

Abstract
Phagocytosis of IgG-opsonized microbes via the Fcγ receptor (FcγR) requires the precise coordination of a number of signaling molecules, including the low-molecular mass GTPases. Little is known about the Ras-family GTPase Rap1 in this process. We therefore investigated its importance in mediating FcγR-dependent phagocytosis in NR8383 rat alveolar macrophages. Pulldown of active Rap1 and fluorescence microscopic analysis of GFP-RalGDS (Ral guanine dissociation stimulator)-transfected macrophages revealed that Rap1 is indeed activated by FcγR crosslinking. Inhibition of Rap1 activity, both by Rap1GAP (GTPase-activating protein) expression and liposome-delivered blocking Ab, severely impaired the ability of cells to ingest IgG-opsonized targets. FcγR-induced Rap1 activation was found to be independent of both cAMP and Ca2+, suggesting a role for the second messenger-independent guanosine exchange factor, C3G. This was supported by the facts that 1) liposome-delivered blocking Ab against C3G inhibited both FcγR-dependent phagocytosis and Rap1 activation, and 2) both active Rap1GTP and C3G were found to translocate to the phagosome. Taken together, our data demonstrate a novel role for Rap1 and its exchange factor C3G in mediating FcγR-dependent phagocytosis.
Alveolar macrophages (AMs)3 serve as the resident immune effector cells in the lung, protecting the alveolar epithelial surfaces from invading microorganisms (1). One of the primary means by which they perform this function is through the Fcγ receptor (FcγR)-dependent recognition, engulfment, and eventual elimination of IgG-opsonized pathogens. Defects in such clearance mechanisms compromise the sterility of the lung, resulting in respiratory infections such as pneumonia.
Phagocytosis through the FcγR is well known to require the activation of two Rho-family GTPases, Rac1 and Cdc42 (2). These low-molecular mass GTPases cycle between an inactive GDP-bound conformation and an active GTP-bound conformation; guanosine exchange factors (GEFs) catalyze the exchange of GDP for GTP and thereby activate these GTPase proteins, while GTPase-activating proteins (GAPs) stimulate GTP hydrolysis, resulting in the return of the GTPases to their inactive, GDP-bound state. Activation of another low-molecular mass GTPase, Rap1 (smg21/Krev1), has been demonstrated to be essential for cell adhesion (3), complement-mediated phagocytosis (4), and maintenance of T cell anergy (5), but its participation in FcγR phagocytosis has not been fully investigated. Rap1 can be activated by several GEFs, including C3G (6), calcium diacylglycerol (CalDAG)-GEF (7), and the exchange protein directly activated by cAMP (Epac)-1 or -2 (8).
In this study, we sought to 1) biochemically and microscopically characterize Rap1 activation upon FcγR ligation, and 2) further elucidate the functional role and molecular mechanism by which Rap1 regulates FcγR-mediated phagocytosis. Herein, we report an unexpected role for Rap1 in mediating phagocytosis, and demonstrate that its activation is mediated by C3G.
Materials and Methods
Reagents and plasmids
RPMI 1640, penicillin/streptomycin solution, glutathione agarose beads, Lipofectin, Lipofectamine LTX, Lipofectamine Plus reagent, geneticin, and mouse IgG-conjugated 3-μm paramagnetic beads were purchased from Invitrogen. FBS was obtained from HyClone. ECL and BL21 Escherichia coli were from Amersham Biosciences. Ca2+ chelator BAPTA-AM and adenylyl cyclase inhibitor SQ22536 were supplied by BIOMOL. PP2 was purchased from Calbiochem. Polyclonal rabbit anti-sRBC IgG Ab was obtained from Cappel/Organon Teknika. Mouse monoclonal anti-Rap1, mouse monoclonal anti-flotillin-1, and mouse monoclonal PY20 Abs were purchased from BD Transduction Laboratories. Rabbit polyclonal anti-Rap1 was from Upstate Biotechnology, while rabbit polyclonal anti-C3G, mouse monoclonal anti-C3G, rabbit polyclonal anti-CrkL, and rabbit polyclonal anti-Cbl Abs were from Santa Cruz Biotechnology. Rap1GAP and GFP-Ral guanine dissociation stimulator (RalGDS) plasmids were kindly provided by Dr. Philip Stork (Oregon Health and Science University). GST-RalGDS plasmid constructed by Dr. Johannes Bos (Utrecht University) was a gift from Dr. Danny Altschuler (University of Pittsburgh) and was used with permission.
Cell culture and transfections
Rat alveolar macrophage-derived NR8383 cells and human leukemia-derived U937 monocytes (American Type Culture Collection) were cultured in RPMI 1640 containing 10% FBS and 1% penicillin/streptomycin. Cells were maintained for up to 20 passages in a humidified incubator at 37°C with 5% CO2.
NR8383 macrophages were transfected with Rap1GAP, pcDNA3.1, GFP, or GFP-RalGDS plasmids using Lipofectamine Plus and Lipofectamine LTX according to the manufacturer’s instructions. Rap1GAP- and pcDNA3.1-transfected cells were selected 24–48 h posttransfection with 500 μg/ml geneticin for 36–48 h and maintained with 25 μg/ml geneticin. GFP- and GFP-RalGDS-transfected macrophages were imaged 36–48 h posttransfection.
U937 monocytes were cotransfected with pcDNA3.1 or Rap1GAP plasmids and an enhanced GFP-encoding pmaxGFP vector (Amaxa) using the W-01 program from a Nucleofector device (Amaxa). Nucleofection protocols were performed according to the manufacturer’s instructions, with transfection efficiencies routinely exceeding 75%.
FcγR crosslinking and flow cytometry
Transfected U937 monocytes were incubated with anti-CD64 (Abcam), an Ab directed against the high affinity Fcγ receptor I, for 15 min at 4°C. Surface-bound Abs were crosslinked using goat anti-mouse antibody-coated beads (BD Biosciences) for 10 min at 37°C and washed in ice-cold PBS. Stimulated cells were blocked and stained for flow cytometric analysis with PE-conjugated CBRM1/5 (eBioscience), a mAb that binds to an activation-specific epitope of Mac-1, or their corresponding isotype controls. Cells were then washed twice with PBS and fixed with 4% paraformaldehyde (Sigma-Aldrich). From this population of U937 monocytes, enhanced GFP positives were sorted by FACS and analyzed for surface staining of activated Mac-1 with MoFlo.
