

Abstract
Protein tyrosine phosphatases have only recently become the focus of attention in the search for novel drug targets despite the fact that they play vital roles in numerous cellular processes and are implicated in many human diseases. The hematopoietic protein tyrosine phosphatase (HePTP) is often found dysregulated in preleukemic myelodysplastic syndrome (MDS) as well as in acute myelogenous leukemia (AML). Physiological substrates of HePTP include the mitogen-activated protein kinases (MAPKs) ERK1/2 and p38. Specific modulators of HePTP catalytic activity will be useful for elucidating mechanisms of MAPK regulation in hematopietic cells and may also provide treatments for hematopoietic malignancies such as AML. Here, we report the discovery of phenoxyacetic acids as inhibitors of HePTP. Structure−activity relationship analysis and in silico docking studies reveal the molecular basis of HePTP inhibition by these compounds. We also show that these compounds are able to penetrate cell membranes and inhibit HePTP in human T lymphocytes.
Keywords: HePTP, PTPN7, inhibitors, p38, myelodysplastic syndrome, myelogenous leukemia
Tyrosine phosphorylation is a key mechanism of signal transduction. Abnormalities in protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) have been reported in many inherited or acquired human diseases.1−5 Although the development of specific PTP inhibitors remains a challenging task, many successful examples of targeting PTPs in cellulo and in vivo have been reported, providing opportunities for new treatment strategies for cancer, as well as metabolic and immunological diseases.3−7
Hematopoietic protein tyrosine phosphatase (HePTP)8,9 is a 38 kDa enzyme expressed in bone marrow, thymus, spleen, lymph nodes, and all myeloid and lymphoid lineages and cell lines.8−10 The HePTP gene ptpn7 is located on chromosome 1q3211 and is often duplicated in bone marrow cells from patients with myelodysplastic syndrome,12,13 which is characterized by disturbed hematopoiesis and an increased risk of acute leukemia. Amplification and overexpression of HePTP were also reported in acute myelogenous leukemia (AML).11 The HePTP protein consists of a tyrosine phosphatase domain and a short N-terminal extension, which contains a region referred to as the kinase interaction motif (KIM). Physiological substrates of HePTP include the extracellular signal-regulated kinases 1/2 (ERK1/2) and the p38 mitogen-activated protein kinase (MAPK),14−16 which associate tightly with HePTP via the KIM. In resting T cells, HePTP ensures dephosphorylation of the positive regulatory phosphotyrosine (pTyr) residue in the activation loop of the kinases14,15 and also prevents them from translocating to the nucleus.17 T cell antigen receptor (TCR) ligation results not only in the activation of the MAPKs but also in the phosphorylation of HePTP at Ser-23 by cAMP-dependent kinase, causing the HePTP-MAPK complex to dissociate.15,18 A fraction of activated ERK/p38 is then able to translocate to the nucleus, initiating signaling events that lead to T cell activation.18
Small-molecule inhibitors of PTPs involved in cytosolic MAPK regulation have been reported for the Vaccinia H1-related phosphatase (VHR)19−21 and MAP kinase phosphatase-3 (MKP-3).22 Both enzymes, along with HePTP, share the common physiological target ERK. However, specific modulators of HePTP activity have not yet been described. Here, we report the discovery of a series of phenoxyacetic acids as inhibitors of HePTP. We show that these compounds are able to penetrate cell membranes and inhibit HePTP in human T cells and provide evidence for how these inhibitors interact with HePTP on a molecular level. We also discuss the specificity of these inhibitors for HePTP versus the related phosphatases VHR and MKP-3.
