolated from the ventricles of 5-day-old SHR pups, as previously described (Qi et al. 2010). Briefly, ventricles were dissociated by mechanical disaggregating and enzymatic digestion with 1% collagenase II (Worthington Biochemical Corporation, Freehold, NJ, USA). Next, cells were plated in the presence of 5% fetal bovine serum (FBS). The attached cells on the dish were mainly cardiac fibroblasts, which were cultured in 10% FBS medium [DMEM supplemented with 10% (v/v) FBS, 1% penicillin/streptomycin and 50 μg/ml ascorbic acid]. More than 95% of cultured cells were cardiac fibroblasts, as determined by positive immunostaining for vimentin (data not shown). Fibroblast cultures were dissociated using trypsin/ethylenediaminetetraacetic acid, which contribute to eliminate any residual cardiomyocytes.
Immunohistochemistry
ACE2 and Ang-(1–7) expression in cultured cardiac fibroblasts from left ventricles and in left ventricular sections of control and XNT-treated SHR were evaluated by immunohistochemistry using the streptavidin-biotin-peroxidase method, as described previously (Yamazato et al. 2009). Five-micrometer sections were obtained from left ventricles fixed in 10% formalin and embedded in paraffin. The primary antibodies used were anti-ACE2 (1:500, Cat#GTX15348, GeneTex, Inc., Irvine, CA) and anti-Ang-(1–7) (1:600; Yamazato et al. 2009). Briefly, endogenous peroxidase was blocked with 0.3% hydrogen peroxide and nonspecific binding of the antibody was blocked by 5% rabbit serum. The primary antibodies diluted in 1% BSA in PBS were applied and incubated overnight at 4°C. Subsequently, biotinylated secondary antibody followed by streptavidin-peroxidase conjugate was applied. Positive staining was visualized using diaminobenzidine and nuclei were counterstained with hematoxylin. Negative controls were obtained by omitting the primary antibody from the incubation procedure. The immunostaining results were quantified according to Lehr et al. (1997).
Statistical Analysis
The results are presented as mean ± SEM. Statistical significance was estimated using unpaired Student’s t-test or one-way ANOVA followed by the Bonferroni post-test. Differences were considered significant at a p≤0.05.
RESULTS
XNT Restores Cardiac ACE2 Protein Levels in SHR
As observed in Figure 1a, the expression of ACE2 protein in SHR hearts was significantly lower when compared with WKY rats (1.00 ± 0.02 vs. 0.87 ± 0.01 ACE2/GAPDH ratio in SHR, p<0.01). Interestingly, treatment with XNT resulted in a complete restoration of cardiac ACE2 levels in SHR (p<0.01). In keeping with this later finding, the ACE2 activity was also observed to be higher in SHR that were chronically infused with XNT compared to SHR that received saline alone (Fig. 1b; p=0.05). Unexpectedly, XNT-induced increases in ACE2 protein and activity was accompanied by a significant increase in cardiac ACE2 mRNA expression (Fig. 1c, p=0.05).
Figure 1.

XNT restores cardiac ACE2 protein levels in SHR. A) Quantification of ACE2 protein expression using western blotting in hearts from WKY rats, vehicle-treated SHR and XNT-treated SHR. ACE2 levels were ~15% lower in SHR compared with WKY rats. XNT treatment restored ACE2 protein levels in SHR. Data were normalized using GAPDH. #p<0.05 vs. WKY; *p<0.05 vs. vehicle-treated SHR (One-way ANOVA). Each column represents the mean ± SEM (n=3 to 6). B) XNT increases ACE2 activity in hearts of SHR. Chronic infusion of XNT caused a significant increase in cardiac ACE2 activity in SHR. *p<0.05 vs. untreated animals (Unpaired Student’s t-test). Each column represents the mean ± SEM (n=4). C) XNT increases ACE2 mRNA expression in SHR. Quantitative real-time PCR was carried out to assess the ACE2 expression in left ventricles of SHR treated or not with XNT. XNT treatment increased the ACE2 expression. *p<0.05 vs. untreated animals (Unpaired Student’s t-test). Each column represents the mean ± SEM (n=4).