Phagocytosis assay
Phagocytosis of IgG-opsonized SRBCs was quantitated as described previously (9). Briefly, NR8383 macrophages stably transfected with either pcDNA3.1 or Rap1GAP were plated in 96-well culture-treated dishes at a density of 2 × 105 cells/well. SRBCs were opsonized with a subagglutinating concentration of polyclonal rabbit anti-SRBC IgG as previously described (10). Cells were then preincubated with or without cytochalasin D (5 μg/ml), which inhibits ingestion and thereby serves as a control for nonspecific adherence of SRBCs. Following preincubation, opsonized SRBCs were added at a cell-to-target cell ratio of 30:1 and incubated for an additional 45 min at 37°C. This cell-to-target cell ratio was determined to be optimal on the basis of preliminary dose-response experiments. Cells were then washed with phosphate buffer solution to remove noningested erythrocytes and lysed in 0.3% SDS. Subsequently, o-phenylenediamine dihydrochloride solution was added to each well as a chromogen. Phagocytosed SRBCs were quantitated by measuring A450 (absorbance at 450 nm) values with an automated reader (VersaMax; Molecular Devices). Values were corrected for cytochalasin D-treated wells (background control) and expressed as a percentage of the phagocytic ability of untreated cells (control). Six replicates for each condition were assessed in each of at least four independent experiments.
Liposome-delivered Ab blockade
Liposome-based Ab complexes were introduced into NR8383 macrophages as described previously (11, 12). Briefly, cells were treated with Lipofectin reagent (1% v/v) for 3 h in serum-free medium, with or without anti-Rap1 (1/500), anti-RhoA (1/500), anti-C3G (1/500), or isotype control mouse monoclonal IgG (1/500). Cells were replenished with fresh serum-free medium and allowed to stabilize for at least 1 h before phagocytosis assays were performed. Results are expressed as a percentage of control, to which only vehicle was added.
Phagosome isolation
Phagosomes were isolated as described previously (13). Briefly, cells were held on ice for 10 min before the addition of 30:1 IgG-conjugated paramagnetic beads so that phagocytosis could be synchronized and followed kinetically. Cells were then incubated at 37°C and IgG-bead phagosomes were isolated according to a published method (14) 5, 10, 15 and 30 min after addition of beads. To isolate IgG-bead-containing phagosomes, macrophages were rinsed twice in PBS and lysed in ice-cold homogenization buffer (250 mM sucrose, 10 mM HEPES, 1 mM EDTA (pH 7.2), Roche protease inhibitor cocktail, 10 μg/ml aprotinin and leupeptin, 1 mM PMSF, and 1% Triton X-100). Bead phagosomes were isolated from cell lysates using a magnet (Qiagen) and washed twice in homogenization buffer without Triton X-100. Phagosomal proteins were removed from the beads by sonication followed by boiling for 3 min. The beads were removed with a magnet, and the solubilized material from phagosomes was used as a source of phagosomal proteins for subsequent electrophoresis and immunoblot analysis. Plasma membrane contamination was determined by quantitating the integral plasma membrane protein CD45 (data not shown).
GST-RalGDS glutathione-agarose bead generation
GST-RalGDS coupled to glutathione agarose beads was generated as described previously (13). Briefly, BL21 E. coli transformed with the GST-Ral-GDS-expressing vector were inoculated in Luria-Bertani medium-ampicillin containing 0.1 mM isopropyl β-d-1-thiogalactopyranoside. Bacteria were pelleted, resuspended, and sonicated; the resulting proteins were solubilized in 1% Triton X-100 and incubated with glutathione agarose beads. Beads were then washed and diluted in PBS to generate a 50% slurry, which was added 1:1 to glycerol and snap frozen in liquid nitrogen.
Rap1 pulldown assay
Active levels of Rap1 were measured as described previously (13). Briefly, cells were lysed on ice in Rap1 lysis buffer (25 mM Tris-HCl (pH 7.5), 1% Nonidet P-40, 5 mM MgCl2, 150 mM NaCl, 0.1 mM DTT, 5% glycerol, Roche protease inhibitor cocktail, 10 μg/ml aprotinin and leupeptin, 1 mM PMSF). Lysates were incubated with glutathione agarose beads coupled to GST-RalGDS. Beads were washed three times in lysis buffer and resuspended in 1× sample buffer. Samples were resolved with SDS-PAGE, and the membranes were probed with anti-Rap1 rabbit polyclonal Ab (1/500). Total Rap1 levels were determined by removing aliquots from cell lysates before incubation with beads and blotting for Rap1. Bound primary Ab was visualized with HRP-conjugated secondary Ab and developed with ECL reagent. Western blot band intensities were quantified with Scion Image (National Institutes of Health, Frederick, MD).
Immunoprecipitation and Western blotting
NR8383 macrophages were lysed using ice-cold lysis buffer (25 mM Tris (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM PMSF, Roche protease inhibitor cocktail). Lysates were precleared with protein A-Sepharose beads for 30 min and incubated overnight at 4°C with either anti-CrkL or anti-C3G Ab (1/80). Protein A-Sepharose beads were added the subsequent day and incubated for 4 h. Beads were washed at least three times in lysis buffer without Triton X-100 and resuspended in 1× sample buffer. Proteins were eluted by boiling and submitted to SDS-PAGE. Membranes were probed with anti-CrkL (1/1000), anti-C3G (1/500), anti-PY20 (1/500), or anti-Cbl (1/1000) Abs. Bands were detected by ECL.
Immunofluorescence microscopy
NR8383 macrophages were grown on four-chamber slides and incubated with opsonized RBCs for varying time points. Slides were fixed in 4% paraformaldehyde in PBS for 30 min at room temperature and permeabilized with 0.5% Triton X-100 in PBS for 3 min. The cells were then blocked with 1% BSA in PBS and incubated with mouse anti-C3G Ab (1/200) in blocking buffer for 1 h at room temperature. Slides were washed in PBS and incubated with rhodamine-conjugated goat anti-mouse secondary Ab (1/200) in blocking buffer for 1 h. Mounting was done in fluorescent mounting medium (Vector Laboratories). Macrophages were visualized and imaged using a Nikon E600 microscope equipped for epifluorescence and digital image capture using a SPOT RT camera.
Statistical analysis
Data are represented as means ± SE and were analyzed with the Prism 5.0 statistical program (GraphPad Software). Comparisons between two experimental groups were performed with the Student’s t test. Comparisons among three or more groups were performed with ANOVA followed by Dunnett’s multiple comparison test. Differences were considered significant if p < 0.05.
Results
Rap1 is activated by FcγR crosslinking and localizes to the phagosome
To determine whether Rap1 is activated in response to FcγR crosslinking, we performed pulldown assays to precipitate active, GTP-bound Rap1. Addition of IgG-opsonized SRBCs to NR8383 macrophages induced a rapid activation of Rap1 (Fig. 1A), while unopsonized SRBCs had no effect (Fig. 1C). Densitometric analysis indicated that activity exhibited a slight increase at 1 min, reached statistical significance and peaked 5–10 min after ligation of the FcγR, and persisted at significant levels for least 30 min (Fig. 1B).