To identify inhibitors of the phosphatase VHR, we recently performed a high-throughput screen (HTS) using the Molecular Libraries Probe Production Centers network (MLPCN, http://mli.nih.gov/mli/mlpcn/). HePTP and MKP-3 were used for counterscreens, which led us to the discovery of the HePTP-selective inhibitors described herein. In the primary HTS, the MLPCN collection of 291018 compounds was screened using a colorimetric phosphatase assay,23 resulting in 1524 actives, that is, compounds that demonstrated an inhibition of VHR of ≥50% at a concentration of 13.3 μM. Detailed results of the screening were uploaded to the PubChem Web site (http://pubchem.ncbi.nlm.nih.gov/; AID 1654). Secondary single-concentration and dose−response assays employed the fluorescein-based substrate 3-O-methylfluorescein phosphate (OMFP). These assays confirmed that 195 hits inhibited VHR in a dose-dependent manner (PubChem AIDs 1878 and 1957). The confirmed hits were then tested in counterscreen assays against HePTP and MKP-3, using the same substrate OMFP. Surprisingly, we identified a compound scaffold that exhibited more potent activity toward HePTP than toward either VHR or MKP-3 (Table 1). This scaffold contained a 2H-thiazolo[3,2-a]pyrimidin-3(5H)-one core structure and a phenoxyacetic acid headgroup. Subsequent structure−activity relationship (SAR) analysis revealed that the phenoxyacetic acid group of these compounds was essential for PTP inhibition, probably by mimicking the tyrosine phosphate group of the natural substrate. Analogue 6, which did not contain the phenoxyacetic acid group, did not inhibit any of the three PTPs at 50 μM concentration. We also found that the substitution pattern at the phenyl ring in the 5-position of the thiazolo-pyrimidinone heterocycle altered the inhibitory activity of the compounds. Inhibitors with small substituents, such as a chlorine atom in the para-position (compounds 1 and 2), performed best against HePTP. In fact, compound 1 was the most selective inhibitor for HePTP, showing 19.5-fold selectivity over VHR and 42-fold selectivity over MKP-3. To evaluate the selectivity of 1 in a wider context of tyrosine-specific PTPs such as HePTP, we performed additional experiments using a standard phosphatase assay and a panel of six recombinant PTPs that are also present in T cells. In these experiments, we found that compound 1 selectively inhibits HePTP over CD45 (8.8-fold), Shp2 (4.1-fold), TCPTP (2.6-fold), Lyp (1.4-fold), LAR (>50-fold), and also PTP1B (4.3-fold). In addition, Michaelis−Menten kinetic studies revealed that compound 1 is a competitive inhibitor with a Ki of 0.69 ± 0.20 μM for HePTP.