Restoration of Cardiac ACE2 Levels in SHR is Associated with Reduction in Fibrosis
The total ventricular collagen content was evaluated by the hydroxyproline assay method. Chronic administration of XNT (260 ng/kg/min for 28 days) considerably decreased (approximately 15%) the total collagen content in the hearts of SHR (Fig. 2, p<0.05), suggesting that the anti-fibrotic effect of XNT may be associated with increased cardiac ACE2 activity and levels.
Figure 2.

XNT reduces total collagen content in hearts of SHR. Collagen content was evaluated by hydroxyproline assay in hearts of SHR treated or not with XNT. XNT promotes a significant reduction in collagen content. *p<0.05 vs. untreated animals (Unpaired Student’s t-test). Each column represents the mean ± SEM (n=5 to 6).
Effects of XNT on ACE2 and Ang-(1–7) Expression in Cardiac Fibroblasts
To evaluate the effects of XNT on ACE2 and Ang-(1–7) expression in cardiac fibroblasts, the main cellular type involved in collagen deposition, we performed immunohistochemical analysis using histological sections from left ventricles of SHR. We found that XNT treatment increased ACE2 expression by approximately 16% (p>0.05) in both interstitial (Fig. 3b) and perivascular (Fig. 3d) fibroblasts. In addition, similar effects were observed for Ang-(1–7) expression, i.e. the expression of Ang-(1–7) was increased by about 16% (p>0.05) in interstitial and perivascular fibroblasts of XNT-administered SHR (Figs. 4b and 4d). To further confirm these data, we incubated isolated fibroblasts with XNT. In keeping with our immunohistochemical results, isolated fibroblasts treated with XNT also exhibited higher expression of both ACE2 (Fig. 3g) and Ang-(1–7) (Fig. 4g).
Figure 3.
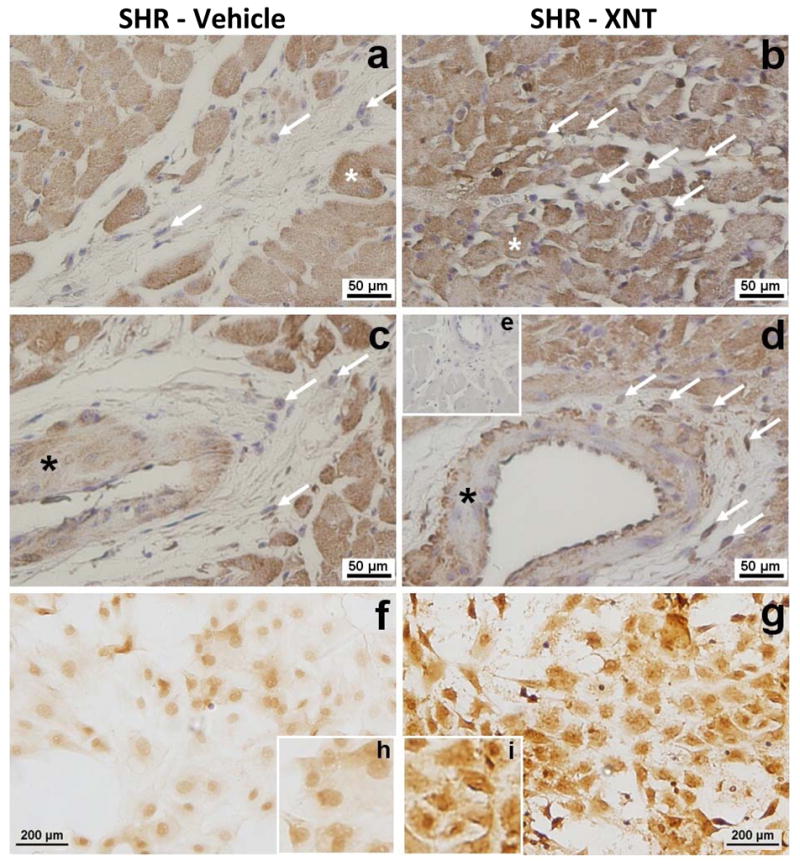
XNT increases ACE2 protein expression in cardiac fibroblasts of SHR. Immunohistochemical analysis was used to detect the expression of ACE2 in paraffin-embedded sections of left ventricles and in isolated cardiac fibroblasts of SHR. XNT increased the expression of ACE2 in interstitial (b) and perivascular (d) fibroblasts (arrows) when compared with untreated animals (a; control interstitial area and c; control perivascular area). (e) Negative control. (*); wall vessels. XNT treatment also increased the ACE2 expression in isolated cardiac fibroblasts (g) (d; untreated fibroblasts). Higher magnification of cardiac fibroblasts is shown in h (untreated cells) and in i (treated cells). (n=3 different animals).