FIGURE 1.
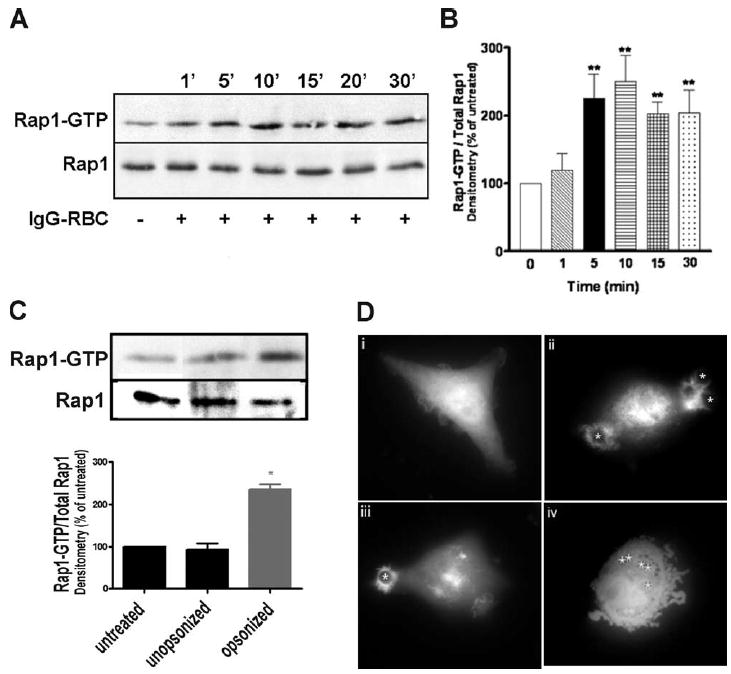
FcγR ligation activates Rap1. NR8383 macrophages were incubated with 30/1 (A) IgG-opsonized or (C) unopsonized SRBCs for varying time points, lysed, and assayed for GTP-bound Rap1 as described in Materials and Methods. Total levels of Rap1 in cell lysates are shown (bottom blot). No crossreactivity was observed between the Rap1 Ab and SRBC proteins (data not shown). B, Densitometric analysis of Rap1 pulldowns from cells challenged with IgG-opsonized SRBCs was performed as described in Materials and Methods. Results represent the means (±SE) for 10 independent experiments. **, p < 0.01 compared with control by ANOVA followed by Dunnett’s multiple comparison test. D, GFP-RalGDS-transfected macrophages were overlaid with 10/1 IgG-opsonized SRBCs (denoted by asterisks) and imaged using fluorescence microscopy at (i) 0 min, (ii) 5 min, and (iii) 10 min. Negative control GFP-transfected macrophages were overlaid with IgG-opsonized latex beads and imaged at (iv) 10 min. Images are representative of at least three separate experiments.
As a complementary approach, we examined the spatial distribution of active Rap1. Because there are no commercially available Abs that distinguish between active and inactive forms of Rap1, we transiently transfected NR8383 macrophages with GFP-RalGDS, a fluorescent probe specific for active Rap1GTP (15). In resting cells, GFP-RalGDS was diffusely spread throughout the entire cell (Fig. 1Di). However, upon challenge with IgG-opsonized targets, active Rap1 appeared to localize near the phagosome both at 5 min (Fig. 1Dii) and 10 min (Fig. 1Diii). This was in sharp contrast to negative control GFP-transfected NR8383 cells, which remained uniformly fluorescent when challenged with IgG-opsonized latex beads (Fig. 1Div). These observations further support the utility of GFP-RalGDS as a marker for detecting Rap1GTP, and suggest that ligation of the FcγR results in a spatial redistribution of active Rap1.
Rap1 activation is essential for FcγR-mediated phagocytosis
The findings that Rap1 is rapidly activated by FcγR crosslinking and localizes near the phagosome suggested that it may participate in FcγR-mediated phagocytosis. To assess whether active Rap1 is functionally necessary for this process to occur, we stably transfected NR8383 macrophages with Rap1GAP, a GTPase-activating protein that specifically promotes the conversion of Rap1GTP to Rap1GDP (Fig. 2A). Rap1GAP has been reported to indiscriminately target and inactivate both isoforms of Rap1 (Rap1a, Rap1b) (16). We chose to use the Rap1GAP expression vector rather than the Rap1N17 dominant-negative construct in light of previous reports that Rap1N17 is unable to inhibit C3G-mediated Rap1 activation (17). The relevance of this limitation will become apparent. As expected, expression of Rap1GAP abrogated the increase in Rap1GTP induced by challenging macrophages with IgG-opsonized targets, while expression of the control vector pcDNA3.1 did not (Fig. 2A, top panel). This was confirmed by densitometric analysis (Fig. 2A, bottom panel). We consistently found that Rap1GAP-transfected macrophages had higher basal levels of Rap1 activity, for reasons that remain unclear. As indicated in Fig. 2B, Rap1GAP expressing NR8383 cells were severely inhibited in their capacity to ingest IgG-opsonized RBCs, with the rate of ingestion being reduced by 72.0 ± 4.5%.
FIGURE 2.