Table 1. In Vitro Activity Profiling of the Inhibitors against the Phosphatases HePTP, VHR, and MKP-3.
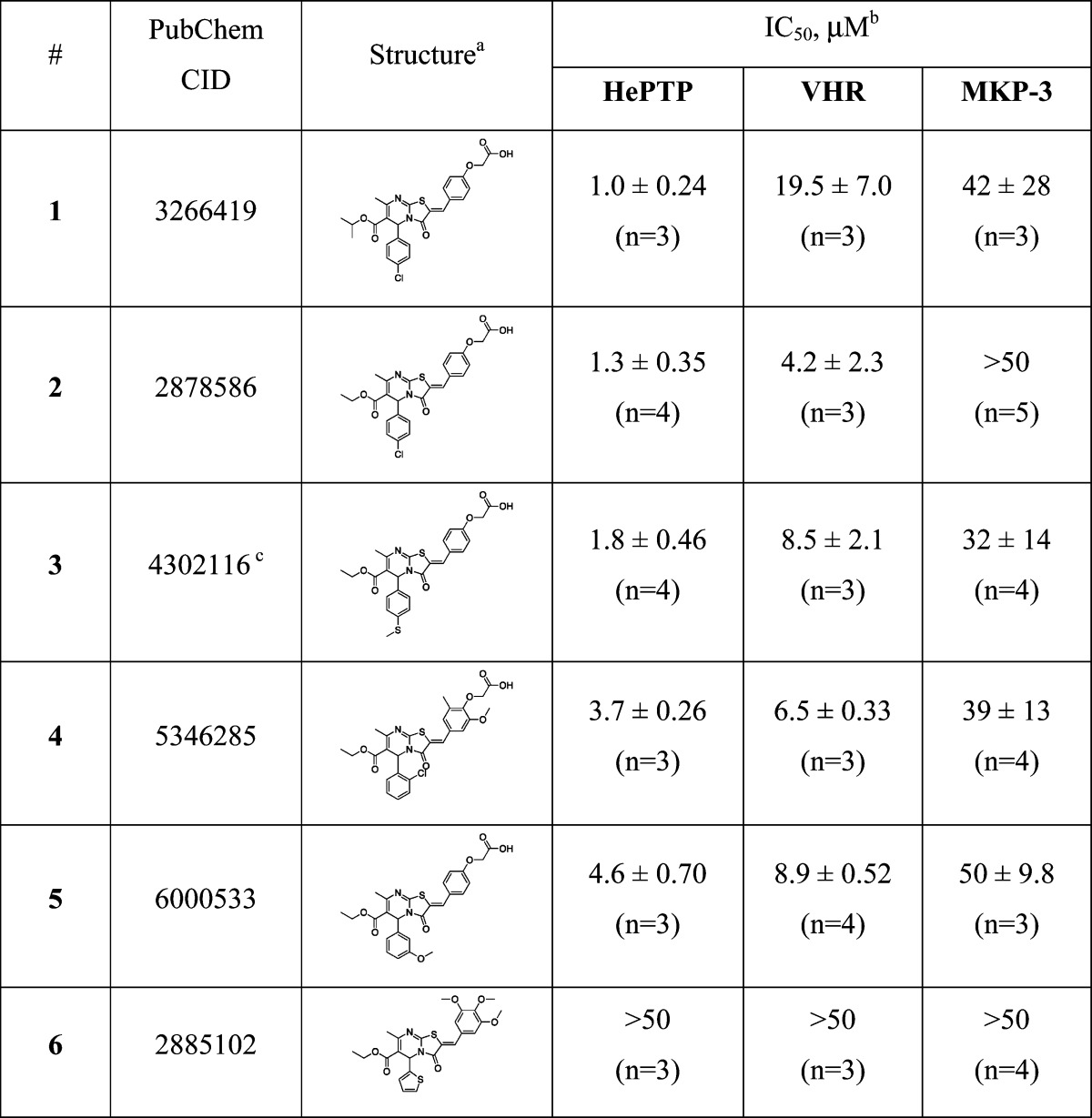 |
All inhibitors were racemic mixtures of a 50:50 ratio of R- and S-enantiomers.
IC50 values are reported as means ± SEMs.
This compound was nominated as molecular probe and has the PubMed probe number ML120.
To understand how the phenoxyacetic acids inhibit HePTP on a molecular level, we used in silico docking to explore potential binding modes of these inhibitors with HePTP. Flexible ligand docking was carried out with the ICM docking algorithm as implemented in the ICM-Pro program (v3.7-1g, Molsoft, LLC). The coordinates of the three-dimensional (3D) structure of HePTP in complex with a phosphorylated peptide corresponding to the ERK activation loop were used for the docking studies (PDB code 3D44, ref (24)). The binding pocket in HePTP was defined as an 8 Å radius around the pTyr residue of the phosphopeptide. Because all inhibitors were racemic mixtures containing both R- and S-enantiomers at a 50:50 ratio, ICM docking scores were calculated for each isomer (Table 2). Structures (R)-1, (R)-2, (R)-3, (R)-5, (S)-1, (S)-2, (S)-3, and (S)-4 docked into the HePTP active site with very good (−34) to excellent (−65) scores (Table 2; scores below −32 are generally regarded as sound). Conversely, structures (R)-4, (R)-6, (S)-5, and (S)-6 failed to dock with reasonable scores and, in fact, were not able to bind into the catalytic pocket. Correlating these docking results with the IC50 values for HePTP obtained from the racemic mixtures of the compounds (Table 1) suggest that the inhibitors fall into three groups. In the first group (compounds 1, 2, and 3), both the R- and S-enantiomers docked very well into the active site, while also exhibiting very low IC50 values, suggesting that both enantiomers inhibit HePTP. In the second group (compounds 4 and 5), the docking scores varied substantially for the individual enantiomers despite the low IC50 values exhibited by the compounds, suggesting that only one isomer inhibited HePTP activity in the biochemical assay. In group three (compound 6), both enantiomers failed to dock into HePTP, consistent with the complete absence of inhibitory activity for this compound.