Figure 4.
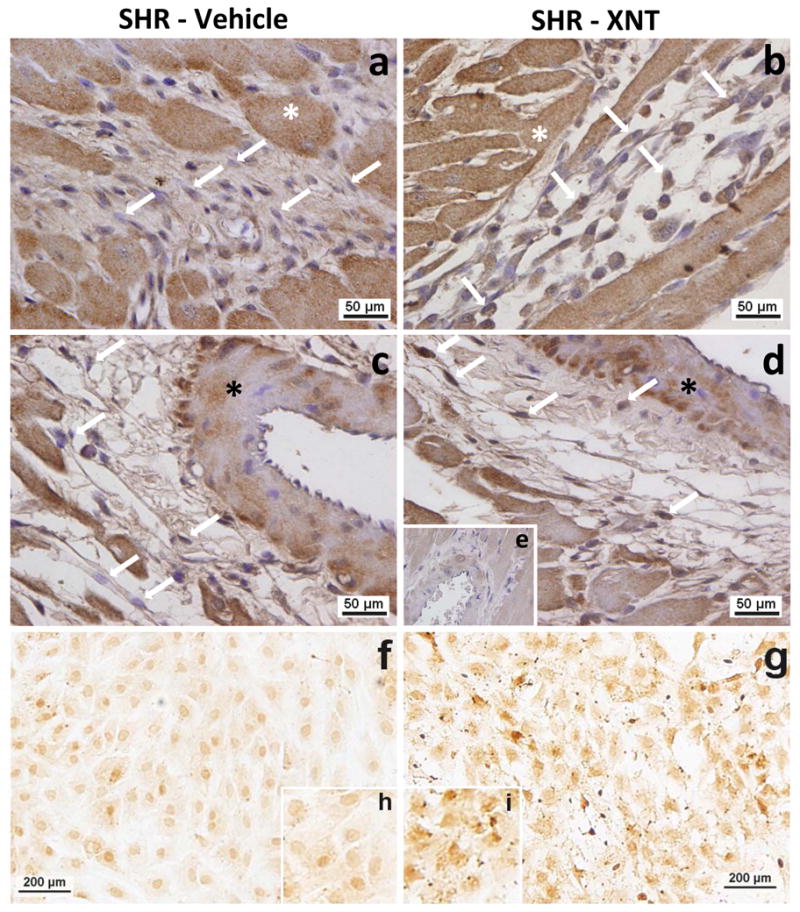
XNT increases Ang-(1–7) expression in cardiac fibroblasts of SHR. Immunohistochemical analysis was used to detect the expression of Ang-(1–7) in paraffin-embedded sections of left ventricles and in isolated cardiac fibroblasts of SHR. XNT increased the expression of Ang-(1–7) in interstitial (b) and perivascular (d) fibroblasts (arrows) when compared with untreated animals (a; control interstitial area and c; control perivascular area). (e) Negative control. (*); wall vessels. XNT treatment also increased the Ang-(1–7) expression in isolated cardiac fibroblasts (g) (d; untreated fibroblasts). Higher magnification of cardiac fibroblasts is shown in h (untreated cells) and in i (treated cells). (n=3 different animals).
The anti-fibrotic effect of XNT was accompanied by ERKs reduction
It has been well established that ERK activation is one of the downstream signaling events that participates in collagen synthesis. Therefore, we examined if the beneficial anti-fibrotic effect of XNT would involve ERKs. As shown in Figures 5a and 5b, phosphorylation of ERKs (p44 and p42) was higher in SHR compared with WKY rats. XNT treatment restored or in fact, reduced ERK phosphorylation in SHR (WKY: 1.00 ± 0.04; Control SHR: 1.46 ± 0.25; SHR-treated: 0.86 ± 0.02 phospho ERK/total ERK ratio, p<0.05). No significant changes were observed in the levels of cardiac AT1R expression (Fig. 5c).