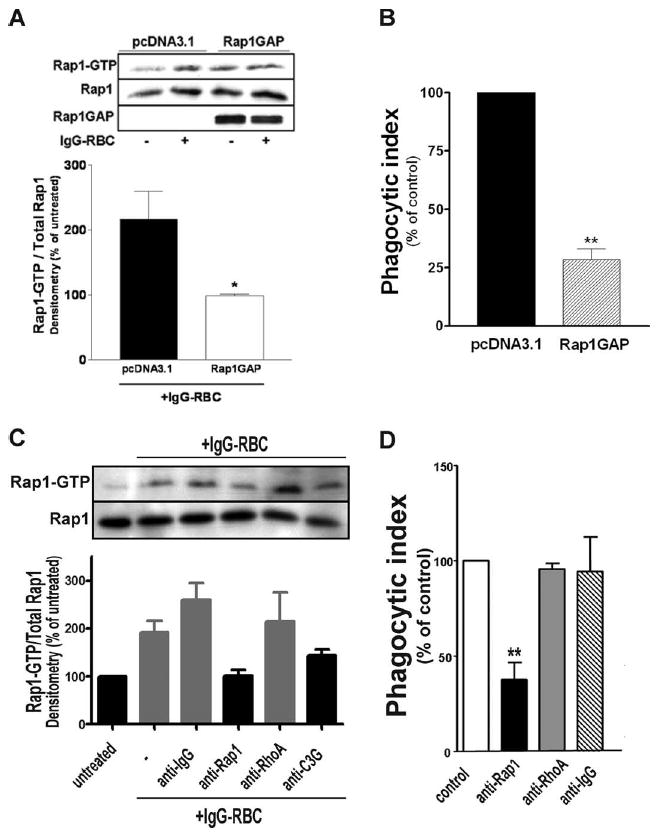
Inhibition of Rap1 activity inhibits FcγR-mediated phagocytosis. A, pcDNA3.1-and Rap1GAP-transfected NR8383 macrophages were stimulated with 30/1 IgG-opsonized SRBCs for 7 min, lysed, and assayed for GTP-bound Rap1 as described in Materials and Methods. Aliquots were removed from lysates before incubation with beads and immunoblotted for Rap1 and Rap1GAP. Densitometric analysis was performed for each Western blot as described in Materials and Methods. Data shown represent values relative to untreated pcDNA3.1-and Rap1GAP-transfected macrophages, and are the means ± SE of three independent experiments. *, p < 0.05 compared with vehicle macrophages by Student’s t test. B, pcDNA3.1- and Rap1GAP-transfected macrophages were assessed for phagocytic ability as described in Materials and Methods. NR8383 macrophages preincubated with liposome constructs containing anti-Rap1, anti-RhoA, anti-C3G, or isotype control Ab as detailed in Materials and Methods were assessed for (C) GTP-bound Rap1 and (D) phagocytic ability. Data shown are the means ± SE of three independent experiments. **, p < 0.01 compared with control by Student’s t test.
As an alternative approach, we sought to functionally “knock down” the activity of Rap1 without affecting its expression levels. Both monoclonal anti-Rap1 IgG and its corresponding isotype control IgG were complexed with Lipofectin to facilitate their delivery into the cytosol of the cell before target challenge. Preincubation with anti-Rap1containing liposomes significantly decreased the levels of Rap1 activity induced by target challenge, while incubation with isotype control had no effect (Fig. 2C). Furthermore, consistent with the impairment in phagocytosis of Rap1GAP-expressing macrophages, NR8383 cells incubated with anti-Rap1containing liposomes manifested a 64.0 ± 7.5% reduction in their capacity to phagocytose (Fig. 2D). Neither the vehicle alone nor the isotype control antibody-containing liposomes had any effect on phagocytic ability, suggesting that the observed effect of the Rap1-blocking Ab was specific. To further verify the specificity of our approach, we introduced mAbs against RhoA, a GTPase reported to be unimportant for FcγR-mediated phagocytosis (2, 18), into NR8383 cells. Not surprisingly, these macrophages were unaffected both in their ability to increase Rap1 activity upon FcγR ligation (Fig. 2C) and to ingest IgG-opsonized targets (Fig. 2D). Taken together, these data suggest a crucial role for Rap1 in facilitating FcγR-mediated phagocytosis.
Rap1 activation by FcγR crosslinking is independent of cAMP and Ca2+, but is downstream of Src kinase
The conversion of Rap1GDP to Rap1GTP is catalyzed by GEFs such as cAMP-dependent Epac (8), Ca2+/diacylglycerol-dependent CalDAG-GEF (7), and C3G (6). Activation of Rap1 by ligation of the TCR, which contains intracellular tyrosine activated motifs similar to that of the FcγR, has been shown to be dependent on Ca2+ (19). Therefore, we sought to determine whether this requirement also pertained to FcγR-dependent activation of Rap1 in NR8383 macrophages. Surprisingly, pretreatment with the intracellular calcium chelator BAPTA-AM before stimulation with IgG-opsonized targets was not only unable to block Rap1 activation, but instead increased basal levels of Rap1GTP (Fig. 3A). The adenylyl cyclase inhibitor SQ22536 demonstrated the same inability to block Rap1 activation. Not surprisingly, the simultaneous use of both compounds also failed to block the increase in GTP-bound Rap1 (Fig. 3A, left panel). This was confirmed by densitometric analysis, as the effect of each pharmacological inhibitor on the fold increase in Rap1 activity induced by FcγR ligation was found to be statistically insignificant by ANOVA (Fig. 3A, right panel). Taken together, these data strongly suggest that Rap1 activation by FcγR crosslinking is independent of both the cAMP-dependent GEF Epac-1 and the Ca2+-activated GEF CalDAG.
FIGURE 3.
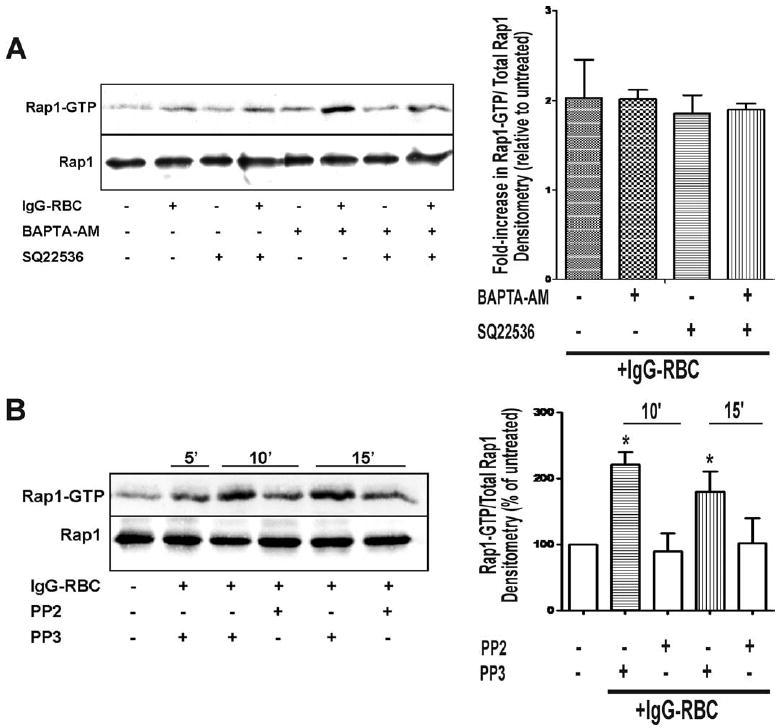
Rap1 activation by FcγR ligation is independent of both cAMP and Ca2+ but is downstream of Src kinase. NR8383 macrophages were pretreated with (A) calcium chelator BAPTA-AM (10 μM), adenylyl cyclase inhibitor SQ22536 (10 μM), both BAPTA-AM and SQ22536 (10 μM each), or (B) Src kinase inhibitor PP2 and negative control PP3 for 30 min before the addition of 30/1 IgG-opsonized SRBCs. Cells were lysed and assayed for GTP-bound Rap1 as described in Materials and Methods. Total levels of Rap1 in cell lysates are shown (bottom blot). One blot representative of a total of four is shown; densitometric analysis was performed for each of the four Western blots as described in Materials and Methods.