Table 2. ICM Docking Scores of the Inhibitor Stereoisomers with HePTP, VHR, and MKP-3.
| HePTP |
VHR |
MKP-3 |
||||
|---|---|---|---|---|---|---|
| no. | Ra | Sb | Ra | Sb | Ra | Sb |
| 1 | −64.10 | −58.10 | −41.49 | −31.95 | 4.26 | −25.04 |
| 2 | −65.18 | −57.65 | −40.61 | −34.87 | −1.37 | −24.50 |
| 3 | −60.39 | −59.00 | −40.12 | −33.80 | 1.71 | −24.54 |
| 4 | −13.62 | −42.45 | −26.91 | −11.01 | −21.24 | −4.09 |
| 5 | −34.19 | −10.00 | −39.28 | −33.46 | −3.86 | −21.33 |
| 6 | −13.96 | −2.29 | −9.36 | −8.28 | −0.30 | −4.86 |
ICM score for the R-enantiomer.
ICM score for the S-enantiomer.
To explore if the observed selectivity of the inhibitors for HePTP over VHR and MKP-3 could be explained on the basis of structural differences within the active sites of the three enzymes, we also docked the compounds into the crystal structures of VHR (PDB code 1J4X) and MKP-3 (PDB code 1MKP). While all of the compounds failed to dock into MKP-3, the majority of the compounds could be docked into the catalytic center of VHR, although with significantly lower docking scores as compared to HePTP (Table 2). Thus, as seen for HePTP, the flexible ligand docking results correlated very well with the biochemical assay data, in which low or no inhibitory activity was found toward MKP-3, and moderate inhibitory activity was observed toward VHR.
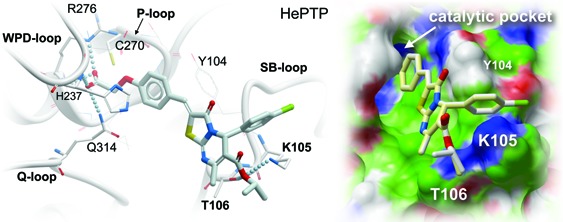
The docking results also suggest how these compounds might interact with residues at the HePTP active site. SAR demonstrated that the phenoxyacetic acid group is critical for inhibition. Indeed, in all successfully docked compounds, this group formed multiple hydrogen bonds with multiple residues in the HePTP catalytic pocket, perfectly mimicking the binding of a phosphate moiety (Figure 1). Specifically, hydrogen bonds were formed between the guanidinium nitrogens and the backbone nitrogen of the invariant Arg-276 of the phosphate-binding loop (P-loop), the backbone nitrogen of His-237 of the WPD-loop, and the side chain of the conserved Gln-314 of the Q-loop. In addition, the phenyl ring of the phenoxyacetic acid group was involved in π−π stacking interactions with Tyr-104 of the substrate-binding loop (SB-loop), which is not present in MKP-3 or VHR. Critically, the inhibitors were also found to interact with two other residues of the SB-loop, Lys-105 and Thr-106 (Figure 1). In all successfully docked molecules, the ε-amino group of Lys-105 and/or the hydroxyl group of Thr-106 formed a hydrogen bond with the oxygen atom of the ester group in the 6-position of the heterocycle. Neither Lys-105 nor Thr-106 is conserved in VHR or MKP-3 (the corresponding loop containing these residues is missing in MKP-3 and much shorter in VHR), suggesting that these interactions may contribute to the selective inhibition of HePTP. In fact, a threonine residue at position 106 is only found in the three KIM-containing PTPs,25 including HePTP, striatum-enriched phosphatase (STEP), and the STEP-like PTP (PTP-SL/PCPTP1), the latter two of which are not expressed in T cells as they are expressed predominantly in the brain.24 To give further evidence for such a binding mode in which the inhibitor specifically interacts with these two residues, we used site-directed mutagenesis to mutate Lys-105 and Thr-106 to alanines and tested compound 1 against the recombinant HePTP double mutant. Using 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) as small artificial substrate, kinetic parameters including Km and Vmax were similar between wild type and mutant protein, which allowed us to directly compare the IC50 values of the inhibitor. Interestingly, compound 1 was 2.1-fold less active against the K105A/T106A mutant enzyme, clearly demonstrating that the two residues are involved in inhibitor binding. The fact that the difference in inhibition is modest is likely due to favorable hydrophobic interactions between the ethyl ester group of compound 1 and the methyl groups of the mutated alanine residues in HePTP. Taken together, our computational studies revealed molecular details about a possible binding mode for the phenoxyacetic acids with HePTP, as well as interactions that may confer selectivity of these compounds for HePTP. Importantly, the results obtained from the docking studies were in excellent agreement with the biochemical data, including mutagenesis studies.