Figure 5.

XNT reverses the activated state of ERKs (p44 and p42) in hearts of SHR. The phosphorylation of ERKs was determined by western blotting in left ventricle samples of WKY rats, control SHR and XNT-treated SHR. A) Representative gel of western blotting. B) Quantification of the western blotting data. The data were normalized using total ERKs. *p<0.05 vs. vehicle-treated SHR (One-way ANOVA). Each column represents the mean ± SEM (n=3 to 6). C) AT1 receptor expression was unaffected by XNT treatment when evaluated by western blotting. The data were normalized using GAPDH. Each column represents the mean ± SEM (One-way ANOVA; n=3 to 6).
DISCUSSION
The major finding of this study is that the synthetic ACE2 activator XNT stimulates endogenous cardiac ACE2 to inhibit ventricular fibrosis. These effects are associated with increases in Ang-(1–7) and inhibition of cardiac ERK signaling.
The pro-fibrotic role of Ang II acting via the AT1 receptor is well established (Bader at al. 2010; Leask et al. 2010). Activation of AT1 receptors is associated with a range of deleterious effects including vasoconstriction, inflammation, hypertrophy and extracellular matrix protein synthesis (Bader at al. 2010; Rosenkranz, 2004). These actions can induce tissue-specific remodeling effects including cardiac hypertrophy and fibrosis, as well as arterial stiffness, all of which are key factors in cardiovascular diseases. On the other hand, the role of ACE2/Ang-(1–7)/Mas axis on the fibrotic process has only recently been described. We have previously reported that cardiac overexpression of ACE2 using lentiviral gene transfer prevents both hypertrophy and fibrosis in SHR and Ang II-infused rat model of hypertension (Diez-Freire et al. 2006; Huentelman et al. 2005). Additionally, pharmacological activation of ACE2 reduces hypertension-induced cardiac and renal fibrosis in SHR (Hernández Prada et al. 2008), as well as prevents pulmonary vascular remodeling and right ventricular hypertrophy and fibrosis in rats with pulmonary hypertension (Ferreira et al. 2009). In the present study, we further confirm the cardiac anti-fibrotic effects of XNT using a well established biochemical assay.
The mechanisms, by which ACE2 reduces cardiac fibrosis remain poorly understood. The beneficial effect of ACE2 might be, at least in part, due to reduction in Ang II and subsequent augmentation of Ang-(1–7) levels. Indeed, it has been reported that Ang-(1–7) acts as an important anti-fibrotic agent (Katovich et al. 2008). Grobe et al. (2006) have reported that Ang-(1–7) infusion prevents cardiac collagen deposition without decreasing blood pressure, indicating that the anti-fibrotic effect of Ang-(1–7) is independent of blood pressure changes. Furthermore, the Ang-(1–7) receptor, Mas which is expressed on cardiac fibroblasts plays an important role against cardiac collagen deposition (Iwata et al. 2007; Santos et al. 2006). In keeping with these findings, we found that the hearts from XNT-treated SHR exhibit a significant increase in Ang-(1–7) immunolabeling, suggesting that Ang-(1–7) participates in the anti-fibrotic effects of XNT.
Cardiac fibroblasts are the main cell type involved in extracellular matrix protein deposition in the heart. Several studies have reported that functional AT1 receptors are expressed on cardiac fibroblasts and activation of these receptors stimulate synthesis of extracellular matrix proteins (Qi et al. 2010; Gonzalez et al. 2002; Kawano et al. 2000). Ang II acting through the AT1 receptor has been shown to trigger signaling cascades that stimulate mitogen activated protein kinases (MAPKs), including extracellular signal-regulated kinases (ERK1/2), to induce fibrosis (Mehta et al. 2007). In the current study, XNT restored cardiac ACE2 protein levels and reduced ventricular collagen deposition.