In light of these negative data, we next hypothesized that C3G, a GEF known to be activated by Src kinases (20) but independent of the second messengers cAMP and Ca2+, is responsible for mediating Rap1 activation under our experimental conditions. Such a hypothesis would be consistent with a previous report showing that knocking down CrkII, an adapter molecule for C3G, in RAW 264.7 macrophages significantly inhibits FcγR-mediated phagocytosis (21). We therefore pretreated NR8383 macrophages with the Src kinase inhibitor PP2, a well-known inhibitor of FcγR-mediated phagocytosis (22), before the addition of IgG-opsonized RBCs and found that the compound substantially attenuated, albeit not completely, the levels of GTP-bound Rap1 (Fig. 3B, left panel). This was confirmed by densitometric analysis, as the inhibitory effect of PP2 on FcγR-induced Rap1 activation was found to be statistically significant at both 10 and 15 min (Fig. 3B, right panel). The negative control PP3 did not have any effects on Rap1 activation (Fig. 3B, left panel), suggesting that the observed effect of PP2 was specific. We were also able to verify that pretreatment of NR8383 macrophages with PP2, but not PP3, significantly inhibited FcγR-mediated phagocytosis (data not shown). PP2 inhibited IgG-opsonized sRBC ingestion by 73.11 ± 6.54%, while PP3 had no effect. These data indicate that Rap1 activation was dependent on a Src kinase, consistent with a role for the Src kinase-dependent GEF, C3G.
C3G is activated by FcγR crosslinking and localizes to the phagosome
C3G has been reported to be in its activated state when it forms a complex with CrkL and tyrosine-phosphorylated Cbl (5, 23). To confirm that stimulation of NR8383 macrophages results in C3G activation, we sought evidence for formation of such complexes by performing immunoprecipitation experiments with various Abs. As illustrated in Fig. 4A, FcγR ligation resulted in a rapid and transient increase in interaction between CrkL and C3G. Furthermore, there was a more persistent association of a 116-kDa tyrosine phosphorylated protein with CrkL upon binding of target to the FcγR (Fig. 4B, top panel). This band co-migrated with Cbl (Fig. 4B, middle panel). These data are consistent with the hypothesis that C3G is activated by FcγR crosslinking.
FIGURE 4.
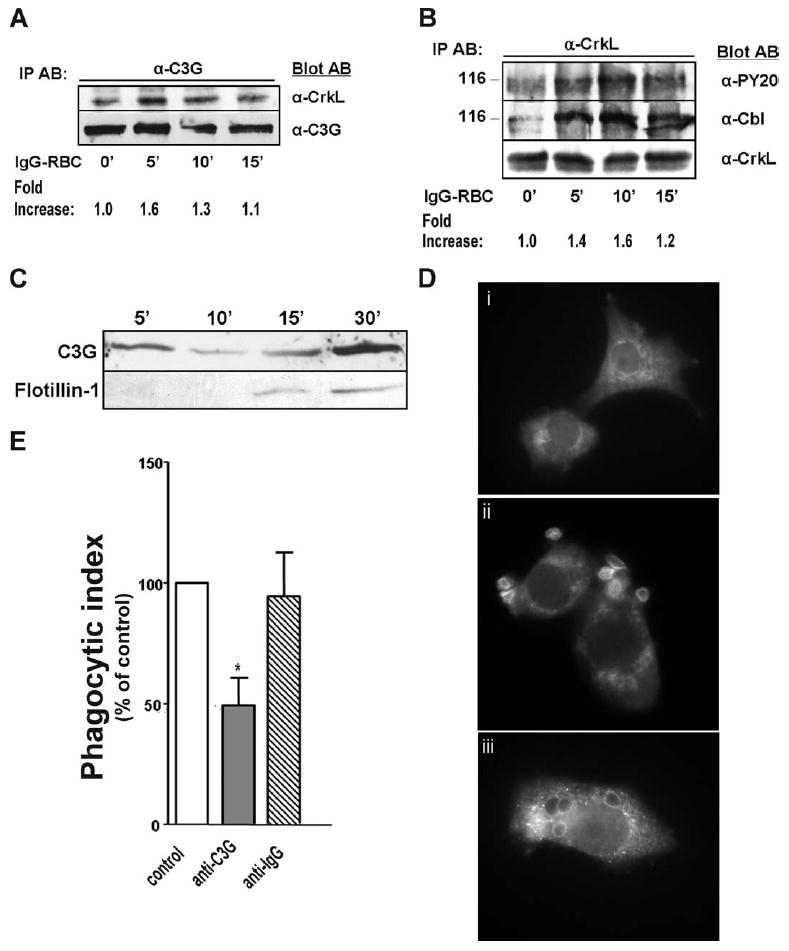
C3G is activated by FcγR ligation and translocates to the phagosome. NR8383 macrophages were incubated with 30/1 IgG-opsonized SRBCs for varying time points, lysed, and immunoprecipitated with (A) anti-C3G and (B) anti-CrkL Abs. Bound proteins were immunoblotted with Abs indicated. Fold increase in (A) CrkL or (B) phosphorylated tyrosine at 116 kDa was measured by densitometric analysis for each of the three Western blots and normalized to unstimulated cells. One blot representative of a total of three is shown. C, Phagosomal membranes were harvested from NR8383 macrophages stimulated with 30/1 IgG-opsonized magnetic beads as described in Materials and Methods. As a late phagosome marker, membranes were probed for flotillin-1 (1/1000, BD Biosciences). D, NR8383 macrophages were overlaid with 10/1 IgG-opsonized SRBCs for (i) 0 min, (ii) 3 min, and (iii) 15 min and immunostained for C3G. Images are representative of at least three separate experiments. E, NR8383 cells incubated with liposome constructs containing anti-C3G or isotype control Ab as detailed in Materials and Methods were assessed for phagocytic ability. Data shown are the means ± SE of three independent experiments. *, p < 0.05 compared with vehicle macrophages by Student’s t test.
We next determined whether C3G translocates to the phagosome by harvesting phagosomal membrane proteins at different time points after addition of IgG-labeled magnetic beads. We verified the purity of our isolations by confirming the lack of CD45, a plasma membrane protein, in our phagosomal fractions (data not shown). Interestingly, C3G was consistently found to accumulate on phagosomes at early time points (i.e., 5 min), to then become less abundant at 10–15 min, and to reaccumulate at 30 min (Fig. 4C, top panel), suggesting a dynamic and possibly biphasic interaction between C3G and the phagosome. By contrast, the phagosomal maturation marker flotillin-1 was found to gradually accumulate over time (Fig. 4C, bottom panel).