Figure 1.

In silico docking of phenoxyacetic acid enantiomers into the HePTP active site (PDB code 3D44). Upper panel: Surface representation of HePTP active site residues with docked compounds (R)-1 (white), (R)-2 (light yellow), (R)-3 (orange), (R)-5 (blue), (S)-1 (light blue), (S)-2 (light orange), (S)-3 (light green), and (S)-4 (magenta). Surface color code: white, neutral; green, hydrophobic; red, hydrogen bond acceptor potential; and blue, hydrogen bond donor potential. Lower panel: Expanded stereo ribbon diagram of the docking poses for the R- and S-enantiomers of inhibitor 1. Hydrogen-bonding interactions are shown as lines of spheres. HePTP amino acid residues that are involved in hydrogen-bonding interactions, as well as the catalytic Cys-270, are shown in stick representation. Other residues in close proximity to each enantiomer (5 Å) are shown in line representation. Docking and image preparation were done with ICM-Pro (v3.7-1g, Molsoft, LLC).
To assess the ability of these compounds to inhibit HePTP activity in intact human cells, we performed T cell-based assays to probe phosphorylation sites in the direct substrates of HePTP. For the immunoblot assays, we preincubated Jurkat T cells with compound 1 or 2 at concentrations of 5 and 40 μM or vehicle (DMSO) at 37 °C for 45 min. Cells were then TCR-stimulated for 5 min, and reactions were stopped by adding lysis buffer. Immunoblotting was performed with phospho-ERK1/2 (pERK) and phospho-p38 (pp38) specific antibodies. Activation of the c-Jun N-terminal kinase (JNK), which is a target for VHR but not for HePTP, was measured to assess the specificity of the inhibitors in cells. The results show that both compounds 1 and 2 at concentrations of 5 and 40 μM augmented pp38 levels, while pJNK levels were not affected (Figure 2; as can be seen, the pp38 levels increase by 1.35−2.04-fold in the presence of inhibitor). The effect was greater at 40 μM, demonstrating a dose-dependent efficacy of the two inhibitors. Notably, the compounds did not increase ERK activation, suggesting redundancies in ERK dephosphorylation by MKP-3 and/or VHR, the only other two PTPs known to act on ERK at this early time point in TCR signaling. Given the fact that MKP-3 was considerably less or not inhibited by the compounds in the phosphatase assay and that VHR activity was unaffected in cells (as measured by the unchanged pJNK levels), redundancy in deactivating ERK through these two PTPs seems likely and would explain the observed results. Along those lines, knocking down HePTP protein by ∼80% was previously shown to increase pERK levels by only a small amount.26 Importantly, we demonstrated that the phenoxyacetic acid inhibitors were able to penetrate cell membranes and inhibit HePTP in cells. Moreover, these data suggest that it is indeed possible to selectively modulate the p38 MAPK pathway with these small molecules. T cells treated with the inactive analogue 6 did not show any changes in MAP kinase activation, showing that the augmented p38 phosphorylation by compounds 1 and 2 is not due to nonspecific effects (data not shown).