However, no significant change in cardiac AT1 receptor was observed; suggesting that this receptor may not be involved in the anti-fibrotic effects of XNT. On the other hand, XNT decreased the phosphorylation of ERKs (p44 and p42) in the heart. The exact mechanism by which XNT reduces ERK activation has not been addressed in our current study. However, we speculate that Ang-(1–7) is involved, since cardiac level of this peptide is increased with XNT administration and that Ang-(1–7) has been previously shown to modulate ERK activation (Sampaio et al. 2007). Because we did not measure the Ang II levels in the heart, a reduction in the cardiac concentration of Ang II, as a cause of the anti-fibrotic effect of XNT and reduction of the ERKs phosphorylation, can not be excluded at this point. In addition, taking into consideration that ACE2 forms Ang-(1–7) degrading Ang II, both increases in Ang-(1–7) and decreases in Ang II levels may contribute to the down-regulation observed in the ERKs expression.
Our data showing that XNT induces increases in ACE2 mRNA expression suggests that XNT may have other mechanisms of action besides the direct activation of ACE2 enzyme. It has been shown that Ang II may decrease ACE2 mRNA expression in cardiac myocytes and fibroblasts (Gallagher et al. 2008). Thus, it is possible that XNT increases the ACE2 mRNA expression by decreasing the Ang II bioavailability.
One of the limitations of our study is that we have used 12 week old SHR to evaluate the anti-fibrotic effects of XNT. This may not represent the best age group to examine ventricular fibrosis and associated cardiac dysfunction in the SHR. Increased ventricular fibrosis might have been evident if older animals were to be used in our experiments. However, at 12 weeks of age, these animals did show elevated mean arterial pressure (184±5mmHg in SHR vs. 123±4mmHg in WKY rats) and modest increase in ventricular fibrosis (2.78±0.62% area of fibrosis in SHR vs. 1.42±0.17% area of fibrosis in WKY) (Hernández Prada et al. 2008) These observations definitely suggest onset of cardiac damage in the hearts of these rats, thereby, validating our results.
In summary, our study demonstrates that endogenous ACE2 activation restores cardiac ACE2 protein levels resulting in increased ACE2 activity in SHR. These effects correlate with significant decrease in cardiac collagen content, increase in Ang-(1–7) and reduced ERK signaling. Altogether, our findings suggest that XNT, a synthetic ACE2 activator could be a potential lead compound for the treatment of cardiac fibrosis, facilitating the management of hypertensive heart disease.
Acknowledgments
This study was partially supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), FAPEMIG (Fundação de Amparo à Pesquisa de Minas Gerais), and NIH (National Institutes of Health).
References
- Bader M, Peters J, Baltatu O, Müller DN, Luft FC, Ganten D. Tissue renin-angiotensin systems: new insights from experimental animal models in hypertension research. J Mol Med. 2001;79:76–102. doi: 10.1007/s001090100210. [DOI] [PubMed] [Google Scholar]
- Díez-Freire C, Vázquez J, Correa de Adjounian MF, Ferrari MF, Yuan L, Silver X, Torres R, Raizada MK. ACE2 gene transfer attenuates hypertension-linked pathophysiological changes in the SHR. Physiol Genomics. 2006;27:12–19. doi: 10.1152/physiolgenomics.00312.2005. [DOI] [PubMed] [Google Scholar]
- Ferreira AJ, Shenoy V, Yamazato Y, Sriramula S, Francis J, Yuan L, Castellano RK, Ostrov DA, Oh SP, Katovich MJ, Raizada MK. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:1048–1054. doi: 10.1164/rccm.200811-1678OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira AJ, Santos RAS, Bradford CN, Mecca AP, Sumners C, Katovich MJ, Raizada MK. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension. 2010;55:207–213. doi: 10.1161/HYPERTENSIONAHA.109.140145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PE, Ferrario CM, Tallant EA. Regulation of ACE2 in cardiac myocytes and fibroblasts. Am J Physiol Heart Circ Physiol. 2008;295:H2373–H2379. doi: 10.1152/ajpheart.00426.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, Lopez B, Querejeta R, Diez J. Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J Mol Cell Cardiol. 2002;34:1585–1593. doi: 10.1006/jmcc.2002.2081. [DOI] [PubMed] [Google Scholar]