To confirm our biochemical data, we examined the spatial localization of C3G by indirect immunofluorescence microscopy. In resting cells, C3G was found to localize perinuclearly (Fig. 4Di); however, upon addition of IgG-opsonized targets to NR8383 macrophages, C3G rapidly translocated to the site of target binding within 5 min (Fig. 4Dii) and to the fully formed phagosome at 15 min (Fig. 4Diii).
C3G is essential for FcγR-mediated phagocytosis
Our findings that C3G is activated by FcγR crosslinking and localizes near the phagosome suggested that it may play a critical role during FcγR-dependent phagocytosis as the upstream activator of Rap1. To address this possibility, we performed blocking Ab experiments by introducing anti-C3G or its corresponding isotype control into the cytosol of NR8383 macrophages. Preincubation with anti-C3G-containing liposomes significantly decreased the levels of Rap1 activity induced by target challenge, while incubation with isotype control had no effect (Fig. 2C). Consistent with our Rap1 pulldown data, phagocytosis of IgG-opsonized SRBCs by cells incubated with anti-C3G liposome complexes was severely impaired by 51.7 ± 11.3% (Fig. 4E). This was in sharp contrast to NR8383 macrophages incubated with control isotype containing liposomes, which were completely unaffected in their ability to ingest targets (Fig. 4E).
C3G and Rap1 are both recruited to phagosomes
Based on our findings that both Rap1 and C3G are activated by FcγR ligation, we sought to confirm that they are both recruited to the forming phagosome. To do this, we analyzed the spatial distribution of both Rap1GTP and C3G in GFP-RalGDS-transfected NR8383 macrophages at 0, 5, and 10 min postchallenge. In resting cells, C3G was perinuclear (Fig. 5A) while GFP-RalGDS was distributed diffusely throughout the cell (Fig. 5B). Binding of target resulted in the translocation of both C3G (Fig. 5D) and Rap1GTP (Fig. 5E), with enrichment occurring near the phagosome at 5 min. This accumulation was particularly noticeable for C3G, as evidenced by the now weakly stained perinuclear region. Both active Rap1 (Fig. 5G) and C3G (Fig. 5H) appeared to remain associated near the phagosome at 10 min; interestingly, the distribution of C3G was found to be more scattered throughout the cell at 10 min (Fig. 5G) than at 5 min (Fig. 5D). This was consistent with our data obtained using phagosome preparations (Fig. 4C), in which we found more C3G on the phagosomal membrane at 5 min than at 10 min.
FIGURE 5.
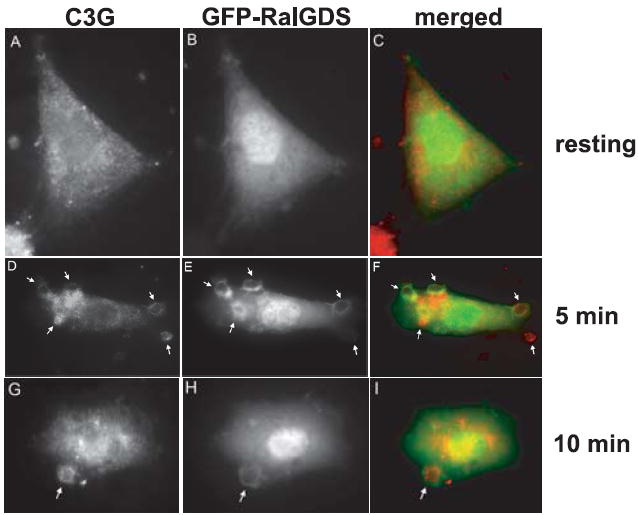
C3G and Rap1GTP both translocate to the phagosome upon FcγR ligation. GFP-RalGDS-transfected macrophages were overlaid with 10/1 IgG-opsonized SRBCs for (B) 0 min, (E) 5 min, and (H) 10 min and immunostained for C3G (A, D, and G). Merged images (C, F, and I) show GFP-RalGDS in green and C3G in red. Arrows indicate location of bound SRBCs on NR8383 cells. Images are representative of at least three separate experiments.
FcγR-dependent Rap1 activation increases Mac-1 affinity
Optimal FcγR-dependent phagocytosis is dependent on the functional activation of Mac-1 (also known as αMβ2, CD11b/CD18, or CR3), a heterodimeric receptor that is a member of the β2 integrin family (24). Rap1 has been reported to be a crucial mediator of LPS and other proinflammatory cytokine-induced integrin activation (4, 25), as reflected by either an increase in affinity or avidity for ligands. We therefore hypothesized that the role of Rap1 during FcγR-dependent phagocytosis is to functionally activate Mac-1 and ensure optimal target ingestion. To assess this possibility, we performed flow cytometric analysis with PE-conjugated CBRM1/5, a mAb that binds to an activation-specific epitope of Mac-1. Initial experiments in U937 human monocytes verified an increase in the availability of the Mac-1 activation epitope upon FcγR ligation. This increase was significantly reduced by expression of the Rap1GAP plasmid, but not by the expression of the control vector, pcDNA3.1 (Fig. 6B). In sharp contrast, and consistent with previous reports that AMs do not express Mac-1 (26), the conformation-specific Ab failed to recognize any epitopes on the AM-derived NR8383 cells, even upon stimulation with PMA (data not shown). Thus, while Mac-1 activation may contribute to Rap1 effects on FcγR-dependent phagocytosis in some macrophage populations, it cannot in AMs.
FIGURE 6.

FcγR-mediated Rap1 activation increases Mac-1 affinity in U937 cells. Flow cytometric analysis with CBRM1/5, an Ab that recognizes an activation epitope of Mac-1, was performed on (A) unstimulated and (B) FcγR-ligated U937 monocytes transfected with pcDNA3.1 or Rap1GAP as described in Materials and Methods. Histograms are representative of five separate experiments.
Discussion
Members of several different low-molecular mass GTPase families, including the Rho and Arf families (27), have been implicated to play essential roles in mediating FcγR-dependent phagocytosis. Herein, we provide evidence for the activation of Rap1, a member of the Ras family of GTPases, as another critical step in facilitating this process. Furthermore, we show that FcγR-dependent Rap1 activation is cAMP- and Ca2+-independent but Src kinase-dependent, and therefore it is likely mediated by C3G, a known exchange factor for Rap1.