Figure 2.

Immunoblot analysis of HePTP inhibitors in Jurkat TAg T cells. Cells were incubated with vehicle (DMSO, 0.2%) or racemic compounds 1 or 2 at 5 or 40 μM concentration for 45 min at 37 °C prior to TCR stimulation with OKT3 for 5 min. Cell lysates were probed for the direct substrates of HePTP with phospho-specific antibodies against phospho-p38 (pp38) and phospho-ERK1/2 (pERK). As a specificity control, lysates were also tested for phospho-JNK (pJNK) levels. Antibodies against ERK, p38, and JNK were used on stripped membranes to control equal loading between lanes.
In conclusion, we identified a series of HePTP-specific inhibitors with low micromolar activities in vitro and in cells. SAR analysis revealed that the phenoxyacetic acid group was essential for the inhibitory activity of the compounds, suggesting that this group mimics the pTyr of the natural substrate. Indeed, in silico docking studies illustrated that this group interacts strongly with the P-loop in the catalytic pocket of HePTP. These studies, in combination with mutagenesis experiments, also identified favorable interactions of the compounds with the less conserved residues Lys-105 and Thr-106 in the HePTP SB-loop and support the strategy of selective PTP inhibition through protein−ligand interactions peripheral to the catalytic pocket.23,27,28 The fact that the docking scores varied substantially between the different enantiomers for some of the inhibitors indicates that future studies with enantiomerically pure compounds are warranted and will be important for further optimization of the compounds. It also indicates that the IC50 values for enantiomerically pure compounds are likely lower than reported. Importantly, we found the HePTP inhibitors to be active in T lymphocytes at concentrations as low as 5 μM. These assays also demonstrated that at the tested concentrations the compounds did not inhibit VHR in cells. In fact, we observed a specific effect on p38 activation, while the pERK substrate was not affected. Thus, these compounds will be useful to specifically modulate p38 signaling, while signaling through the Ras-Raf-MEK-ERK pathway will be unaffected. Future studies with these compounds will establish whether pharmacological inhibition of HePTP will provide treatments for various (pre)-leukemic disorders.
Acknowledgments
We are grateful to Dr. Torkel Vang for discussions and advice. We thank Derek Stonich for helping with the deposition of the data at the PubChem website.
Abbreviations
HePTP, hematopoietic protein tyrosine phosphatase; PTP, protein tyrosine phosphatase; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; MKP, MAP kinase phosphatase; VHR, Vaccinia H1-related; pTyr, phosphotyrosine; JNK, c-Jun N-terminal kinase; SAR, structure−activity relationship; OMFP, 3-O-methylfluorescein phosphate; DiFMUP, 6,8-difluoro-4-methylumbelliferyl phosphate.
Supporting Information Available
Detailed information of the experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
E.V.B. was the lead scientist at CPCCG responsible for confirmatory assays, selectivity assays, and SAR; she oversaw S.C. and J.R, who performed the assays and made editing contributions. W.L. conducted the T cell-based assays in L.T.'s laboratory and made editing contributions. S.V. was responsible for performing the HTS at CPCCG and oversaw C.G., who helped with the HTS. DAC expressed and purified HePTP protein and made editing contributions. R.D. provided analytical chemistry support and quality-controlled commercially available compounds. Y.S. provided cheminformatic support at CPCCG. E.S. was the overseeing PI of the HTS phosphatase assay development at CPCCG, performed assays, and oversaw X.C. T.D.Y.C. was the project manager at CPCCG, coordinating and overseeing all efforts regarding HTS and hit follow-up. T.M. made significant intellectual contributions. R.P. made significant intellectual contributions, made editing contributions, and oversaw D.A.C. L.T. was the main PI of the project; he wrote the manuscript, provided significant intellectual input, helped with the assay development, conducted the in silico docking studies, was responsible for the T cell-based assays, and oversaw W.L.