- Grobe JL, Mecca AP, Mao H, Katovich MJ. Chronic angiotensin-(1–7) prevents cardiac fibrosis in the DOCA-salt model of hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H2417–H2423. doi: 10.1152/ajpheart.01170.2005. [DOI] [PubMed] [Google Scholar]
- Hernández Prada JA, Ferreira AJ, Katovich MJ, Shenoy V, Qi Y, Santos RAS, Castellano RK, Lampkins AJ, Gubala V, Ostrov DA, Raizada MK. Structure-based identification of small-molecule angiotensin- converting enzyme 2 activators as novel antihypertensive agents. Hypertension. 2008;51:1312–1317. doi: 10.1161/HYPERTENSIONAHA.107.108944. [DOI] [PubMed] [Google Scholar]
- Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, Ferrario CM, Raizada MK. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2. Exp Physiol. 2005;90:783–790. doi: 10.1113/expphysiol.2005.031096. [DOI] [PubMed] [Google Scholar]
- Huentelman MJ, Zubcevic J, Katovich MJ, Raizada MK. Cloning and characterization of a secreted form of angiotensin-converting enzyme 2. Regul Pep. 2004;122:61–67. doi: 10.1016/j.regpep.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Iwata M, Cowling RT, Gurantz D, Moore C, Zhang S, Yuan JX, Greenberg BH. Angiotensin-(1–7) binds to specific receptors on cardiac fibroblasts to initiate antifibrotic and antitrophic effects. Am J Physiol Heart Circ Physiol. 2005;289:H2356–H2363. doi: 10.1152/ajpheart.00317.2005. [DOI] [PubMed] [Google Scholar]
- Katovich MJ, Grobe JL, Raizada MK. Angiotensin-(1–7) as an antihypertensive, antifibrotic target. Curr Hypertens Rep. 2008;10:227–232. doi: 10.1007/s11906-008-0043-9. [DOI] [PubMed] [Google Scholar]
- Kawano H, Do YS, Kawano Y, Starnes V, Barr M, Law RE, Hsueh WA. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation. 2000;101:1130–1137. doi: 10.1161/01.cir.101.10.1130. [DOI] [PubMed] [Google Scholar]
- Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- Lehr HA, Mankoff DA, Corwin D, Santeusanio G, Gown AM. Application of photoshop-based image analysis to quantification of hormone receptor expression in breast cancer. J Histochem Cytochem. 1997;45:1559–1565. doi: 10.1177/002215549704501112. [DOI] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- Qi Y, Liu X, Li H, Shenoy V, Li Q, Hauswirth WW, Sumners C, Katovich MJ. Selective tropism of the recombinant adeno-associated virus 9 serotype for rat cardiac tissue. J Gene Med. 2010;12:22–34. doi: 10.1002/jgm.1404. [DOI] [PubMed] [Google Scholar]
- Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–432. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- Sampaio WO, Castro CH, Santos RAS, Schiffrin EL, Touyz RM. Angiotensin-(1–7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension. 2007;50:1093–1098. doi: 10.1161/HYPERTENSIONAHA.106.084848. [DOI] [PubMed] [Google Scholar]
- Santos RAS, Castro CH, Gava E, Pinheiro SV, Almeida AP, Paula RD, Cruz JS, Ramos AS, Rosa KT, Irigoyen MC, Bader M, Alenina N, Kitten GT, Ferreira AJ. Impairment of in vitro and in vivo heart function in angiotensin-(1–7) receptor MAS knockout mice. Hypertension. 2006;47:996–1002. doi: 10.1161/01.HYP.0000215289.51180.5c. [DOI] [PubMed] [Google Scholar]
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- Weber KT. Fibrosis and hypertensive heart disease. Curr Opin Cardiol. 2000;15:264–272. doi: 10.1097/00001573-200007000-00010. [DOI] [PubMed] [Google Scholar]
- Yamazato Y, Ferreira AJ, Hong KH, Sriramula S, Francis J, Yamazato M, Yuan L, Bradford CN, Shenoy V, Oh SP, Katovich MJ, Raizada MK. Prevention of pulmonary hypertension by Angiotensin-converting enzyme 2 gene transfer. Hypertension. 2009;54:365–371. doi: 10.1161/HYPERTENSIONAHA.108.125468. [DOI] [PMC free article] [PubMed] [Google Scholar]