Inhibition of Rap1 activity in NR8383 macrophages, both by expression of Rap1GAP (Fig. 2B) and liposome-mediated introduction of Rap1 blocking Abs (Fig. 2D), severely impaired their ability to ingest IgG-opsonized RBCs. This observation is consistent with a previous report that Rap1 regulates phagocytosis in Dictyostelium discoideum, which internalizes particles by a non-receptor-mediated process that is nevertheless morphologically similar to the zipper model for FcγR-mediated phagocytosis (28). Various biochemical studies have identified direct interactions between Rap1 and cytoskeletal proteins such as IQGAP1 (29), RIAM (30), and AF-6 (31), suggesting the possibility that active GTP-bound Rap1 assists in the polymerization of actin filaments at the site of membrane beneath the forming phagosome.
During the completion of this study, Li et al. (32) reported that bone marrow-derived macrophages from Rap1a-null mice exhibit increased FcγR-mediated phagocytosis. The authors suggested that these differences were attributable to the ability of Rap1b to translocate to proper internal locales without experiencing competition from Rap1a. Because our experimental approaches sought to indiscriminately inhibit the activity of both the Rap1a and Rap1b isoforms of Rap1, these results neither contradict nor support our data. However, if both studies were deemed to be valid, they would suggest that it is the Rap1b isoform that is necessary for FcγR-mediated phagocytosis as demonstrated herein, and, moreover, that the positive role of Rap1b in this regard outweighs any negative role of Rap1a. Consequently, the efficiency of FcγR-mediated phagocytosis would be expected to reflect the outcome of the competition between the activating effects of Rap1b and the inhibitory effects of Rap1a.
Despite its widely accepted role as an exchange factor for Rap1, there is no information (microscopic, biochemical, or functional) on C3G in phagocytes. Our phagosomal membrane (Fig. 4C) and indirect immunofluorescence (Fig. 4D) data establish that C3G is indeed rapidly recruited to the site of target binding upon FcγR ligation. Interestingly, we did not find that C3G gradually accumulates over time on the phagosome, as do maturation markers such as flotillin-1. C3G levels appeared to peak at early (5 min) and late (30 min) time points, perhaps suggesting a dual role for C3G in mediating not only phagocytosis but also later innate immune events, such as the subsequent generation of reactive oxygen intermediates by the NADPH oxidase complex. This possibility will require direct examination in the future.
A recent study by Wang and colleagues hypothesized that members of the Rap1GEF family selectively couple Rap1 to distinct effector pathways (33). This model is supported by our data showing that C3G, but not cAMP-dependent Epac or Ca2+-dependent CalDAG-GEF, was the upstream activator of Rap1 during FcγR-mediated phagocytosis. Delivery of blocking Abs against C3G into the cytosol of NR8383 macrophages not only decreased levels of Rap1 activation (Fig. 2C), but also inhibited their phagocytic ability (Fig. 4E).
In our studies, we provide evidence supporting the notion that the molecular mechanisms by which Rap1 activation facilitates FcγR-mediated phagocytosis are cell type-dependent. Expression of Rap1GAP in U937 human monocytes significantly decreased Mac-1 functional activation induced by FcγR ligation (Fig. 6B), suggesting that one role of Rap1 during FcγR-mediated phagocytosis is to functionally activate Mac-1 and bolster the integrin-dependent events necessary for effecting the phagocytic response. Consistent with an earlier study reporting that FcγR-induced Mac-1 activation is independent of Ca2+ but dependent on Src family tyrosine kinases (24), FcγR-induced Rap1 activation was Ca2+-independent (Fig. 3A) but Src kinase-dependent (Fig. 3B). On the contrary, NR8383 cells, similar in immunophenotype to the primary AMs from which they were derived, failed to induce conformational changes in Mac-1 upon FcγR ligation. This is in keeping with reports that AMs do not express Mac-1 (26); the alternative Mac-1-independent pathway by which Rap1 activation enhances AM FcγR-dependent phagocytosis will require direct examination in the future.
In summary, we have shown that activation of the Ras-family GTPase Rap1 by the exchange factor C3G is an essential step during the process of FcγR-mediated phagocytosis. Both C3G and Rap1GTP are recruited near the forming phagosome upon FcγR ligation. These data provide new insights into the molecular mechanisms of host defense in the lung milieu.
Acknowledgments
We thank Dr. Philip Stork for the GFP-RalGDS and Rap1GAP constructs and Drs. Johannes Bos and Danny Altschuler for the GST-RalGDS construct.
Footnotes
This work was supported by National Institutes of Health Grant RO1-HL58897 (to M.P.-G.).
Abbreviations used in this paper: AM, alveolar macrophage; CalDAG, calcium diacylglycerol; GAP, GTPase-activating protein; GEF, guanosine exchange factor; Epac-1, exchange protein directly activated by cAMP; RalGDS, Ral guanine dissociation stimulator.
Disclosures The authors have no financial conflict of interest.
References
- 1.Peters-Golden M. The alveolar macrophage: the forgotten cell in asthma. Am J Respir Cell Mol Biol. 2004;31:3–7. doi: 10.1165/rcmb.f279. [DOI] [PubMed] [Google Scholar]
- 2.Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- 3.Jeon TJ, Lee DJ, Merlot S, Weeks G, Firtel RA. Rap1 controls cell adhesion and cell motility through the regulation of myosin II. J Cell Biol. 2007;176:1021–1033. doi: 10.1083/jcb.200607072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caron E, Self AJ, Hall A. The GTPase Rap1 controls functional activation of macrophage integrin αMβ2 by LPS and other inflammatory mediators. Curr Biol. 2000;10:974–978. doi: 10.1016/s0960-9822(00)00641-2. [DOI] [PubMed] [Google Scholar]
- 5.Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T cell anergy: blocking of IL-2 gene transcription by activated Rap1. Science. 1997;278:124–128. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- 6.Gotoh T, Hattori S, Nakamura S, Kitayama H, Noda M, Takai Y, Kaibuchi K, Matsui H, Hatase O, Takahashi H, et al. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol Cell Biol. 1995;15:6746–6753. doi: 10.1128/mcb.15.12.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawasaki H, Springett GM, Toki S, Canales JJ, Harlan P, Blumenstiel JP, Chen EJ, Bany IA, Mochizuki N, Ashbacher A, et al. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc Natl Acad Sci USA. 1998;95:13278–13283. doi: 10.1073/pnas.95.22.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 9.Smacchia C, Rebulla P, Drago F, Morelati F, Pappalettera M, Sirchia G. A micro colorimetric assay using cryopreserved monocytes to evaluate antibody-mediated red cell-monocyte interaction. Haematologica. 1997;82:526–531. [PubMed] [Google Scholar]