This work was supported by Grants 1R21CA132121 (to L.T.), R03MH084230-01A1 (to L.T.), and U54 HG005033 (to CPCCG) from the National Institutes of Health and Research Scholar Grant RSG-08-067-01-LIB (to R.P.) from the American Cancer Society.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Alonso A.; Sasin J.; Bottini N.; Friedberg I.; Friedberg I.; Osterman A.; Godzik A.; Hunter T.; Dixon J.; Mustelin T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [DOI] [PubMed] [Google Scholar]
- Mustelin T.; Vang T.; Bottini N. Protein tyrosine phosphatases and the immune response. Nat. Rev. Immunol. 2005, 5, 43–57. [DOI] [PubMed] [Google Scholar]
- Tonks N. K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell. Biol. 2006, 7, 833–846. [DOI] [PubMed] [Google Scholar]
- Tautz L.; Pellecchia M.; Mustelin T. Targeting the PTPome in human disease. Expert Opin. Ther. Targets 2006, 10, 157–177. [DOI] [PubMed] [Google Scholar]
- Vang T.; Miletic A. V.; Arimura Y.; Tautz L.; Rickert R. C.; Mustelin T. Protein tyrosine phosphatases in autoimmunity. Annu. Rev. Immunol. 2008, 26, 29–55. [DOI] [PubMed] [Google Scholar]
- Jiang Z. X.; Zhang Z. Y. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev. 2008, 27, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Zhang Z. Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discovery Today 2007, 12, 373–381. [DOI] [PubMed] [Google Scholar]
- Zanke B.; Suzuki H.; Kishihara K.; Mizzen L.; Minden M.; Pawson A.; Mak T. W. Cloning and expression of an inducible lymphoid-specific, protein tyrosine phosphatase (HePTPase). Eur. J. Immunol. 1992, 22, 235–239. [DOI] [PubMed] [Google Scholar]
- Adachi M.; Sekiya M.; Isobe M.; Kumura Y.; Ogita Z.; Hinoda Y.; Imai K.; Yachi A. Molecular cloning and chromosomal mapping of a human protein-tyrosine phosphatase LC-PTP. Biochem. Biophys. Res. Commun. 1992, 186, 1607–1615. [DOI] [PubMed] [Google Scholar]
- Gjorloff-Wingren A.; Saxena M.; Han S.; Wang X.; Alonso A.; Renedo M.; Oh P.; Williams S.; Schnitzer J.; Mustelin T. Subcellular localization of intracellular protein tyrosine phosphatases in T cells. Eur. J. Immunol. 2000, 30, 2412–2421. [DOI] [PubMed] [Google Scholar]
- Zanke B.; Squire J.; Griesser H.; Henry M.; Suzuki H.; Patterson B.; Minden M.; Mak T. W. A hematopoietic protein tyrosine phosphatase (HePTP) gene that is amplified and overexpressed in myeloid malignancies maps to chromosome 1q32.1. Leukemia 1994, 8, 236–244. [PubMed] [Google Scholar]
- Fonatsch C.; Haase D.; Freund M.; Bartels H.; Tesch H. Partial trisomy 1q. A nonrandom primary chromosomal abnormality in myelodysplastic syndromes?. Cancer Genet. Cytogenet. 1991, 56, 243–253. [DOI] [PubMed] [Google Scholar]
- Mamaev N.; Mamaeva S. E.; Pavlova V. A.; Patterson D. Combined trisomy 1q and monosomy 17p due to translocation t(1;17) in a patient with myelodysplastic syndrome. Cancer Genet. Cytogenet. 1988, 35, 21–25. [DOI] [PubMed] [Google Scholar]
- Saxena M.; Williams S.; Brockdorff J.; Gilman J.; Mustelin T. Inhibition of T cell signaling by mitogen-activated protein kinase-targeted hematopoietic tyrosine phosphatase (HePTP). J. Biol. Chem. 1999, 274, 11693–11700. [DOI] [PubMed] [Google Scholar]
- Saxena M.; Williams S.; Tasken K.; Mustelin T. Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase. Nat. Cell Biol. 1999, 1, 305–311. [DOI] [PubMed] [Google Scholar]