- 10.Araki N, Johnson MT, Swanson JA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol. 1996;135:1249–1260. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peres CM, Aronoff DM, Serezani CH, Flamand N, Faccioli LH, Peters-Golden M. Specific leukotriene receptors couple to distinct G proteins to effect stimulation of alveolar macrophage host defense functions. J Immunol. 2007;179:5454–5461. doi: 10.4049/jimmunol.179.8.5454. [DOI] [PubMed] [Google Scholar]
- 12.Canetti C, Serezani CH, Atrasz RG, White ES, Aronoff DM, Peters-Golden M. Activation of phosphatase and tensin homolog on chromosome 10 mediates the inhibition of FcγR phagocytosis by prostaglandin E2 in alveolar macrophages. J Immunol. 2007;179:8350–8356. doi: 10.4049/jimmunol.179.12.8350. [DOI] [PubMed] [Google Scholar]
- 13.Serezani CH, Chung J, Ballinger MN, Moore BB, Aronoff DM, Peters-Golden M. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am J Respir Cell Mol Biol. 2007;37:562–570. doi: 10.1165/rcmb.2007-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrissette NS, Gold ES, Guo J, Hamerman JA, Ozinsky A, Bedian V, Aderem AA. Isolation and characterization of monoclonal antibodies directed against novel components of macrophage phagosomes. J Cell Sci. 1999;112:4705–4713. doi: 10.1242/jcs.112.24.4705. [DOI] [PubMed] [Google Scholar]
- 15.Bivona TG, Wiener HH, Ahearn IM, Silletti J, Chiu VK, Philips MR. Rap1 up-regulation and activation on plasma membrane regulates T cell adhesion. J Cell Biol. 2004;164:461–470. doi: 10.1083/jcb.200311093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Mitra RS, Henson BS, Datta NS, McCauley LK, Kumar P, Lee JS, Carey TE, D’Silva NJ. Rap1GAP inhibits tumor growth in oropharyngeal squamous cell carcinoma. Am J Pathol. 2006;168:585–596. doi: 10.2353/ajpath.2006.050132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Berghe N, Cool RH, Horn G, Wittinghofer A. Biochemical characterization of C3G: an exchange factor that discriminates between Rap1 and Rap2 and is not inhibited by Rap1A(S17N) Oncogene. 1997;15:845–850. doi: 10.1038/sj.onc.1201407. [DOI] [PubMed] [Google Scholar]
- 18.Olazabal IM, Caron E, May RC, Schilling K, Knecht DA, Machesky LM. Rho-kinase and myosin-II control phagocytic cup formation during CR, but not FcγR, phagocytosis. Curr Biol. 2002;12:1413–1418. doi: 10.1016/s0960-9822(02)01069-2. [DOI] [PubMed] [Google Scholar]
- 19.Reedquist KA, Bos JL. Costimulation through CD28 suppresses T cell receptor-dependent activation of the Ras-like small GTPase Rap1 in human T lymphocytes. J Biol Chem. 1998;273:4944–4949. doi: 10.1074/jbc.273.9.4944. [DOI] [PubMed] [Google Scholar]
- 20.Radha V, Rajanna A, Swarup G. Phosphorylated guanine nucleotide exchange factor C3G, induced by pervanadate and Src family kinases localizes to the Golgi and subcortical actin cytoskeleton. BMC Cell Biol. 2004;5:31. doi: 10.1186/1471-2121-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee WL, Cosio G, Ireton K, Grinstein S. Role of CrkII in Fcγ receptor-mediated phagocytosis. J Biol Chem. 2007;282:11135–11143. doi: 10.1074/jbc.M700823200. [DOI] [PubMed] [Google Scholar]
- 22.Huang ZY, Barreda DR, Worth RG, Indik ZK, Kim MK, Chien P, Schreiber AD. Differential kinase requirements in human and mouse Fc-γ receptor phagocytosis and endocytosis. J Leukocyte Biol. 2006;80:1553–1562. doi: 10.1189/jlb.0106019. [DOI] [PubMed] [Google Scholar]
- 23.Amsen D, Kruisbeek A, Bos JL, Reedquist K. Activation of the Ras-related GTPase Rap1 by thymocyte TCR engagement and during selection. Eur J Immunol. 2000;30:2832–2841. doi: 10.1002/1521-4141(200010)30:10<2832::AID-IMMU2832>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 24.Jongstra-Bilen J, Harrison R, Grinstein S. Fcγ-receptors induce Mac-1 (CD11b/CD18) mobilization and accumulation in the phagocytic cup for optimal phagocytosis. J Biol Chem. 2003;278:45720–45729. doi: 10.1074/jbc.M303704200. [DOI] [PubMed] [Google Scholar]
- 25.Bos JL. Linking Rap to cell adhesion. Curr Opin Cell Biol. 2005;17:123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Garn H, Siese A, Stumpf S, Wensing A, Renz H, Gemsa D. Phenotypical and functional characterization of alveolar macrophage subpopulations in the lungs of NO2-exposed rats. Respir Res. 2006;7:4. doi: 10.1186/1465-9921-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Garcia E, Rosales C. Signal transduction during Fc receptor-mediated phagocytosis. J Leukocyte Biol. 2002;72:1092–1108. [PubMed] [Google Scholar]
- 28.Seastone DJ, Zhang L, Buczynski G, Rebstein P, Weeks G, Spiegelman G, Cardelli J. The small Mr Ras-like GTPase Rap1 and the phospholipase C pathway act to regulate phagocytosis in Dictyostelium discoideum. Mol Biol Cell. 1999;10:393–406. doi: 10.1091/mbc.10.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeong HW, Li Z, Brown MD, Sacks DB. IQGAP1 binds Rap1 and modulates its activity. J Biol Chem. 2007;282:20752–20762. doi: 10.1074/jbc.M700487200. [DOI] [PubMed] [Google Scholar]
- 30.Lafuente EM, van Puijenbroek AA, Krause M, Carman CV, Freeman GJ, Berezovskaya A, Constantine E, Springer TA, Gertler FB, Boussiotis VA. RIAM, an Ena/VASP and profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev Cell. 2004;7:585–595. doi: 10.1016/j.devcel.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 31.Boettner B, Govek EE, Cross J, Van Aelst L. The junctional multidomain protein AF-6 is a binding partner of the Rap1A GTPase and associates with the actin cytoskeletal regulator profilin. Proc Natl Acad Sci USA. 2000;97:9064–9069. doi: 10.1073/pnas.97.16.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Yan J, De P, Chang HC, Yamauchi A, Christopherson KW, 2nd, Paranavitana NC, Peng X, Kim C, Munugulavadla V, et al. Rap1a null mice have altered myeloid cell functions suggesting distinct roles for the closely related rap1a and 1b proteins. J Immunol. 2007;179:8322–8331. doi: 10.4049/jimmunol.179.12.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Dillon TJ, Pokala V, Mishra S, Labudda K, Hunter B, Stork PJ. Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol Cell Biol. 2006;26:2130–2145. doi: 10.1128/MCB.26.6.2130-2145.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]