- Gronda M.; Arab S.; Iafrate B.; Suzuki H.; Zanke B. W. Hematopoietic protein tyrosine phosphatase suppresses extracellular stimulus-regulated kinase activation. Mol. Cell. Biol. 2001, 21, 6851–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettiford S. M.; Herbst R. The protein tyrosine phosphatase HePTP regulates nuclear translocation of ERK2 and can modulate megakaryocytic differentiation of K562 cells. Leukemia 2003, 17, 366–378. [DOI] [PubMed] [Google Scholar]
- Nika K.; Hyunh H.; Williams S.; Paul S.; Bottini N.; Tasken K.; Lombroso P. J.; Mustelin T. Haematopoietic protein tyrosine phosphatase (HePTP) phosphorylation by cAMP-dependent protein kinase in T-cells: dynamics and subcellular location. Biochem. J. 2004, 378, 335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Vossius S.; Rahmouni S.; Miletic A. V.; Vang T.; Vazquez-Rodriguez J.; Cerignoli F.; Arimura Y.; Williams S.; Hayes T.; Moutschen M.; Vasile S.; Pellecchia M.; Mustelin T.; Tautz L. Multidentate small-molecule inhibitors of vaccinia H1-related (VHR) phosphatase decrease proliferation of cervix cancer cells. J. Med. Chem. 2009, 52, 6716–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z.; Tabassum S.; Jiang W.; Zhang J.; Mathur S.; Wu J.; Shi Y. Identification of a potent inhibitor of human dual-specific phosphatase, VHR, from computer-aided and NMR-based screening to cellular effects. ChemBioChem 2007, 8, 2092–2099. [DOI] [PubMed] [Google Scholar]
- Usui T.; Kojima S.; Kidokoro S.; Ueda K.; Osada H.; Sodeoka M. Design and synthesis of a dimeric derivative of RK-682 with increased inhibitory activity against VHR, a dual-specificity ERK phosphatase: Implications for the molecular mechanism of the inhibition. Chem. Biol. 2001, 8, 1209–1220. [DOI] [PubMed] [Google Scholar]
- Vogt A.; Cooley K. A.; Brisson M.; Tarpley M. G.; Wipf P.; Lazo J. S. Cell-active dual specificity phosphatase inhibitors identified by high-content screening. Chem. Biol. 2003, 10, 733–742. [DOI] [PubMed] [Google Scholar]
- Tautz L.; Mustelin T. Strategies for developing protein tyrosine phosphatase inhibitors. Methods 2007, 42, 250–260. [DOI] [PubMed] [Google Scholar]
- Critton D. A.; Tortajada A.; Stetson G.; Peti W.; Page R. Structural basis of substrate recognition by hematopoietic tyrosine phosphatase. Biochemistry 2008, 47, 13336–13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. N.; Mortensen O. H.; Peters G. H.; Drake P. G.; Iversen L. F.; Olsen O. H.; Jansen P. G.; Andersen H. S.; Tonks N. K.; Moller N. P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 2001, 21, 7117–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nika K.; Charvet C.; Williams S.; Tautz L.; Bruckner S.; Rahmouni S.; Bottini N.; Schoenberger S. P.; Baier G.; Altman A.; Mustelin T. Lipid raft targeting of hematopoietic protein tyrosine phosphatase by protein kinase C theta-mediated phosphorylation. Mol. Cell. Biol. 2006, 26, 1806–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr A. J.; Ugochukwu E.; Lee W. H.; King O. N.; Filippakopoulos P.; Alfano I.; Savitsky P.; Burgess-Brown N. A.; Muller S.; Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidovic D.; Schurer S. C. Knowledge-based characterization of similarity relationships in the human protein-tyrosine phosphatase family for rational inhibitor design. J. Med. Chem. 2009, 52, 6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.